
Design, Synthesis and Cellular Characterization of a Dual Inhibitor of 5-Lipoxygenase and Soluble Epoxide Hydrolase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
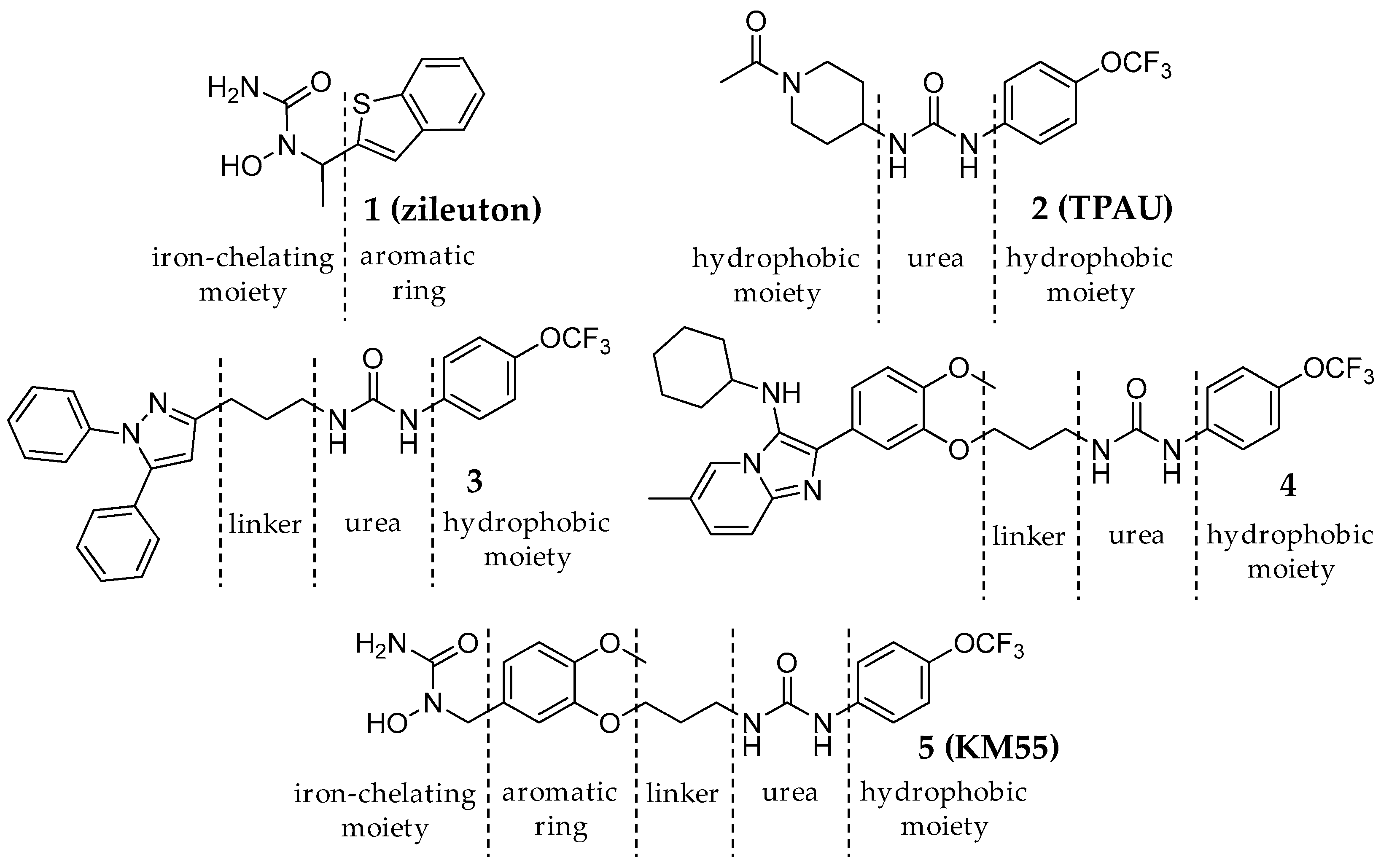
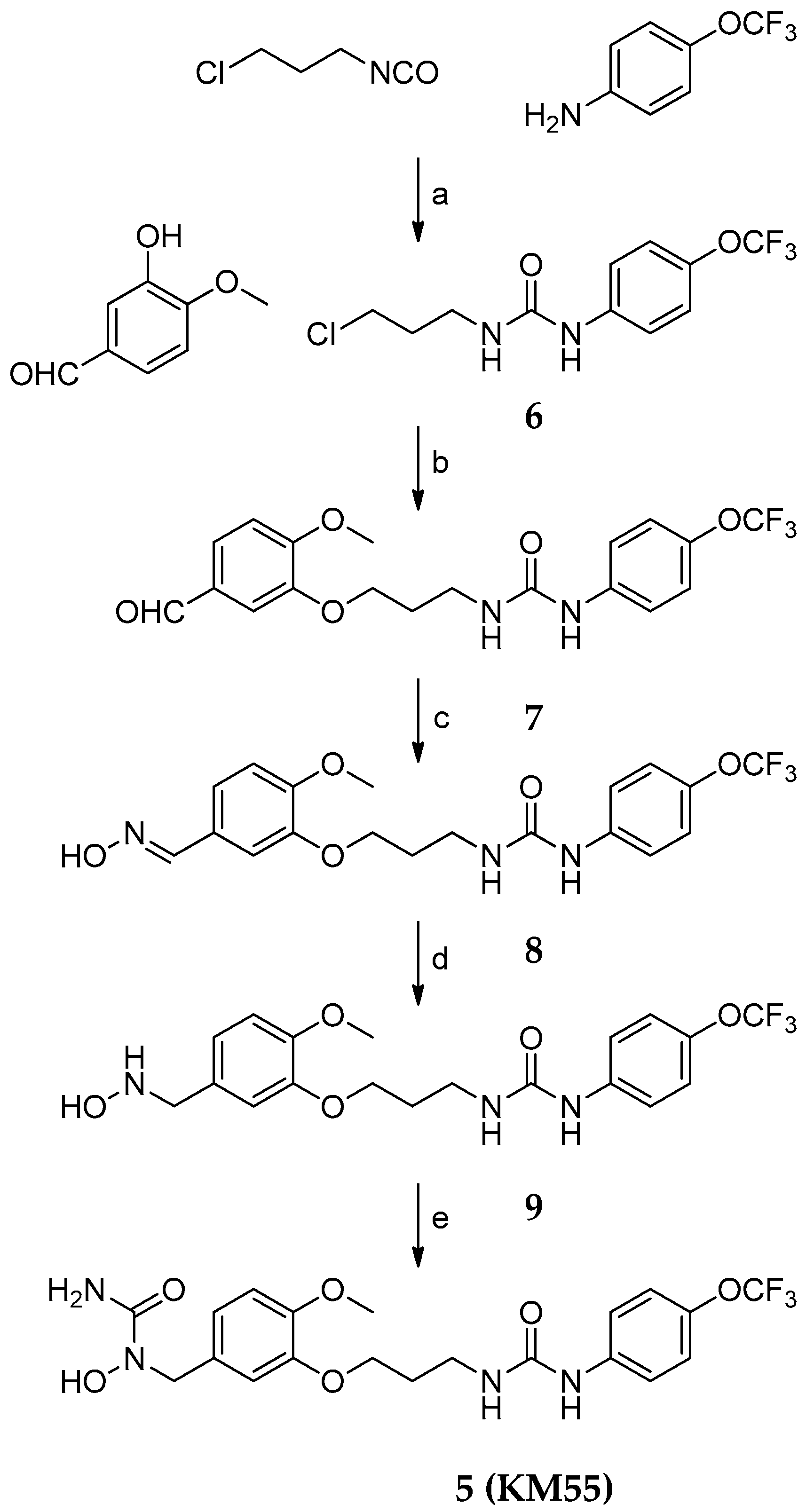
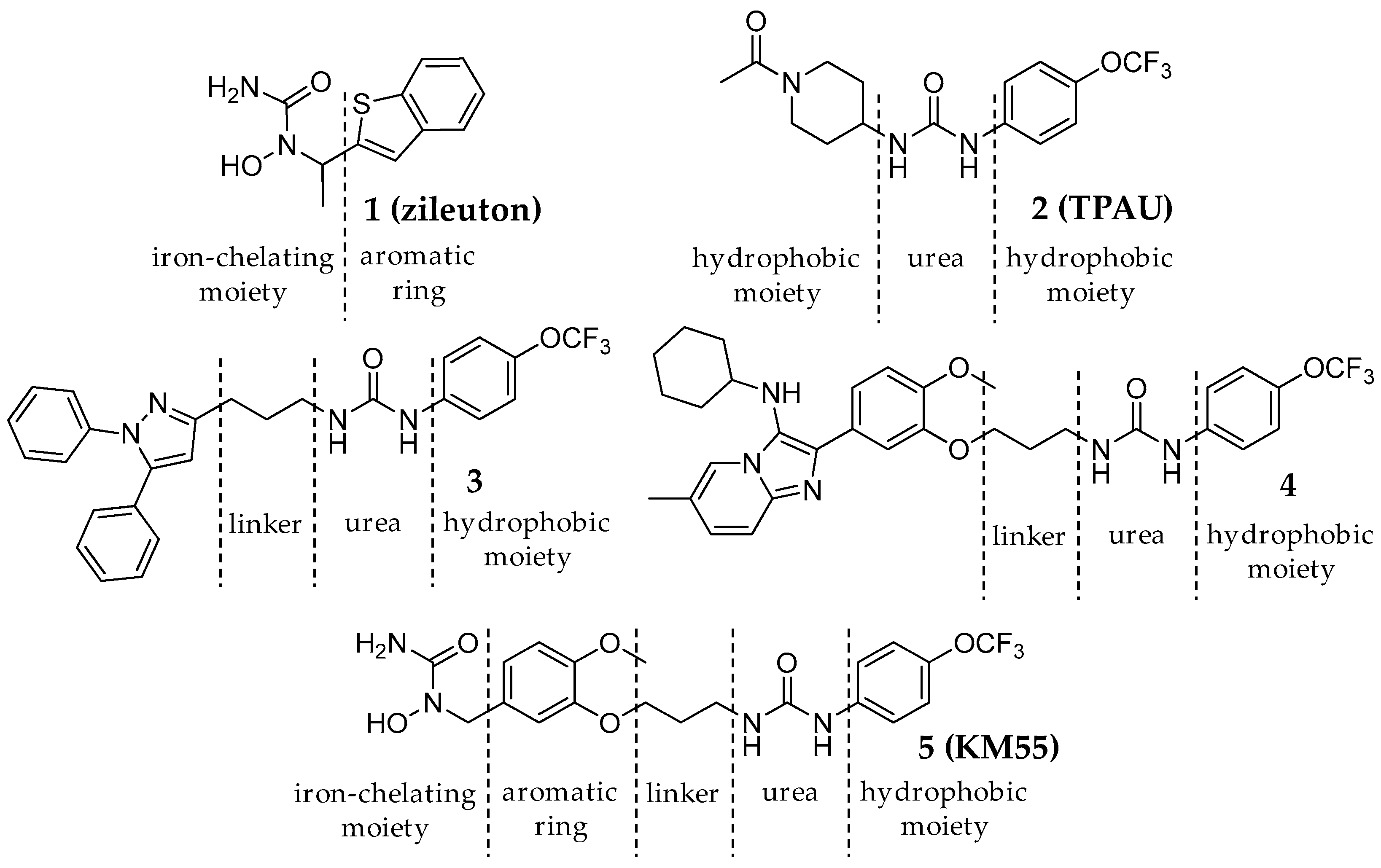
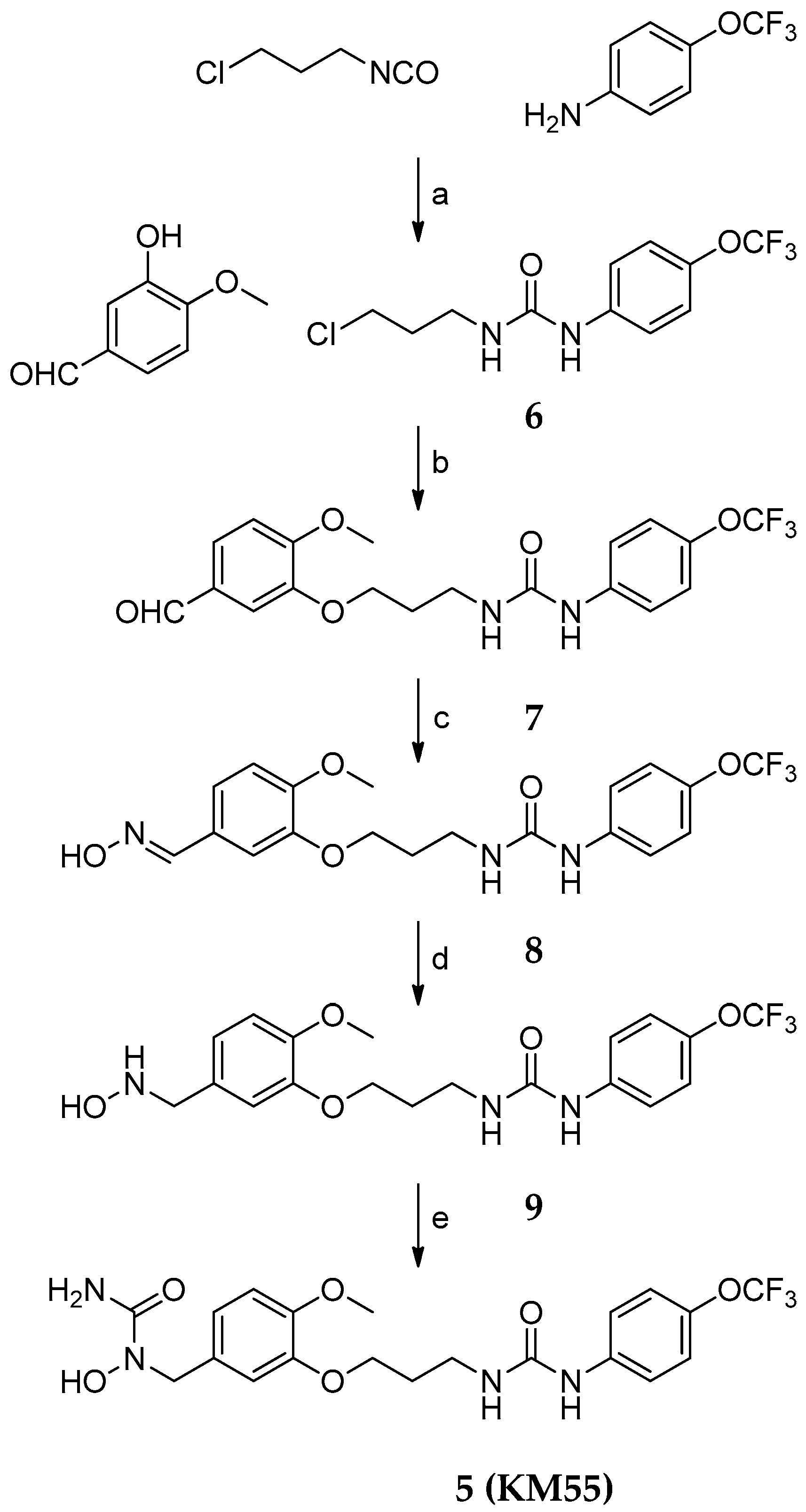
2.1. Design and Synthesis of KM55, a Dual sEH/5-LO Inhibitor
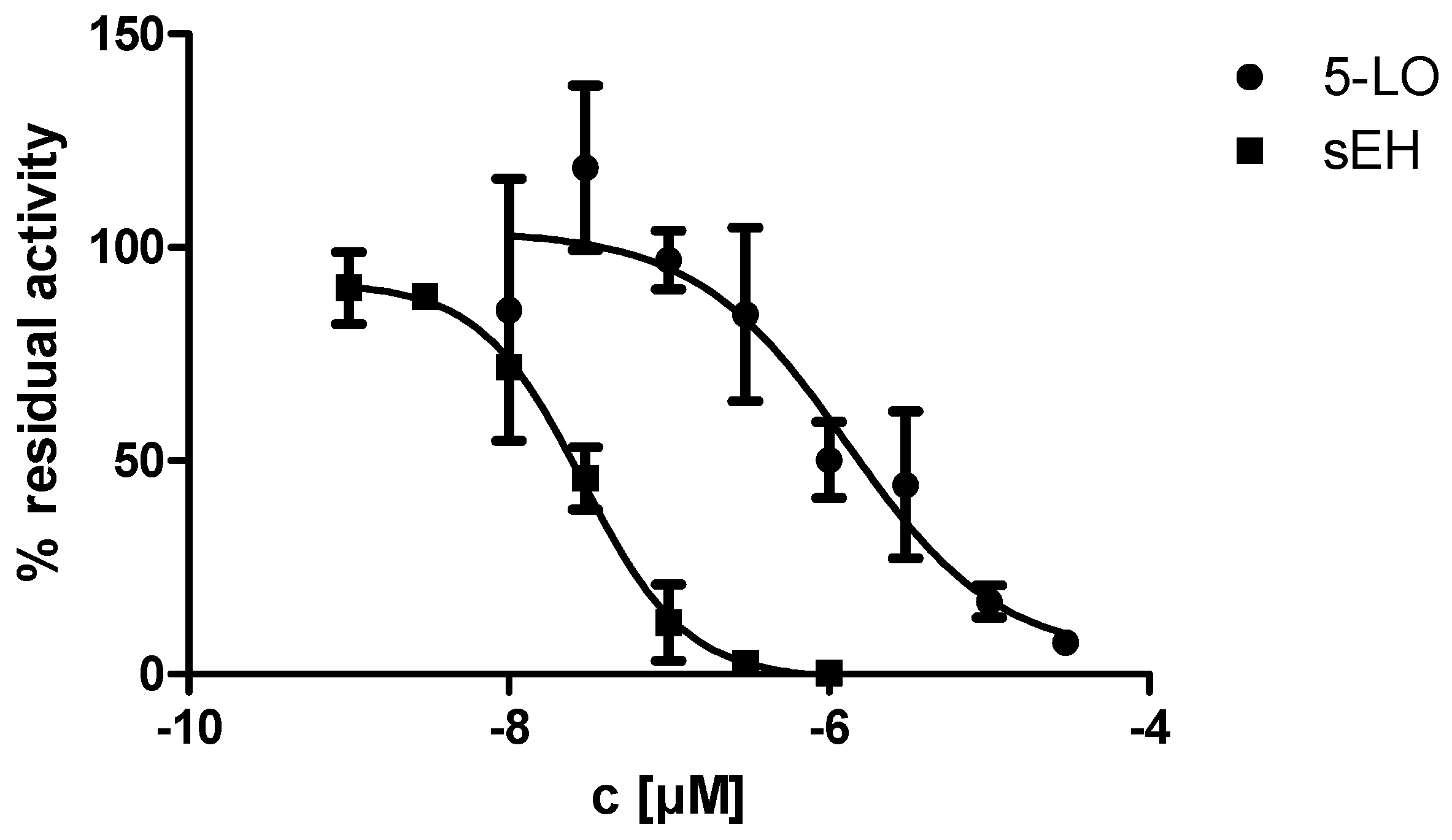
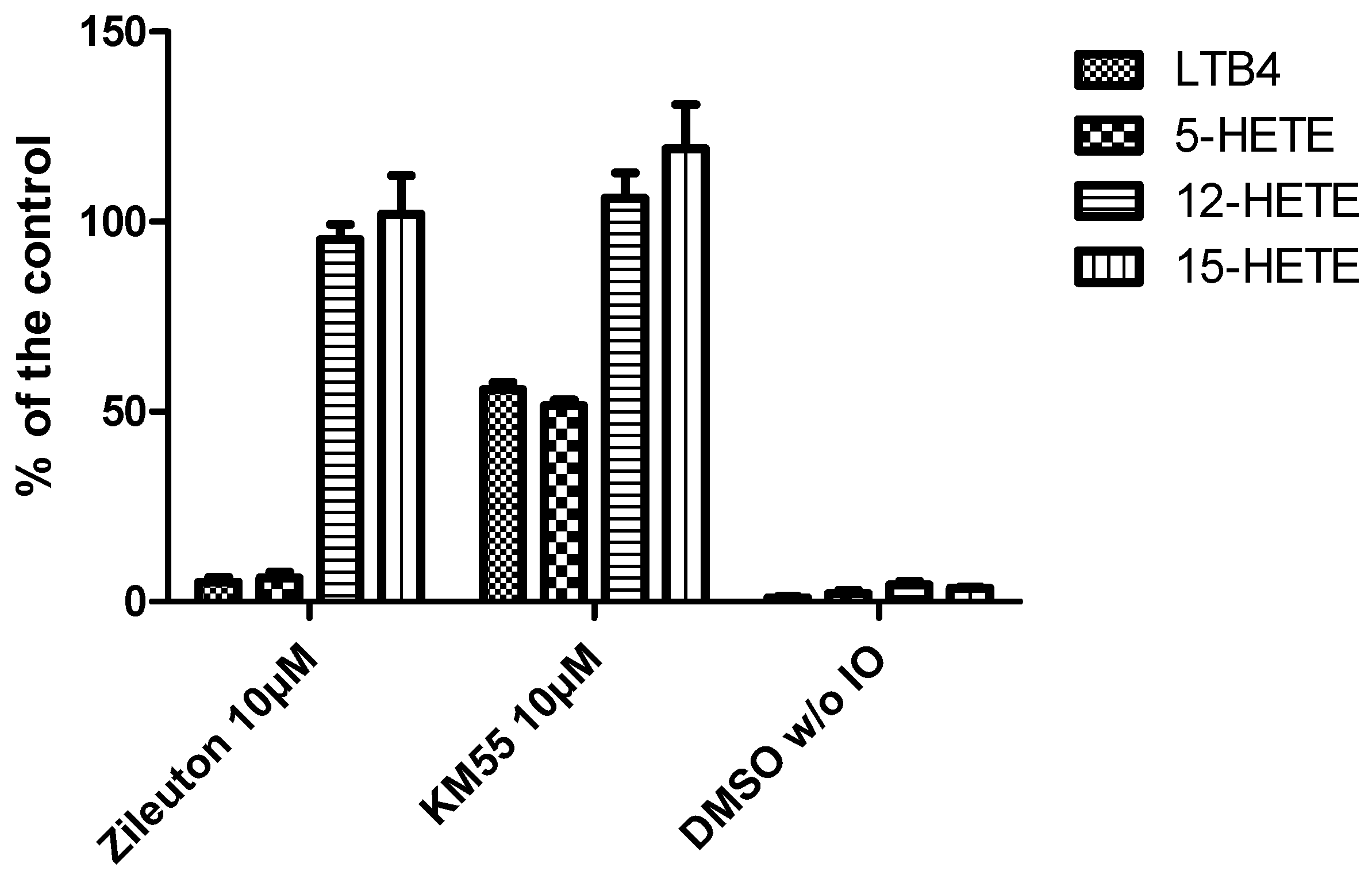
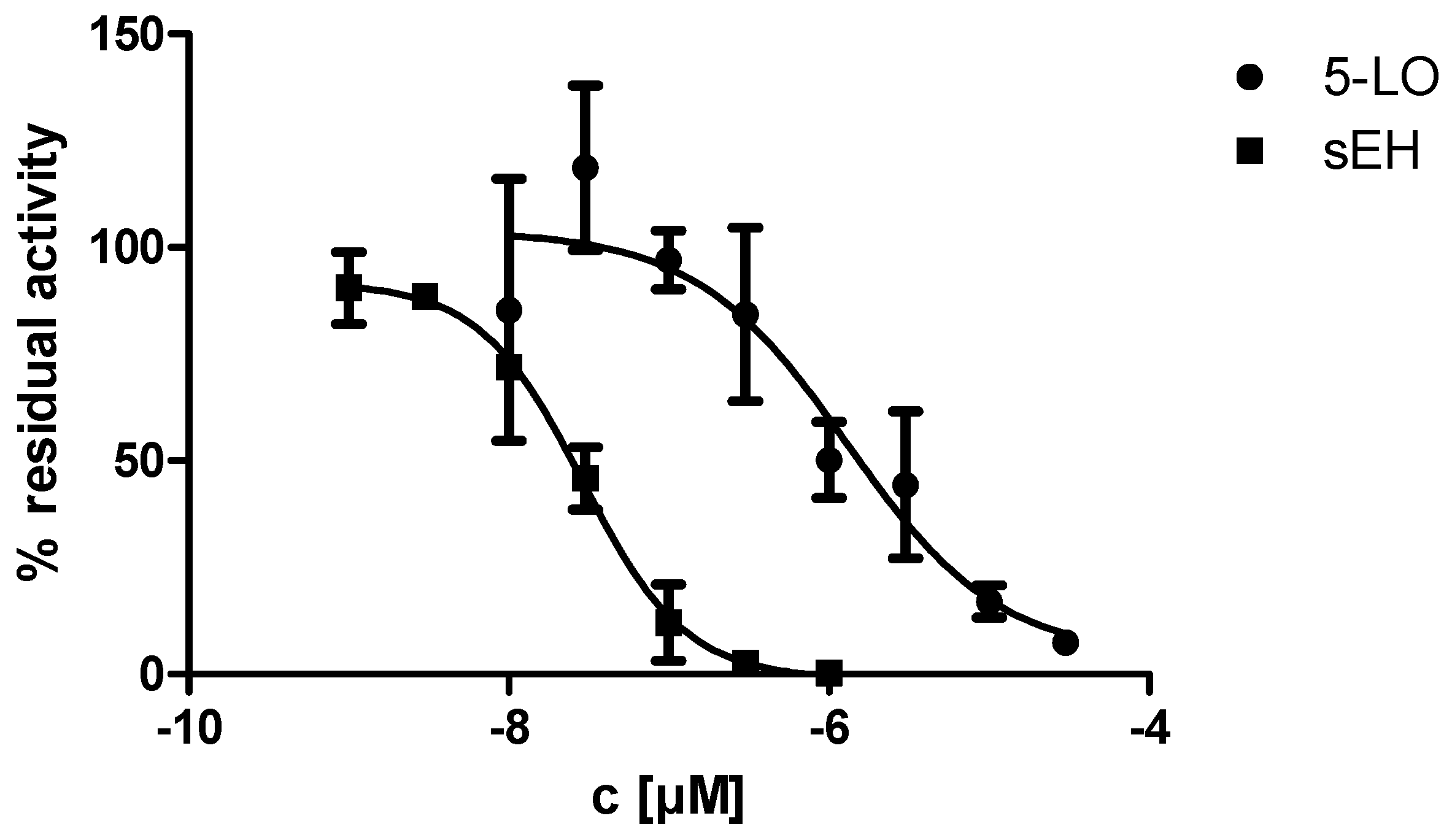
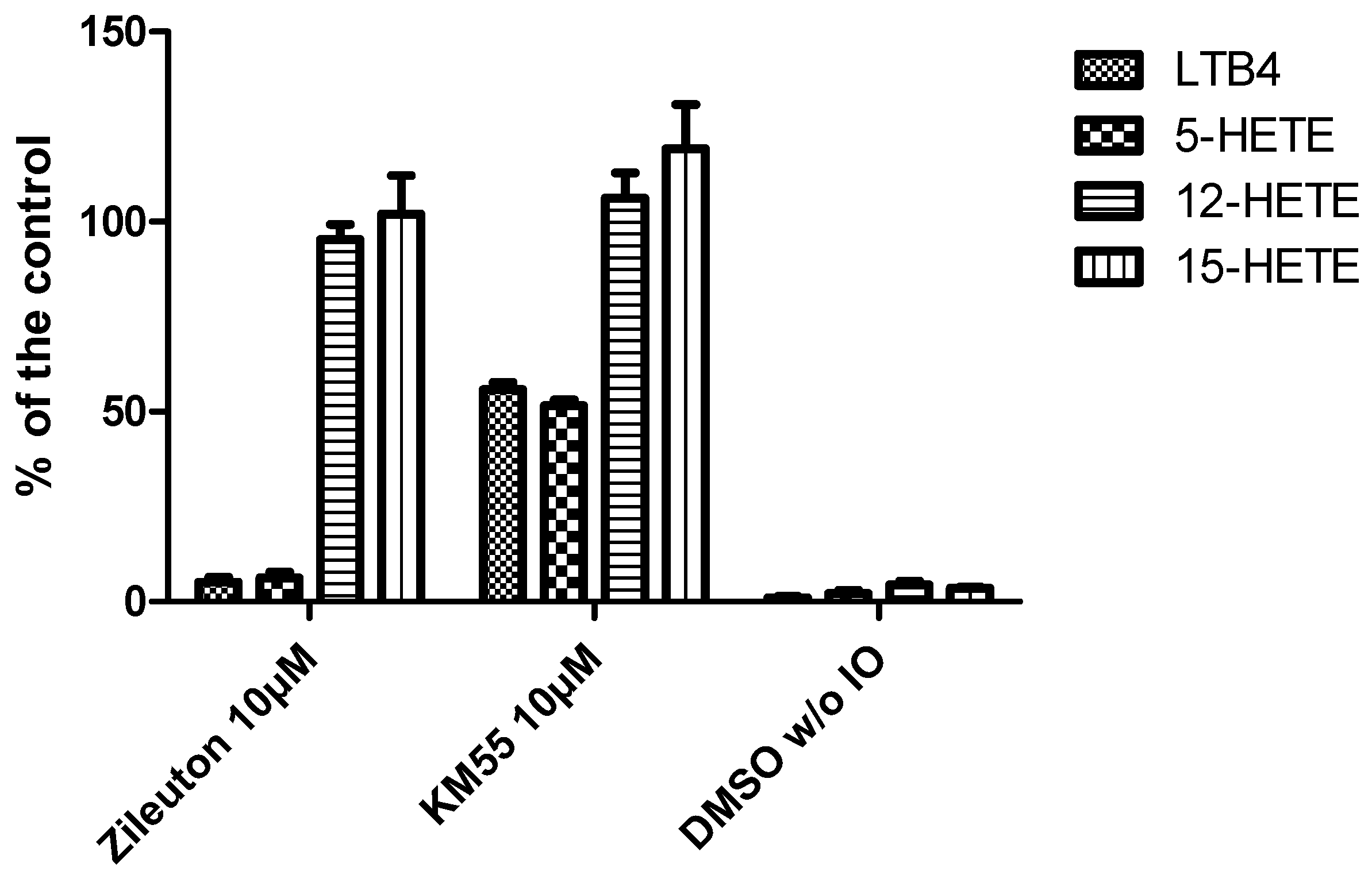
2.2. In Vitro Evalulation of KM55 on Recombinant Enzymes and in Human Whole Blood
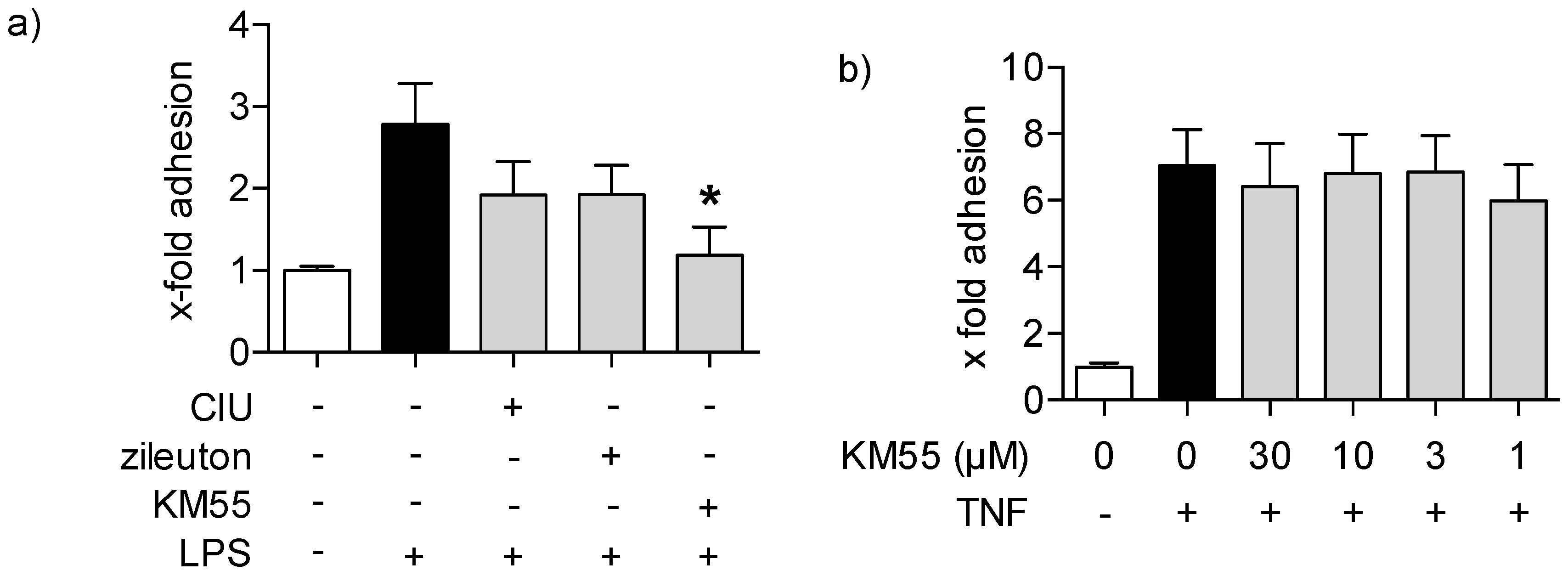
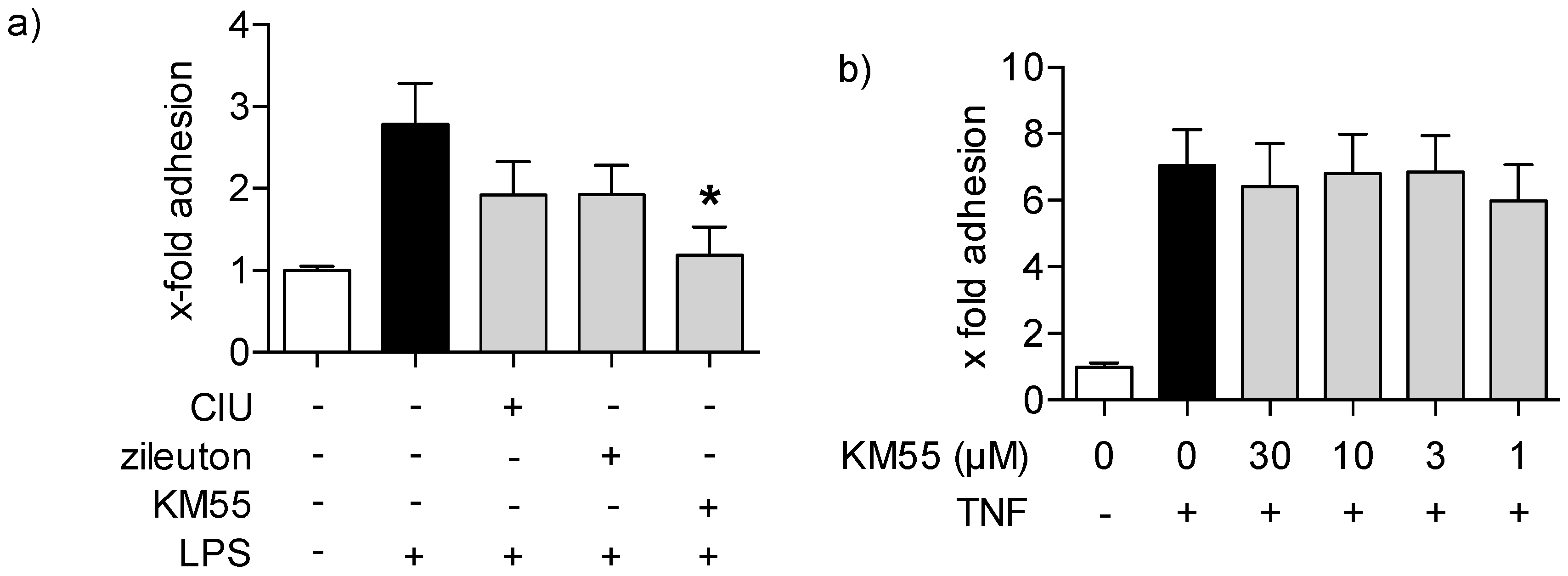
2.3. Effects of KM55 on Leukocyte-Endothelial Cell Interaction
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Synthesis
4.3. Recombinant sEH Enzyme Activity Assay
4.4. Recombinant 5-LO Activity Assay
4.5. Human Whole Blood Assay
4.6. Cell Culture
4.7. Cell Adhesion Assay
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dennis, E.A.; Cao, J.; Hsu, Y.-H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed]
- Piper, P.; Vane, J. The release of prostaglandins from lung and other tissues. Ann. N. Y. Acad. Sci. 1971, 180, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, B.; Dahlén, S.E.; Lindgren, J.A.; Rouzer, C.A.; Serhan, C.N. Leukotrienes and lipoxins: Structures, biosynthesis, and biological effects. Science 1987, 237, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. The pharmacology of the cytochrome P450 epoxygenase/soluble epoxide hydrolase axis in the vasculature and cardiovascular disease. Pharmacol. Rev. 2014, 66, 1106–1140. [Google Scholar] [CrossRef] [PubMed]
- Higashi, N.; Taniguchi, M.; Mita, H.; Yamaguchi, H.; Ono, E.; Akiyama, K. Aspirin-intolerant asthma (AIA) assessment using the urinary biomarkers, leukotriene E4 (LTE4) and prostaglandin D2 (PGD2) metabolites. Allergol. Int. 2012, 61, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.L.; Dunford, P.J.; Xue, X.; Jiang, X.; Lundeen, K.A.; Coles, F.; Riley, J.P.; Williams, K.N.; Grice, C.A.; Edwards, J.P.; et al. Anti-inflammatory activity of a potent, selective leukotriene A4 hydrolase inhibitor in comparison with the 5-lipoxygenase inhibitor zileuton. J. Pharmacol. Exp. Ther. 2007, 321, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, C.; Homann, J.; Ball, A.-K.; Blöcher, R.; Kleinschmidt, T.K.; Basavarajappa, D.; Angioni, C.; Ferreirós, N.; Häfner, A.-K.; Rådmark, O.; et al. Lipoxin and resolvin biosynthesis is dependent on 5-lipoxygenase activating protein. FASEB J. 2015, 29, 5029–5043. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, K.R.; Kubala, L.; Newman, J.W.; Kim, I.-H.; Eiserich, J.P.; Hammock, B.D. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9772–9777. [Google Scholar] [CrossRef] [PubMed]
- Jung, O.; Jansen, F.; Mieth, A.; Barbosa-Sicard, E.; Pliquett, R.U.; Babelova, A.; Morisseau, C.; Hwang, S.H.; Tsai, C.; Hammock, B.D.; et al. Inhibition of the soluble epoxide hydrolase promotes albuminuria in mice with progressive renal disease. PLoS ONE 2010, 5, e11979. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Y.; Yang, J.; Inceoglu, B.; Qiu, H.; Ulu, A.; Hwang, S.-H.; Chiamvimonvat, N.; Hammock, B.D. Inhibition of soluble epoxide hydrolase enhances the anti-inflammatory effects of aspirin and 5-lipoxygenase activation protein inhibitor in a murine model. Biochem. Pharmacol. 2010, 79, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rankovic, Z. The physicochemical challenges of designing multiple ligands. J. Med. Chem. 2006, 49, 4961–4970. [Google Scholar] [CrossRef] [PubMed]
- Meirer, K.; Steinhilber, D.; Proschak, E. Inhibitors of the arachidonic acid cascade: Interfering with multiple pathways. Basic Clin. Pharmacol. Toxicol. 2014, 114, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, A.; Werz, O. Multi-target approach for natural products in inflammation. Drug Discov. Today 2014, 19, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.H.; Wecksler, A.T.; Wagner, K.; Hammock, B.D. Rationally designed multitarget agents against inflammation and pain. Curr. Med. Chem. 2013, 20, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Moser, D.; Wisniewska, J.M.; Hahn, S.; Achenbach, J.; Buscató, E.L.; Klingler, F.-M.; Hofmann, B.; Steinhilber, D.; Proschak, E. Dual-target virtual screening by pharmacophore elucidation and molecular shape filtering. ACS Med. Chem. Lett. 2012, 3, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Meirer, K.; Rödl, C.B.; Wisniewska, J.M.; George, S.; Häfner, A.-K.; Buscató, E.; Klingler, F.-M.; Hahn, S.; Berressem, D.; Wittmann, S.K.; et al. Synthesis and structure-activity relationship studies of novel dual inhibitors of soluble epoxide hydrolase and 5-lipoxygenase. J. Med. Chem. 2013, 56, 1777–1781. [Google Scholar] [CrossRef] [PubMed]
- Achenbach, J.; Klingler, F.-M.; Blöcher, R.; Moser, D.; Häfner, A.-K.; Rödl, C.B.; Kretschmer, S.; Krüger, B.; Löhr, F.; Stark, H.; et al. Exploring the chemical space of multitarget ligands using aligned self-organizing maps. ACS Med. Chem. Lett. 2013, 4, 1169–1172. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.L.; Young, P.R.; Albert, D.; Lanni, C.; Summers, J.B.; Brooks, D.W.; Rubin, P.; Carter, G.W. The discovery and development of zileuton: An orally active 5-lipoxygenase inhibitor. Int. J. Immunopharmacol. 1992, 14, 505–510. [Google Scholar] [CrossRef]
- Kim, I.-H.; Morisseau, C.; Watanabe, T.; Hammock, B.D. Design, synthesis, and biological activity of 1,3-disubstituted ureas as potent inhibitors of the soluble epoxide hydrolase of increased water solubility. J. Med. Chem. 2004, 47, 2110–2122. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.E.; Morisseau, C.; Liu, J.-Y.; Inceoglu, B.; Jones, P.D.; Sanborn, J.R.; Hammock, B.D. 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: Structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J. Med. Chem. 2010, 53, 7067–7075. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.H.; Wagner, K.M.; Morisseau, C.; Liu, J.-Y.; Dong, H.; Wecksler, A.T.; Hammock, B.D. Synthesis and structure-activity relationship studies of urea-containing pyrazoles as dual inhibitors of cyclooxygenase-2 and soluble epoxide hydrolase. J. Med. Chem. 2011, 54, 3037–3050. [Google Scholar] [CrossRef] [PubMed]
- Malamas, M.S.; Carlson, R.P.; Grimes, D.; Howell, R.; Glaser, K.; Gunawan, I.; Nelson, J.A.; Kanzelberger, M.; Shah, U.; Hartman, D.A. Azole phenoxy hydroxyureas as selective and orally active inhibitors of 5-lipoxygenase. J. Med. Chem. 1996, 39, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Gomez, G.A.; Morisseau, C.; Hammock, B.D.; Christianson, D.W. Structure of Human Epoxide Hydrolase Reveals Mechanistic Inferences on Bifunctional Catalysis in Epoxide and Phosphate Ester Hydrolysis. Biochemistry 2004, 43, 4716–4723. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.C.; Hammock, B.D. Discovery of inhibitors of soluble epoxide hydrolase: A target with multiple potential therapeutic indications. J. Med. Chem. 2012, 55, 1789–1808. [Google Scholar] [CrossRef] [PubMed]
- Morisseau, C.; Goodrow, M.H.; Dowdy, D.; Zheng, J.; Greene, J.F.; Sanborn, J.R.; Hammock, B.D. Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc. Natl. Acad. Sci. USA 1999, 96, 8849–8854. [Google Scholar] [CrossRef] [PubMed]
- Claesson, H.E.; Haeggström, J. Human endothelial cells stimulate leukotriene synthesis and convert granulocyte released leukotriene A4 into leukotrienes B4, C4, D4 and E4. Eur. J. Biochem. 1988, 173, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Revtyak, G.E.; Johnson, A.R.; Campbell, W.B. Cultured bovine coronary arterial endothelial cells synthesize HETEs and prostacyclin. Am. J. Physiol. 1988, 254, C8–C19. [Google Scholar] [PubMed]
- Wolf, N.M.; Morisseau, C.; Jones, P.D.; Hock, B.; Hammock, B.D. Development of a high-throughput screen for soluble epoxide hydrolase inhibition. Anal. Biochem. 2006, 355, 71–80. [Google Scholar] [CrossRef] [PubMed]
- M Kretschmer, S.B.; Woltersdorf, S.; Rödl, C.B.; Vogt, D.; Häfner, A.-K.; Steinhilber, D.; Stark, H.; Hofmann, B. Development of novel aminothiazole-comprising 5-LO inhibitors. Future Med. Chem. 2016, 8, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.J.; Tausch, L.; Hoernig, M.; Coste, O.; Schmidt, R.; Angioni, C.; Metzner, J.; Groesch, S.; Pergola, C.; Steinhilber, D.; et al. Celecoxib inhibits 5-lipoxygenase. Biochem. Pharmacol. 2008, 76, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compound KM55 are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meirer, K.; Glatzel, D.; Kretschmer, S.; Wittmann, S.K.; Hartmann, M.; Blöcher, R.; Angioni, C.; Geisslinger, G.; Steinhilber, D.; Hofmann, B.; et al. Design, Synthesis and Cellular Characterization of a Dual Inhibitor of 5-Lipoxygenase and Soluble Epoxide Hydrolase. Molecules 2017, 22, 45. https://doi.org/10.3390/molecules22010045
Meirer K, Glatzel D, Kretschmer S, Wittmann SK, Hartmann M, Blöcher R, Angioni C, Geisslinger G, Steinhilber D, Hofmann B, et al. Design, Synthesis and Cellular Characterization of a Dual Inhibitor of 5-Lipoxygenase and Soluble Epoxide Hydrolase. Molecules. 2017; 22(1):45. https://doi.org/10.3390/molecules22010045
Chicago/Turabian StyleMeirer, Karin, Daniel Glatzel, Simon Kretschmer, Sandra K. Wittmann, Markus Hartmann, René Blöcher, Carlo Angioni, Gerd Geisslinger, Dieter Steinhilber, Bettina Hofmann, and et al. 2017. "Design, Synthesis and Cellular Characterization of a Dual Inhibitor of 5-Lipoxygenase and Soluble Epoxide Hydrolase" Molecules 22, no. 1: 45. https://doi.org/10.3390/molecules22010045
APA StyleMeirer, K., Glatzel, D., Kretschmer, S., Wittmann, S. K., Hartmann, M., Blöcher, R., Angioni, C., Geisslinger, G., Steinhilber, D., Hofmann, B., Fürst, R., & Proschak, E. (2017). Design, Synthesis and Cellular Characterization of a Dual Inhibitor of 5-Lipoxygenase and Soluble Epoxide Hydrolase. Molecules, 22(1), 45. https://doi.org/10.3390/molecules22010045