
Spectroscopic, DFT, and XRD Studies of Hydrogen Bonds in N-Unsubstituted 2-Aminobenzamides

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
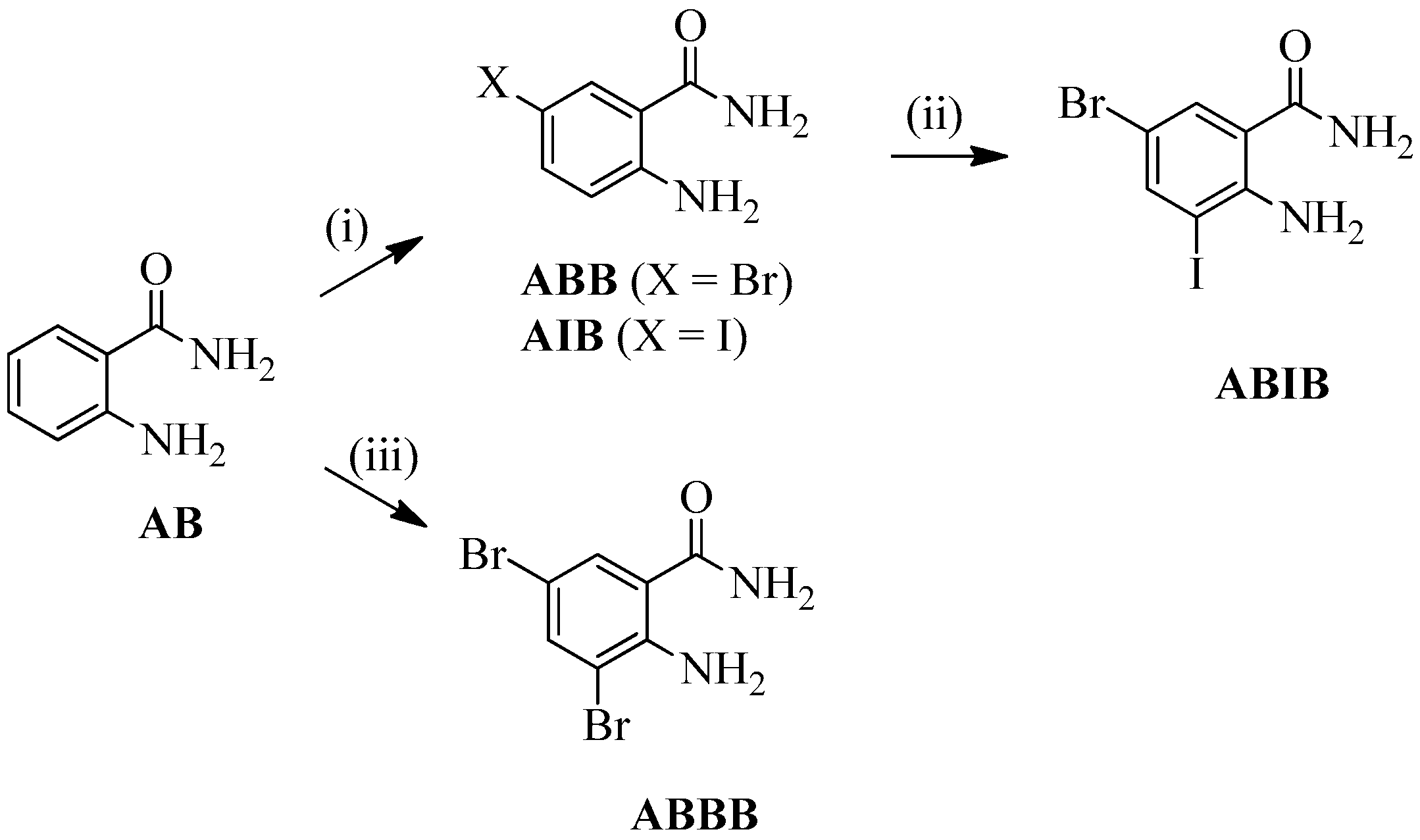
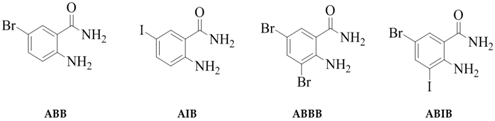
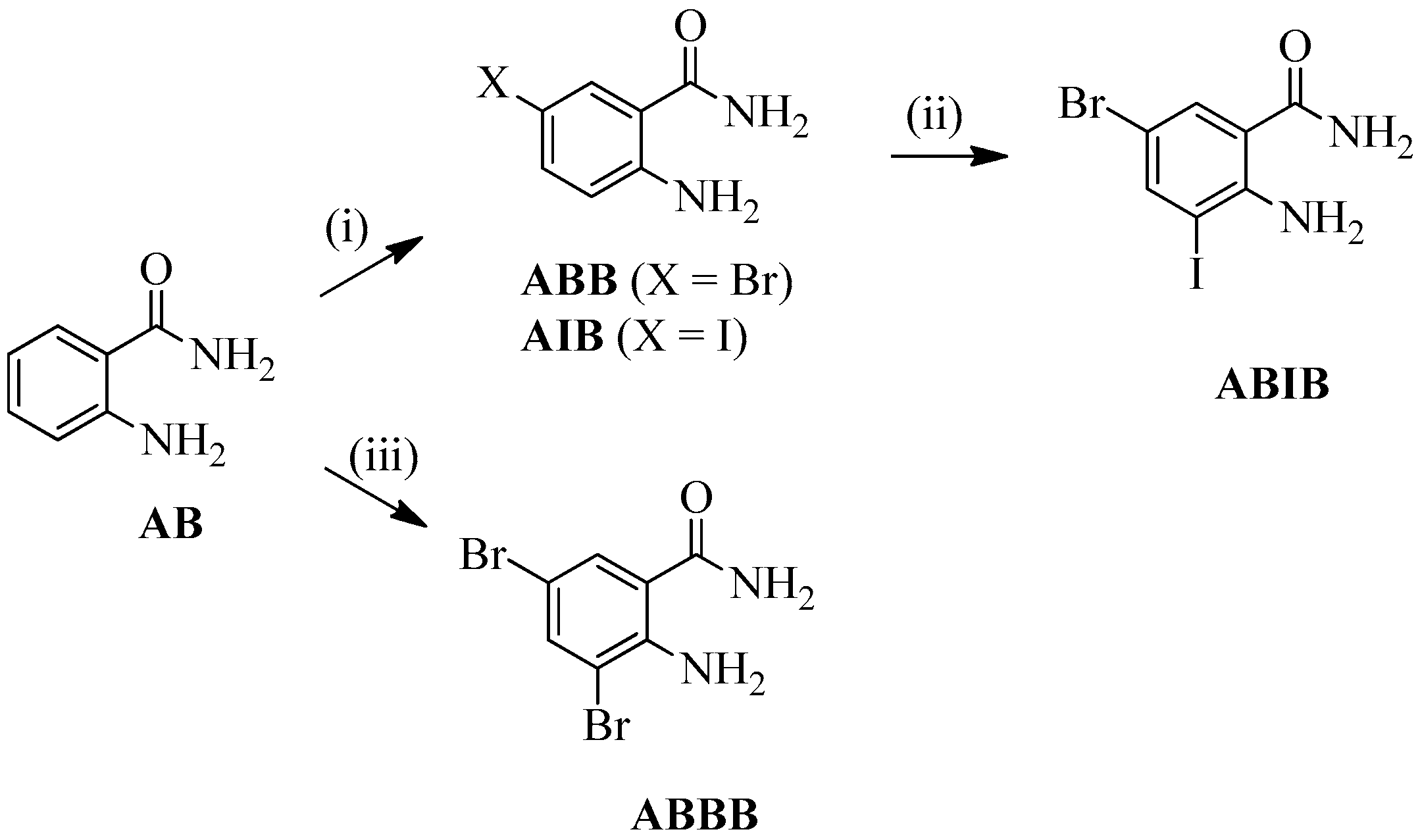
2.1. Synthesis
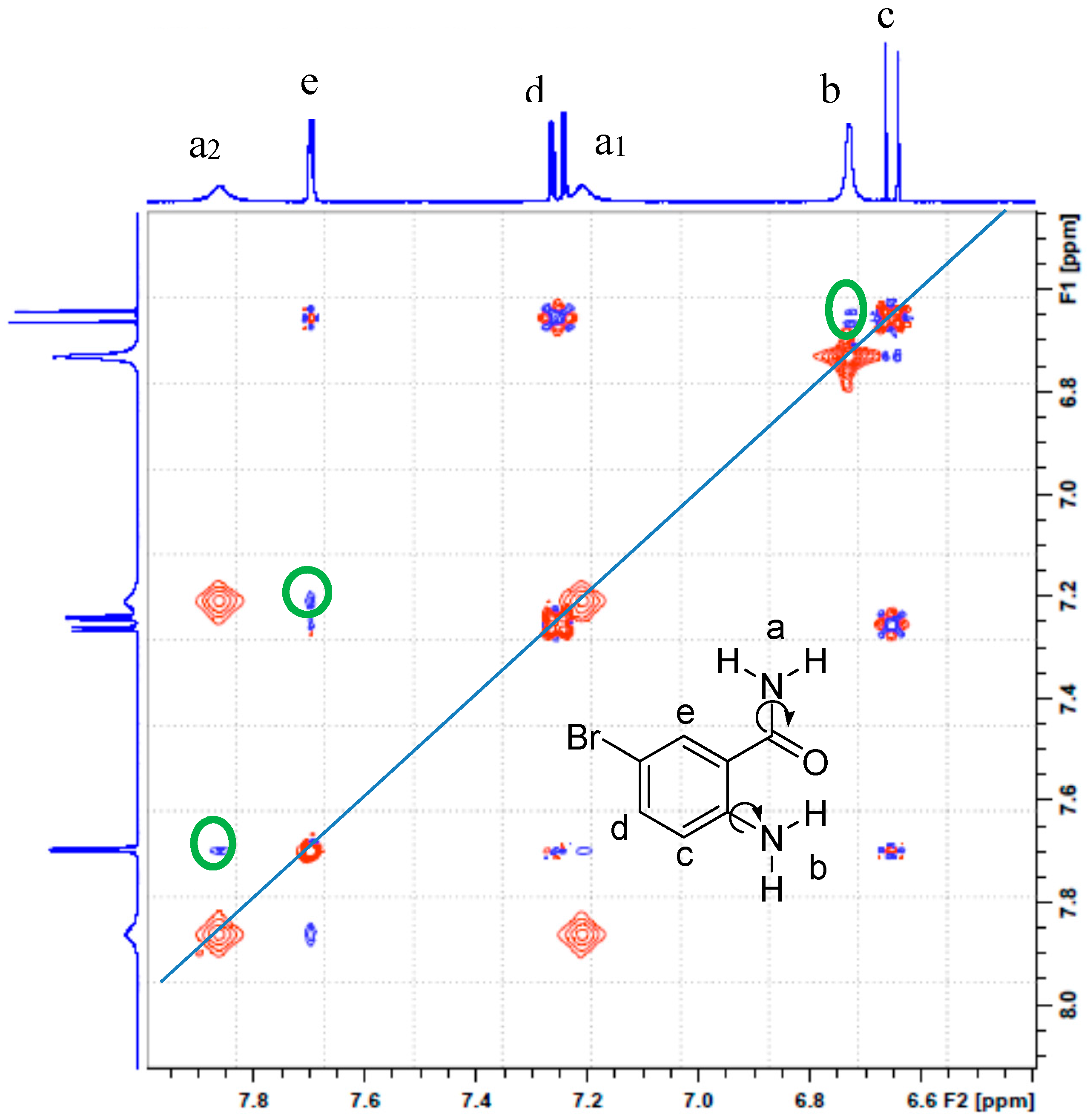
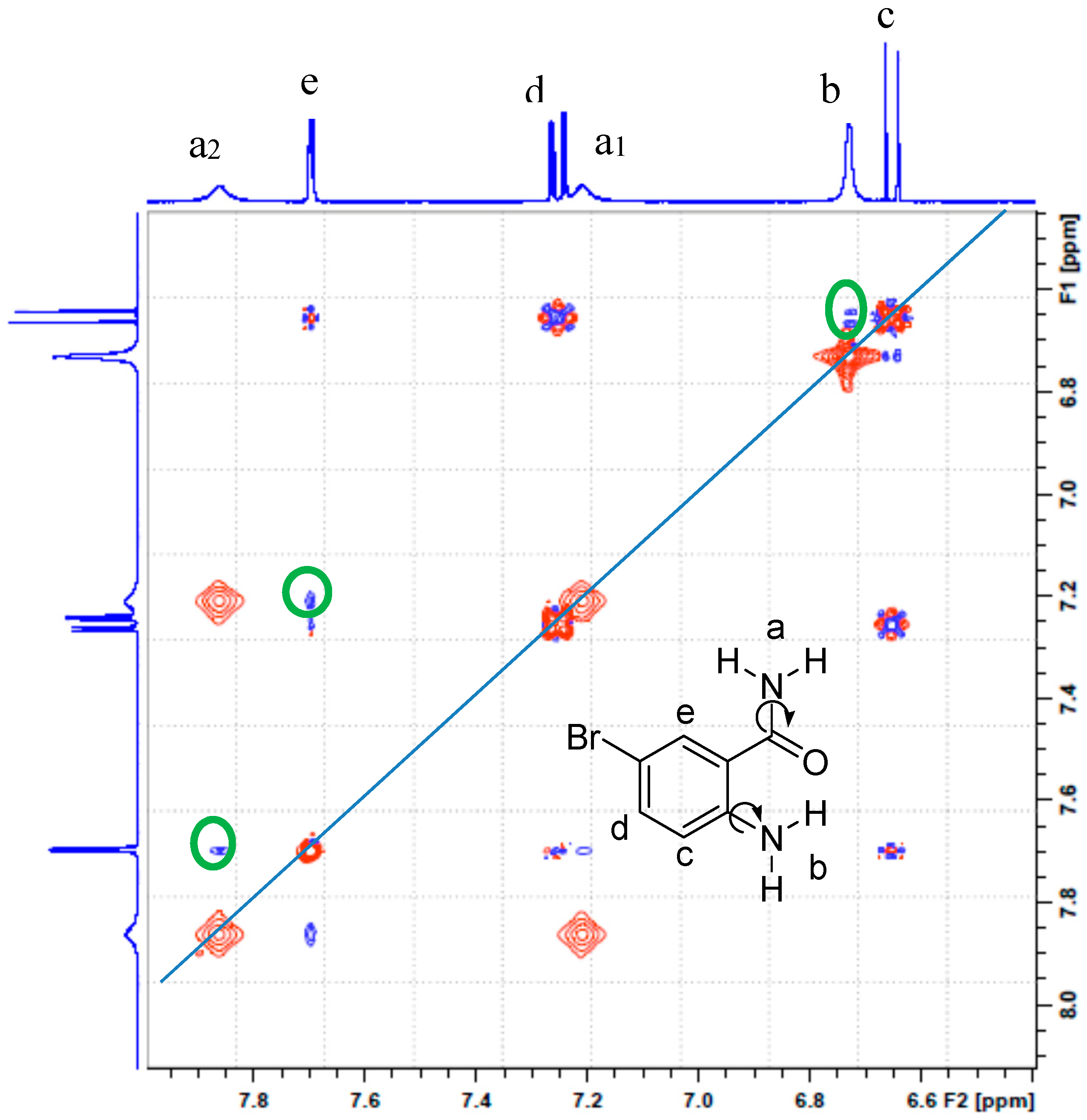
2.2. Solution Phase Studies Using 1H-NMR and UV-Vis Spectroscopy
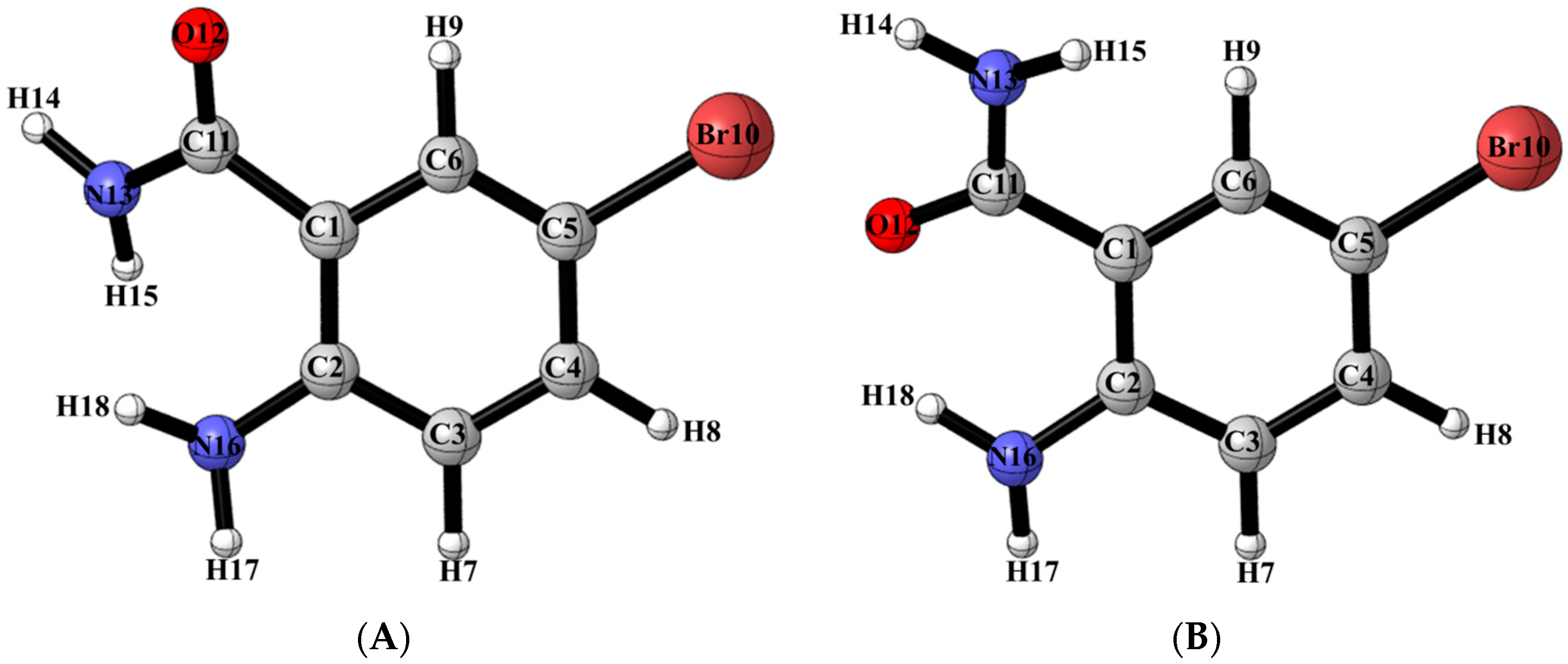
2.3. DFT Studies
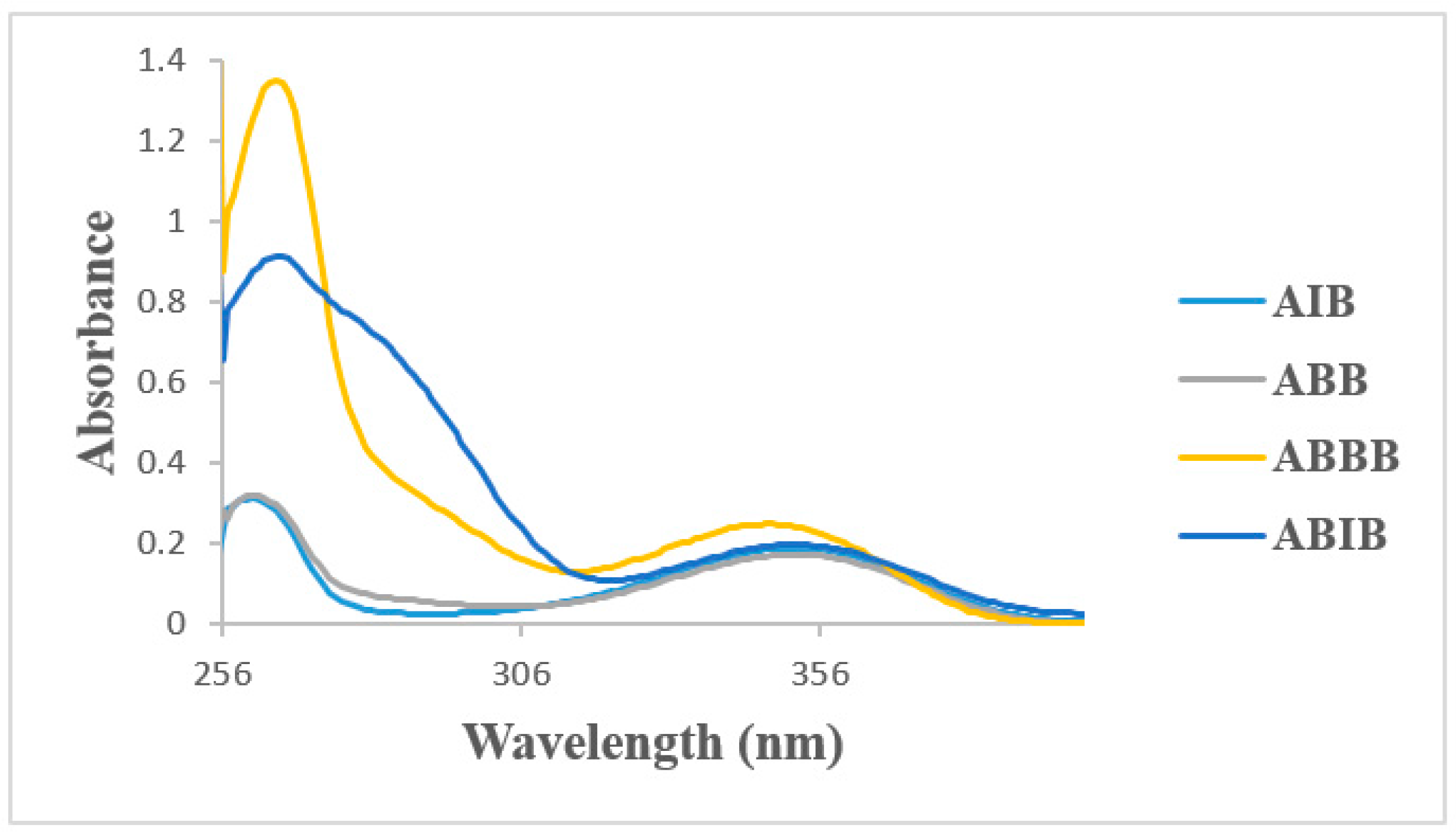
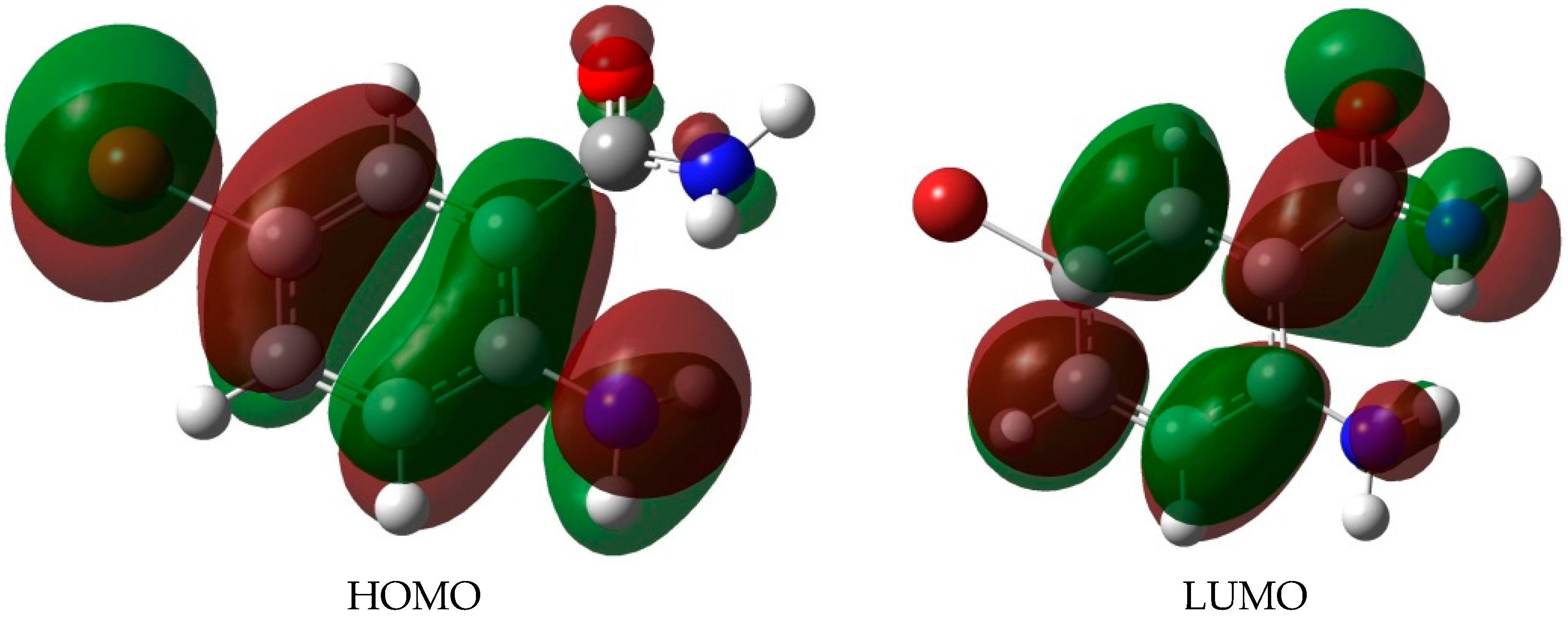
2.4. UV-VIS Spectroscopic Studies
2.5. Solid State Studies Using IR and Raman Spectroscopy
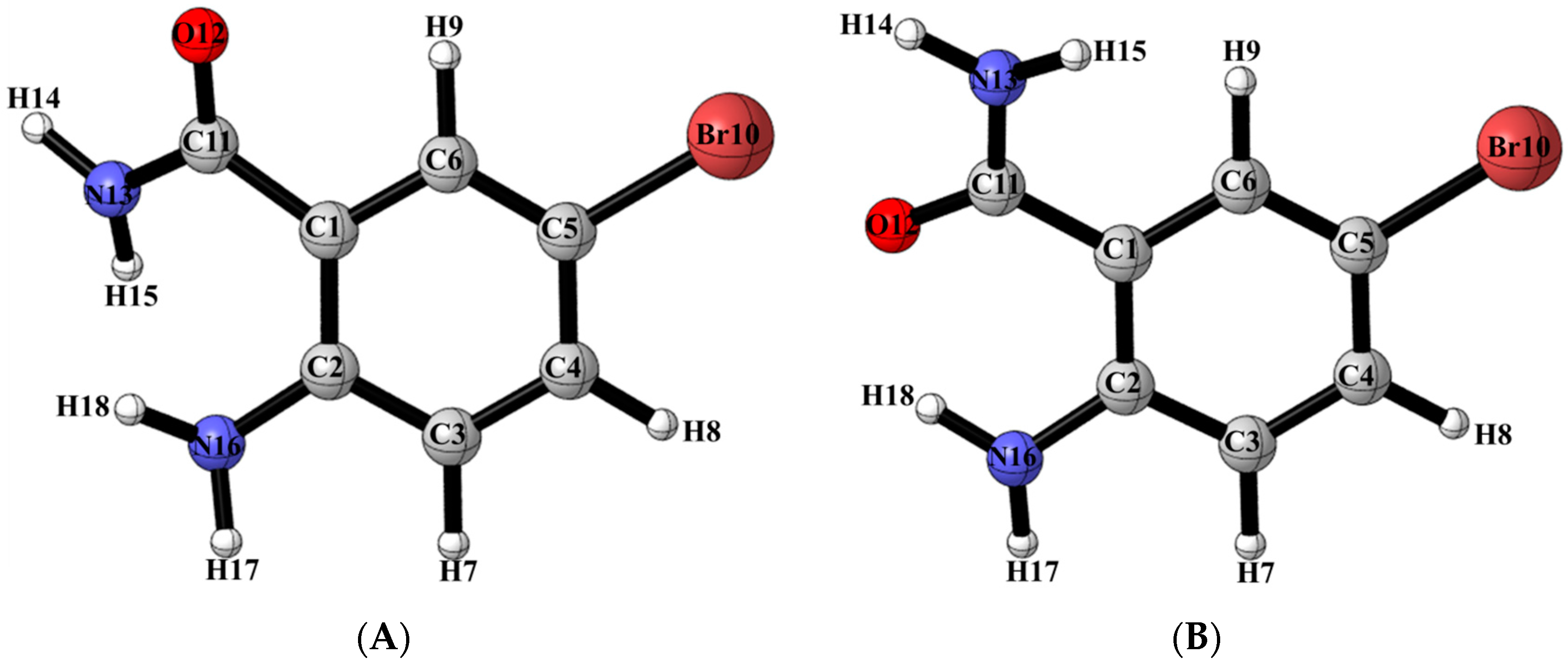
2.6. Solid State Studies of ABB Using X-ray Crystallography
3. Experimental
3.1. Spectroscopic Analysis
3.2. Single X-ray Data Collection and Processing
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hadzi, D.; Kidric, J.; Koller, J.; Mavrij, J. The role of hydrogen bonding in drug-receptor interactions. J. Mol. Struct. 1990, 237, 139–150. [Google Scholar] [CrossRef]
- Kool, E.T. Hydrogen bonding, base stacking, and steric effects in DNA replication. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Gorobets, N.Y.; Yermolayev, S.A.; Gurley, T.; Gurinov, A.A.; Tolstoy, P.M.; Shenderovich, I.G.; Leadbeater, N.E. Difference between 1H-NMR signals of primary amide protons as a simple spectral index of the amide intramolecular hydrogen bond strength. J. Phys. Org. Chem. 2012, 25, 287–295. [Google Scholar] [CrossRef]
- Dado, G.P.; Gellman, S.H. Intramolecular hydrogen bonding in derivatives of beta-alanine and gamma-amino butyric acid; model studies for the folding of unnatural polypeptide backbones. J. Am. Chem. Soc. 1994, 116, 1054–1062. [Google Scholar] [CrossRef]
- Fu, H.L.; Liu, Y.; Zeng, H.Q. Shape-persistent H-bonded macrocyclic aromatic pentamers. Chem. Commun. 2013, 49, 4127–4144. [Google Scholar] [CrossRef]
- Yan, Y.; Zhao, W.W.; Bhagavathy, G.V.; Faurie, A.; Misey, N.J.; Petitjean, A. Controlled synthesis and alkaline earth ion binding of switchable formamidoxime-based crown ether analogs. Chem. Commun. 2012, 48, 7829–7835. [Google Scholar] [CrossRef] [PubMed]
- Hamuro, K.; Geib, S.J.; Hamilton, A.D. Novel molecular scaffolds: Formation of helical secondary structure in a family of oligoanthranilamides. Angew. Chem. Int. Ed. 1994, 33, 446–448. [Google Scholar] [CrossRef]
- Hamuro, K.; Geib, S.J.; Hamilton, A.D. Oligoanthranilamides. Non-peptide subunits that show formation of specific secondary structure. J. Am. Chem. Soc. 1996, 118, 7529–7541. [Google Scholar] [CrossRef]
- Hamuro, K.; Geib, S.J.; Hamilton, A.D. Novel folding patterns in a family of oligoanthranilamides: Non-peptide oligomers that form extended helical secondary structures. J. Am. Chem. Soc. 1997, 119, 10587–10593. [Google Scholar] [CrossRef]
- Etter, M.C. Hydrogen bond rearrangements in organic solids. Part 2. Cyclodehydration of O-acetamidobenzamide. J. Chem. Soc. Perkin Trans. 1983, 2, 115–121. [Google Scholar] [CrossRef]
- Koshio, H.; Hirayama, F.; Ishihara, T.; Shiraki, R.; Shigenaga, T.; Taniuchi, Y.; Sato, K.; Moritani, Y.; Iwatsuki, Y.; Kaku, S.; et al. Synthesis and biological activity of novel 1,2-disubstituted benzene derivatives as factor Xa inhibitors. Bioorg. Med. Chem. 2005, 13, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yuan, J.; Fu, X.; Meng, F.; Zhang, S.; Xu, W.; Xu, Y.; Huang, C. Novel anthranilamide-based FXa Inhibitors: Drug design, synthesis and biological evaluation. Molecules 2016, 21, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Inoue, N.; Kutsukake, T.; Matsuki, S.; Takeuchi, M. Labeling conditions using a 2-aminobenzamide reagent for quantitative analysis of sialo-oligosaccharides. Biol. Pharm. Bull. 2000, 23, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Bao, D.; Upadhyayula, S.; Jones, G.; Vullev, V.I. Anthranilamides as bioinspired molecular electrets: Experimental evidence for a permanent ground-state electric dipole moment. J. Org. Chem. 2013, 78, 1994–2004. [Google Scholar] [CrossRef] [PubMed]
- Dharmaraja, J.; Esakkidurai, T.; Subbaraj, P.; Shobana, S. Mixed ligand complex formation of 2-aminobenzamide with Cu(II) in the presence of some amino acids: Synthesis, structural, biological, pH-metric, spectrophotometric and thermodynamic studies. Spectrochim. Acta A 2013, 114, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Dharmaraja, J.; Subbaraj, P.; Esakkidurai, T.; Shobana, S. Coordination behavior and bio-potent aspects of Ni(II) with 2-aminobenzamide and some amino acid mixed ligands—Part II: Synthesis, spectral, morphological, pharmacological and DNA interaction studies. Spectrochim. Acta A 2014, 132, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Mabkhot, Y.N.; Al-Majid, A.M.; Barakat, A.; Al-Showiman, S.S.; Al-Har, M.S.; Radi, S.; Naseer, M.M.; Hadda, T.B. Synthesis and biological evaluation of 2-Aminobenzamide derivatives as antimicrobial agents: Opening/closing pharmacophore site. Int. J. Mol. Sci. 2014, 15, 5115–5127. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-X.; Hu, B.-Q.; Xiang, J.-F.; Cui, J.; Hao, X.; Liang, T.-L.; Tang, Y.-L. N-aryl-substituted anthranilamides with intramolecular hydrogen bonds. Tetrahedron 2014, 70, 8588–8591. [Google Scholar] [CrossRef]
- Agbo, E.N.; Makhafola, T.J.; Choong, Y.S.; Mphahlele, M.J.; Ramasami, P. Synthesis, biological evaluation and molecular docking studies of 6-aryl-2-styrylquinazolin-4(3H)-ones. Molecules 2016, 21, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Sardon, T.; Cottin, T.; Xu, J.; Giannis, A.; Vernos, I. Development and biological evaluation of a novel aurora A kinase inhibitor. Chem. Biol. Chem. 2009, 10, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Mphahlele, M.J.; Maluleka, M.M.; Khoza, T.A. 2-Aryl-6,8-dibromo-2,3-dihydroquinazolin-4(1H)-ones as substrates for the synthesis of 2,6,8-triarylquinazolin-4-ones. Bull. Chem. Soc. Ethiop. 2014, 28, 81–90. [Google Scholar] [CrossRef]
- Ojo, B.; Chowdhury, B.K. Synthesis of {2,3-dihydro-7-halopyrrolo-[(2,1-b)]-quinazolin-9-(1H)-one and 2,3-dihydro-5,7-dihalopyrrolo-[(2,1-b)]-quinazolin-9-(1H)-one}: New analogs of deoxyvasicinone. Synthetic Commun. 2012, 42, 1002–1009. [Google Scholar] [CrossRef]
- Pingali, S.R.K.; Madhav, M.; Jursic, B.S. An efficient regioselective NBS aromatic bromination in the presence of an ionic liquid. Tetrahedron Lett. 2010, 51, 1383–1385. [Google Scholar] [CrossRef]
- Khoza, T.A.; Makhafola, T.J.; Mphahlele, M.J. Novel polycarbo-substituted imidazo [1,2-c] quinazolines: Synthesis and cytotoxicity study. Molecules 2015, 20, 22520–22533. [Google Scholar] [CrossRef] [PubMed]
- Kashimo, S.; Tateno, S.; Tanabe, H.; Haisa, M. Structures of o-aminobenzamide and p-hydroxybenzamide monohydrate. Acta Cryst. 1991, C47, 2236–2239. [Google Scholar]
- Wardella, J.L.; Tiekink, E.R.T. 2-Amino-3-chloro-5-nitrobenzamide. Acta Cryst. 2012, E68, o991. [Google Scholar] [CrossRef] [PubMed]
- Arenas-Garcia, J.I.; Herrera-Ruiz, D.; Monragón-Vásquez, K.; Morales-Rojas, H.; Höpfl, H. Modification of the supramolecular hydrogen-bonding patterns of acetazolamide in the presence of different cocrystal formers: 3:1, 2:1, 1:1, and 1:2 cocrystals from screening with the structural isomers of hydroxybenzoic acids, aminobenzoic acids, hydroxybenzamides, aminobenzamides, nicotinic acids, nicotinamides, and 2,3-dihydroxybenzoic acids. Cryst. Growth Des. 2012, 12, 811–824. [Google Scholar]
- Banik, M.; Gopi, S.P.; Ganguly, S.; Desiraju, G.R. Cocrystal and salt forms of furosemide: Solubility and diffusion variations. Cryst. Growth Des. 2016, 16, 5418–5428. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dooley, R.; Milfeld, K.; Guiang, C.; Pamidighantam, S.; Allen, G. From proposal to production: Lessons learned developing the computational chemistry grid cyber infrastructure. J. Grid Comput. 2006, 4, 195–208. [Google Scholar] [CrossRef]
- Shen, N.; Fan, Y.; Pamidighantam, S. E-science infrastructures for molecular modeling and parametrization. J. Comput. Sci. 2014, 5, 576–589. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Sheikhet, I. Quantum Chemical and Statistical Theory of Solutions—A Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; Schleyer, P.V.R. Initio Molecular Orbital Theory; Wiley-Interscience: New York, NY, USA, 1986. [Google Scholar]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Furche, F.; Rappoport, D. Density Functional Methods for Excited States: Equilibrium Structure and Electronic Spectra; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Papamokos, G.V.; Demetropoulos, I.N. Vibrational frequencies of amides and amide dimers: The assessment of PW91XC functional. J. Phys. Chem. A 2004, 108, 7291–7300. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, B.; Li, X.; Trindle, O.C.; Zhang, G. External heavy-atom effect via orbital interactions revealed by single-crystal X-ray diffraction. J. Phys. Chem. A 2016, 120, 5791–5797. [Google Scholar] [CrossRef] [PubMed]
- Bruker, S. AINT+, Version 6.02 (Includes XPREP and SADABS); Bruker AXS Inc.: Madison, WI, USA, 2004. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. ORTEP-3 for Windows-a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds (ABB, AIB, ABBB and ABIB) are available from the authors.
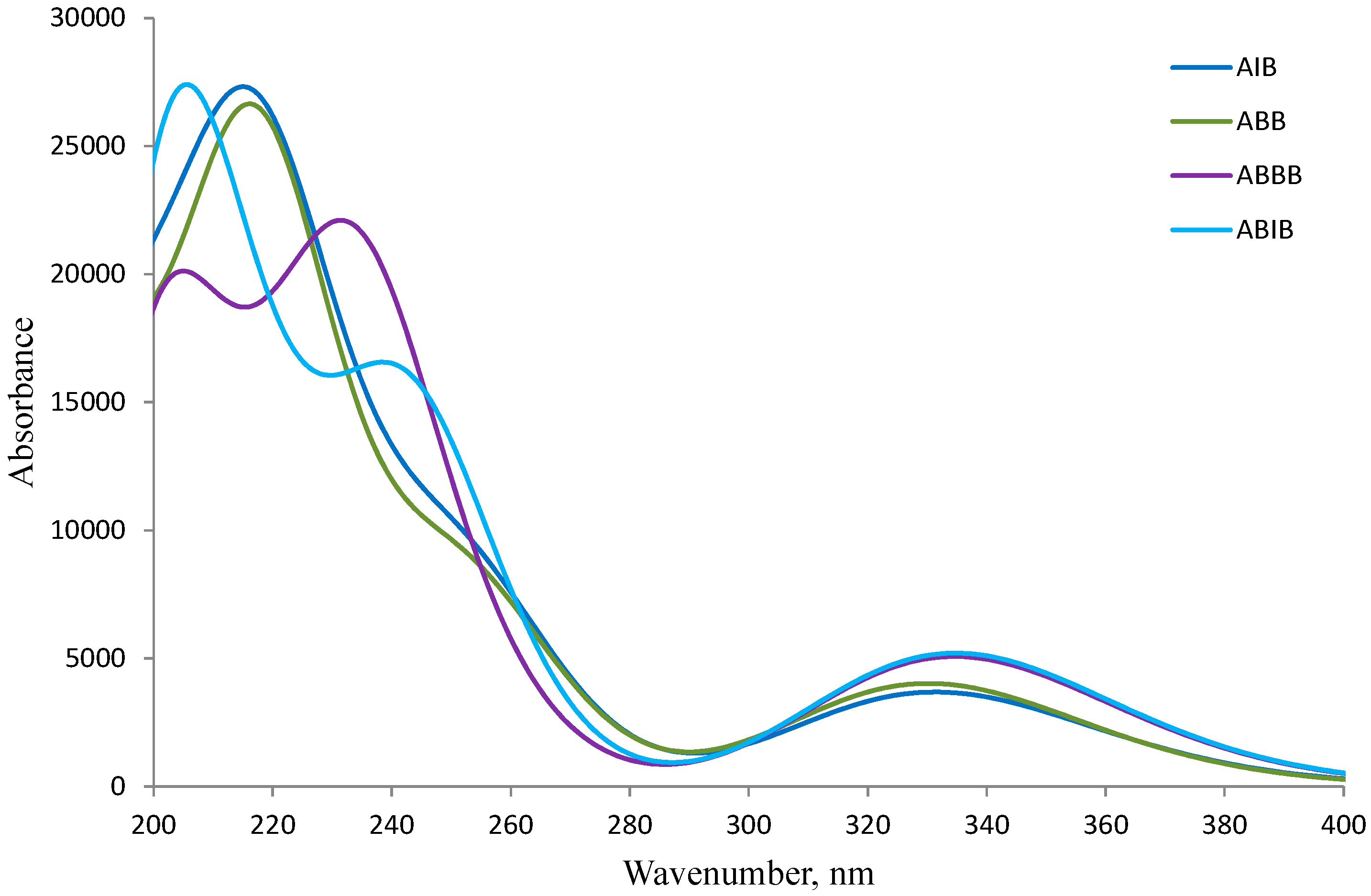
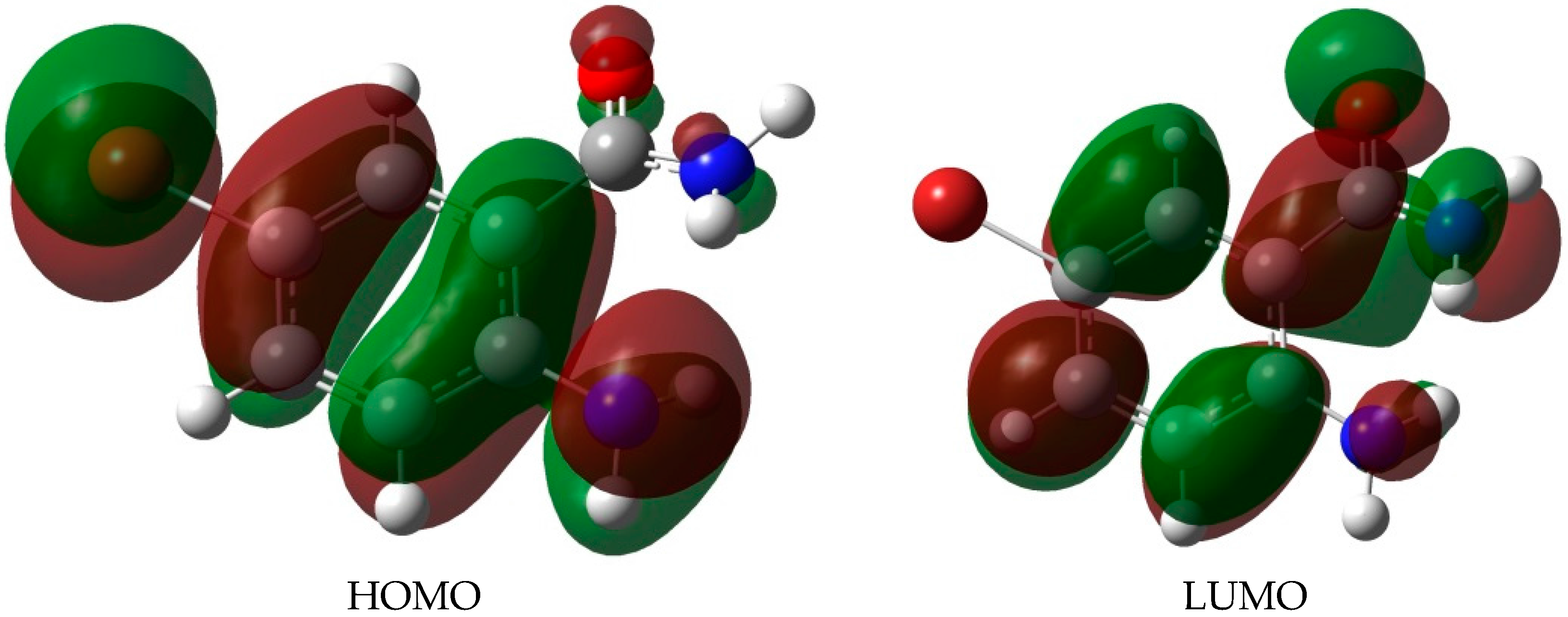
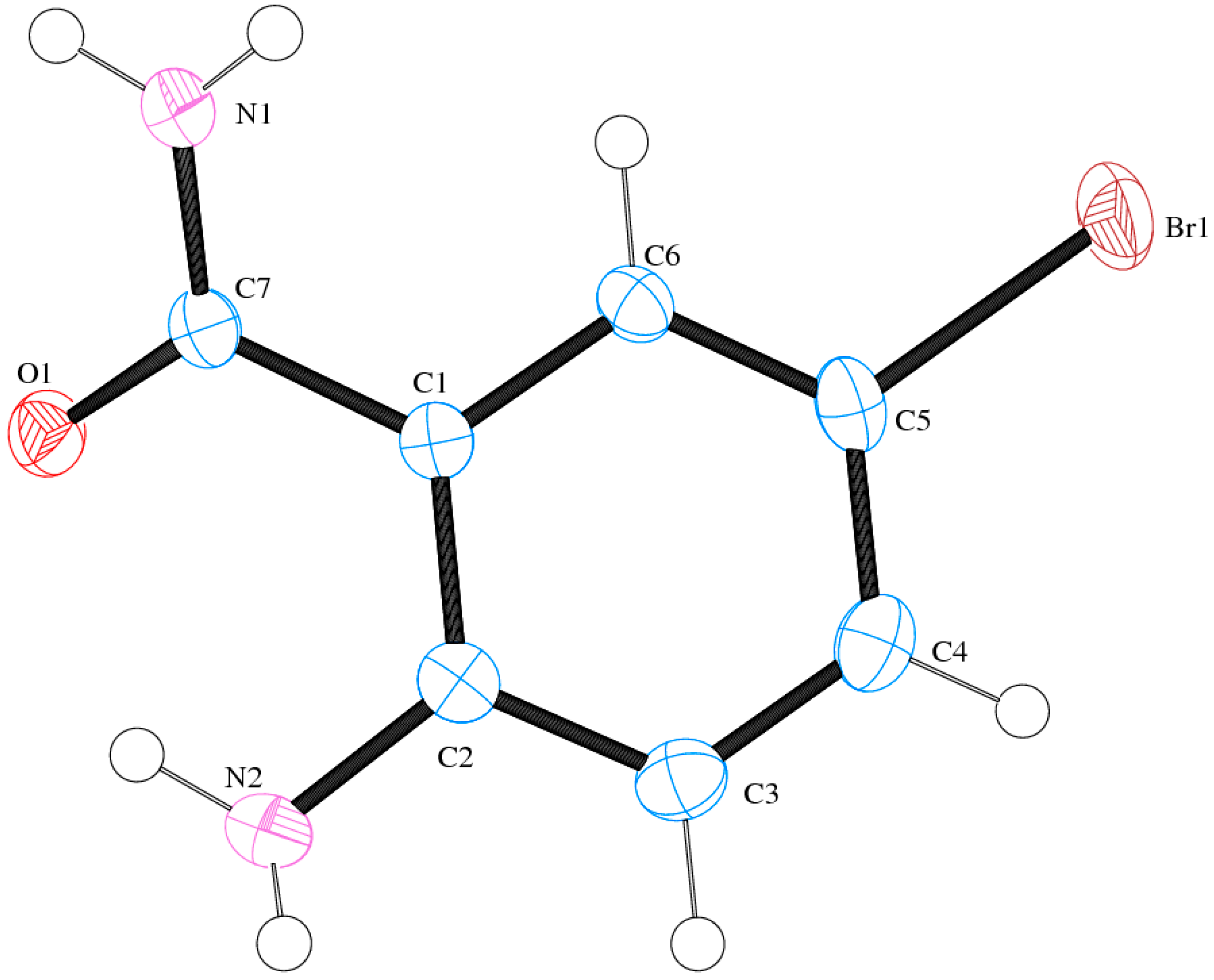


| Gas Phase | DMSO | |||||
| B3LYP | ||||||
| Electronic Energy (Hartree) | ΔE (kJ/mol) | Electronic Energy (Hartree) | ΔE (kJ/mol) | |||
| Conformer (A) | Conformer (B) | Conformer (A) | Conformer (B) | |||
| ABB | −3030.994480 | −3029.988460 | 15.81 | −3030.006409 | −3030.002821 | 9.42 |
| AIB | −467.228178 | −467.222193 | 15.72 | −467.240222 | −467.236665 | 9.34 |
| ABBB | −5603.535596 | −5603.529423 | 16.21 | −5603.546023 | −5603.541927 | 10.75 |
| ABIB | −3040.768917 | −3040.762660 | 16.43 | −3040.779561 | −3040.775398 | 10.93 |
| Gibb Free Energy (Hartree) | ΔG (kJ/mol) | Gibb Free Energy (Hartree) | ΔG (kJ/mol) | |||
| ABB | −3029.897495 | −3029.891223 | 16.47 | −3029.909949 | −3029.906365 | 9.41 |
| AIB | −467.132463 | −467.126178 | 16.50 | −467.144972 | −467.141348 | 9.51 |
| ABBB | −5603.451831 | −5603.445371 | 16.96 | −5603.462814 | −5603.458735 | 10.71 |
| ABIB | −3040.686568 | −3040.679861 | 17.61 | −3040.697530 | −3040.693577 | 10.38 |
| B3LYP-D3 | ||||||
| Electronic Energy (Hartree) | ΔE (kJ/mol) | Electronic Energy (Hartree) | ΔE (kJ/mol) | |||
| Conformer (A) | Conformer (B) | Conformer (A) | Conformer (B) | |||
| ABB | −3030.010097 | −3030.004505 | 14.68 | −3030.022089 | −3030.018918 | 8.32 |
| AIB | −467.244209 | −467.238646 | 14.61 | −467.256320 | −467.253167 | 8.28 |
| ABBB | −5603.554266 | −5603.548575 | 14.94 | −5603.564784 | −5603.561176 | 9.47 |
| ABIB | −3040.788022 | −3040.782271 | 15.10 | −3040.798761 | −3040.795110 | 9.58 |
| Gibb Free Energy (Hartree) | ΔG (kJ/mol) | Gibb Free Energy (Hartree) | ΔG (kJ/mol) | |||
| ABB | −3029.912976 | −3029.907178 | 15.22 | −3029.925554 | −3029.922248 | 8.68 |
| AIB | −467.148366 | −467.142508 | 15.38 | −467.160869 | −467.157580 | 8.64 |
| ABBB | −5603.470457 | −5603.464475 | 15.71 | −5603.481694 | −5603.477920 | 9.91 |
| ABIB | −3040.705570 | −3040.699515 | 15.90 | −3040.716715 | −3040.713097 | 9.50 |
| ABB | ||
|---|---|---|
| Conformer (A) | Conformer (B) | |
| H7 | 7.02 | 7.11 |
| H8 | 7.46 | 7.51 |
| H9 | 7.73 | 8.05 |
| H14 | 5.17 | 5.49 |
| H15 | 5.76 | 6.30 |
| H17 | 4.30 | 4.06 |
| H18 | 7.45 | 4.18 |
| λ (nm) | λ (eV) | Oscillator Strength | Assignment | % Contribution |
|---|---|---|---|---|
| Conformer (A) | ||||
| ABB (HOMO-LUMO gap = 4.38 eV) | ||||
| 216.6 | 5.72 | 0.1174 | HOMO-3→LUMO | 31.6 |
| 330.2 | 3.75 | 0.0993 | HOMO→LUMO | 96.8 |
| AIB (HOMO-LUMO gap = 4.36 eV) | ||||
| 213.6 | 5.80 | 0.2280 | HOMO-4→LUMO | 21.0 |
| 333.0 | 3.72 | 0.0840 | HOMO→LUMO | 93.8 |
| ABBB (HOMO-LUMO gap = 4.34 eV) | ||||
| 234.9 | 5.28 | 0.3589 | HOMO-1→LUMO | 44.7 |
| 334.7 | 3.70 | 0.1252 | HOMO→LUMO | 96.7 |
| ABIB (HOMO-LUMO gap = 4.35 eV) | ||||
| 243.1 | 5.10 | 0.3372 | HOMO→LUMO+2 | 65.7 |
| 334.9 | 3.70 | 0.1186 | HOMO→LUMO | 89.4 |
| Conformer (B) | ||||
| ABB (HOMO-LUMO gap = 4.65 eV) | ||||
| 217.9 | 5.69 | 0.5301 | HOMO-2→LUMO | 35.2 |
| 311.0 | 3.99 | 0.0800 | HOMO→LUMO | 95.2 |
| AIB (HOMO-LUMO gap = 4.62 eV) | ||||
| 218.5 | 5.68 | 0.4237 | HOMO-4→LUMO | 24.5 |
| 313.9 | 3.95 | 0.0758 | HOMO→LUMO | 92.5 |
| ABBB (HOMO-LUMO gap = 4.58 eV) | ||||
| 273.7 | 4.53 | 0.0758 | HOMO-3→LUMO | 8.4 |
| 390.9 | 3.17 | 1.5953 | HOMO→LUMO | 98.5 |
| ABIB (HOMO-LUMO gap = 4.57 eV) | ||||
| 239.1 | 5.19 | 0.4646 | HOMO→LUMO+2 | 43.2 |
| 316.3 | 3.92 | 0.0936 | HOMO→LUMO | 69.6 |
| Compound | IR Frequencies |
|---|---|
| ABB | 508, 534, 581, 652, 694, 818, 857, 1068, 1162, 1257, 1366, 1476, 1538, 1600, 1672, 3161, 3281, 3355, 3395 |
| AIB | 505, 537, 582, 657, 697, 798, 892, 1078, 1159, 1245, 1297, 1390, 1478, 1540, 1605, 1673, 3157, 3287, 3347, 3395 |
| ABBB | 413, 536, 584, 641, 800, 864, 1047, 1127, 1240, 1382, 1408, 1440, 1529, 1561, 1598, 1638, 3186, 3317, 3369, 3412 |
| ABIB | 413, 542, 644, 805, 873, 1054, 1130, 1240, 1276, 1386, 1414, 1451, 1536, 1567, 1602, 1641, 3180, 3326, 3368, 3424 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDCC | 1511089 | |
|---|---|---|
| Empirical formula | C7H7BrN2O | |
| Formula weight | 215.06 | |
| Crystal system | Monoclinic | |
| Space group | C2/c | |
| Unit cell dimensions | a = 30.3359(6) Å | α = 90° |
| b = 6.50570(10) Å | β = 91.4270(10)° | |
| c = 7.7930(2) Å | γ = 90° | |
| Volume | 1537.52(6) Å3 | |
| Z | 8 | |
| Density (calculated) | 1.858 Mg/m3 | |
| Absorption coefficient | 5.285 mm−1 | |
| F(000) | 848 | |
| Crystal size | 0.44 × 0.19 × 0.06 mm3 | |
| Theta range for data collection | 1.34 to 27.99° | |
| Index ranges | −40 < h < 39, −8 < k < 8, −10 < l < 10 | |
| Reflections collected | 11742 | |
| Independent reflections | 1865 [R(int) = 0.0970] | |
| Completeness to theta = 27.99° | 99.9% | |
| Absorption correction | Integration | |
| Max. and min. Transmission | 0.7422 and 0.2045 | |
| Refinement method | Full-matrix least-squares on F2 | |
| Data/restraints/parameters | 1865/0/116 | |
| Goodness-of-fit on F2 | 1.018 | |
| Final R indices [I > 2sigma(I)] | R1 = 0.0327, wR2 = 0.0854 | |
| R indices (all data) | R1 = 0.0408, wR2 = 0.0879 | |
| Largest diff. peak and hole | 1.096 and −0.391 e.Å−3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mphahlele, M.J.; Maluleka, M.M.; Rhyman, L.; Ramasami, P.; Mampa, R.M. Spectroscopic, DFT, and XRD Studies of Hydrogen Bonds in N-Unsubstituted 2-Aminobenzamides. Molecules 2017, 22, 83. https://doi.org/10.3390/molecules22010083
Mphahlele MJ, Maluleka MM, Rhyman L, Ramasami P, Mampa RM. Spectroscopic, DFT, and XRD Studies of Hydrogen Bonds in N-Unsubstituted 2-Aminobenzamides. Molecules. 2017; 22(1):83. https://doi.org/10.3390/molecules22010083
Chicago/Turabian StyleMphahlele, Malose Jack, Marole Maria Maluleka, Lydia Rhyman, Ponnadurai Ramasami, and Richard Mokome Mampa. 2017. "Spectroscopic, DFT, and XRD Studies of Hydrogen Bonds in N-Unsubstituted 2-Aminobenzamides" Molecules 22, no. 1: 83. https://doi.org/10.3390/molecules22010083
APA StyleMphahlele, M. J., Maluleka, M. M., Rhyman, L., Ramasami, P., & Mampa, R. M. (2017). Spectroscopic, DFT, and XRD Studies of Hydrogen Bonds in N-Unsubstituted 2-Aminobenzamides. Molecules, 22(1), 83. https://doi.org/10.3390/molecules22010083