
Stepwise, Protecting Group Free Synthesis of [4]Rotaxanes


Abstract
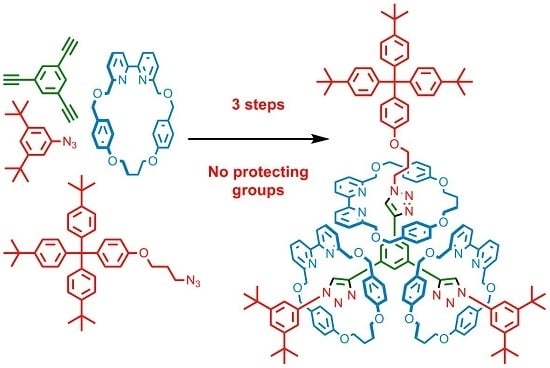
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
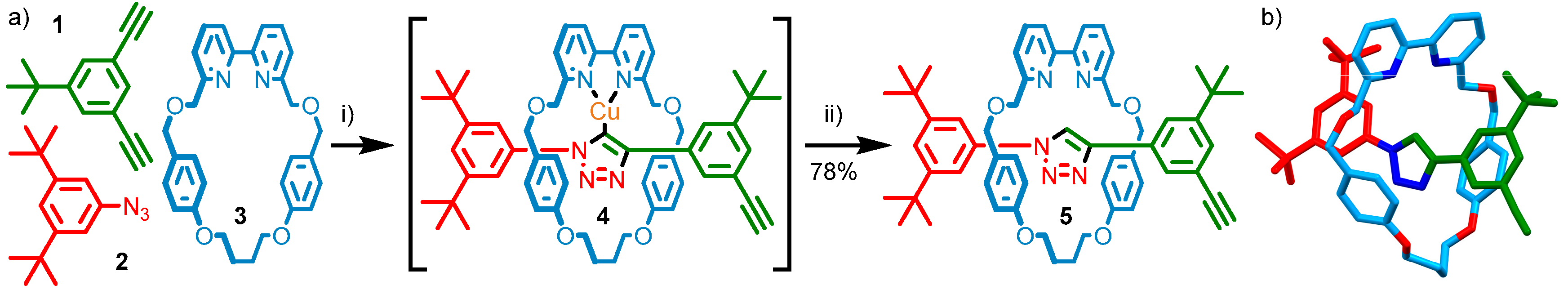
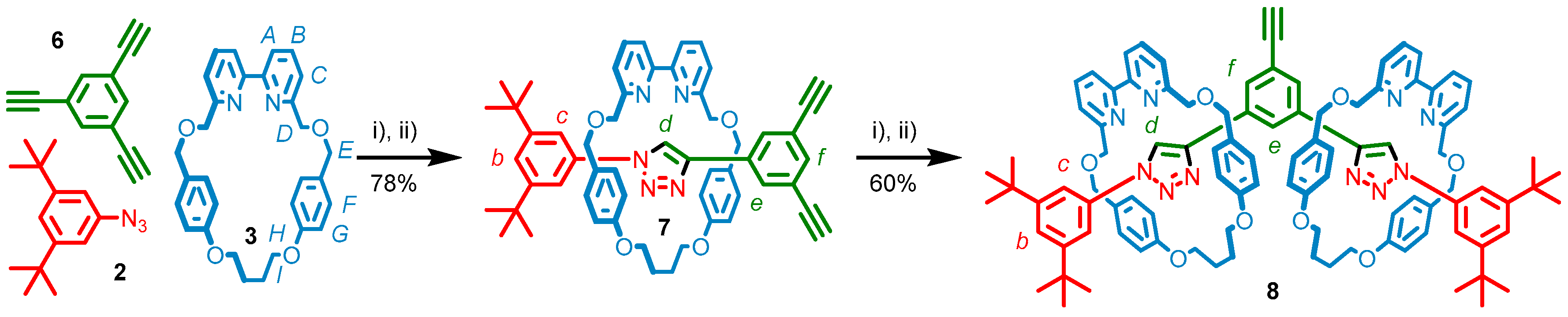
2.1. Stepwise Synthesis of [2]- and [3]Rotaxanes
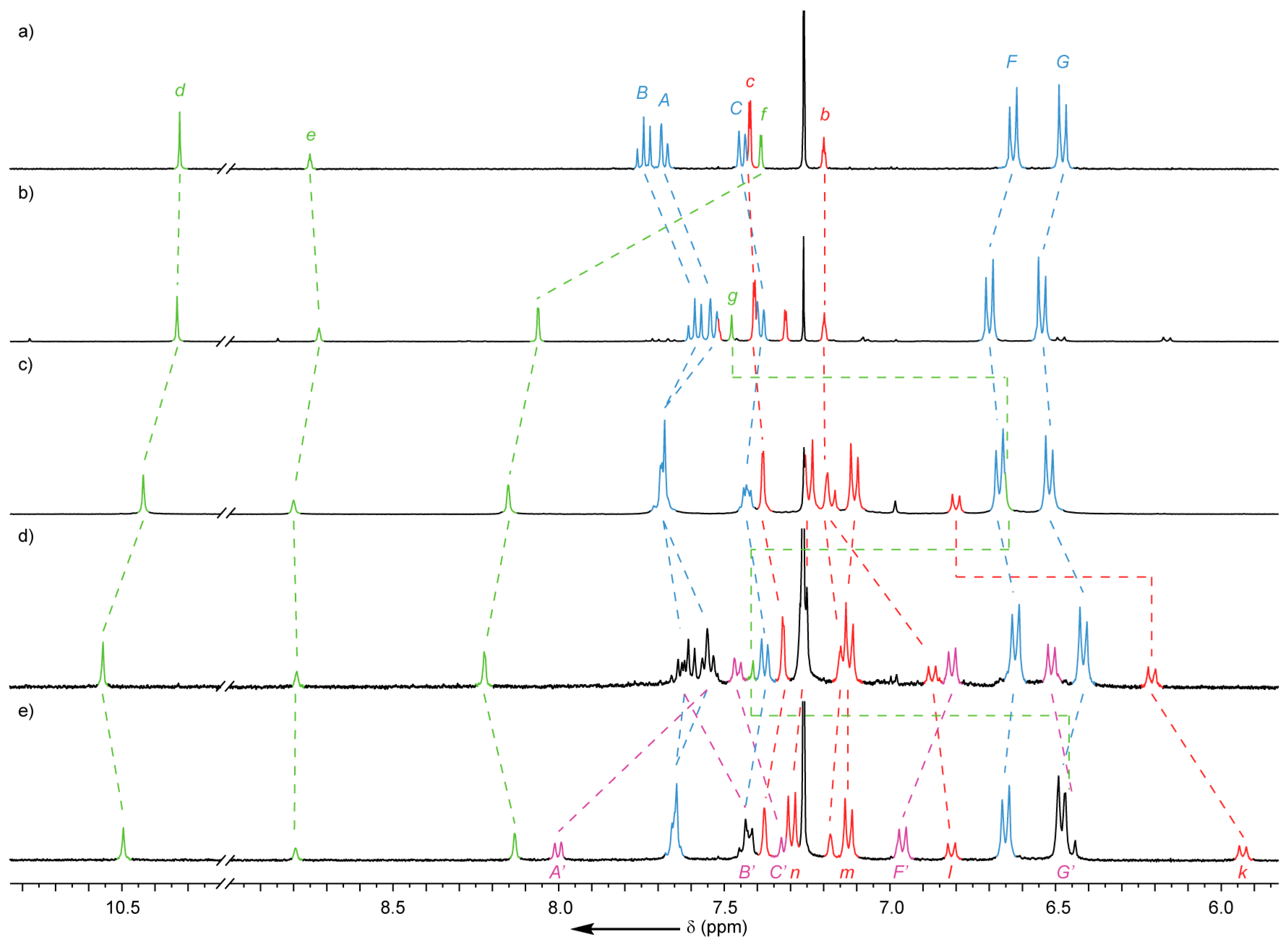
2.2. Analysis of [2]- and [3]Rotaxanes 7 and 8
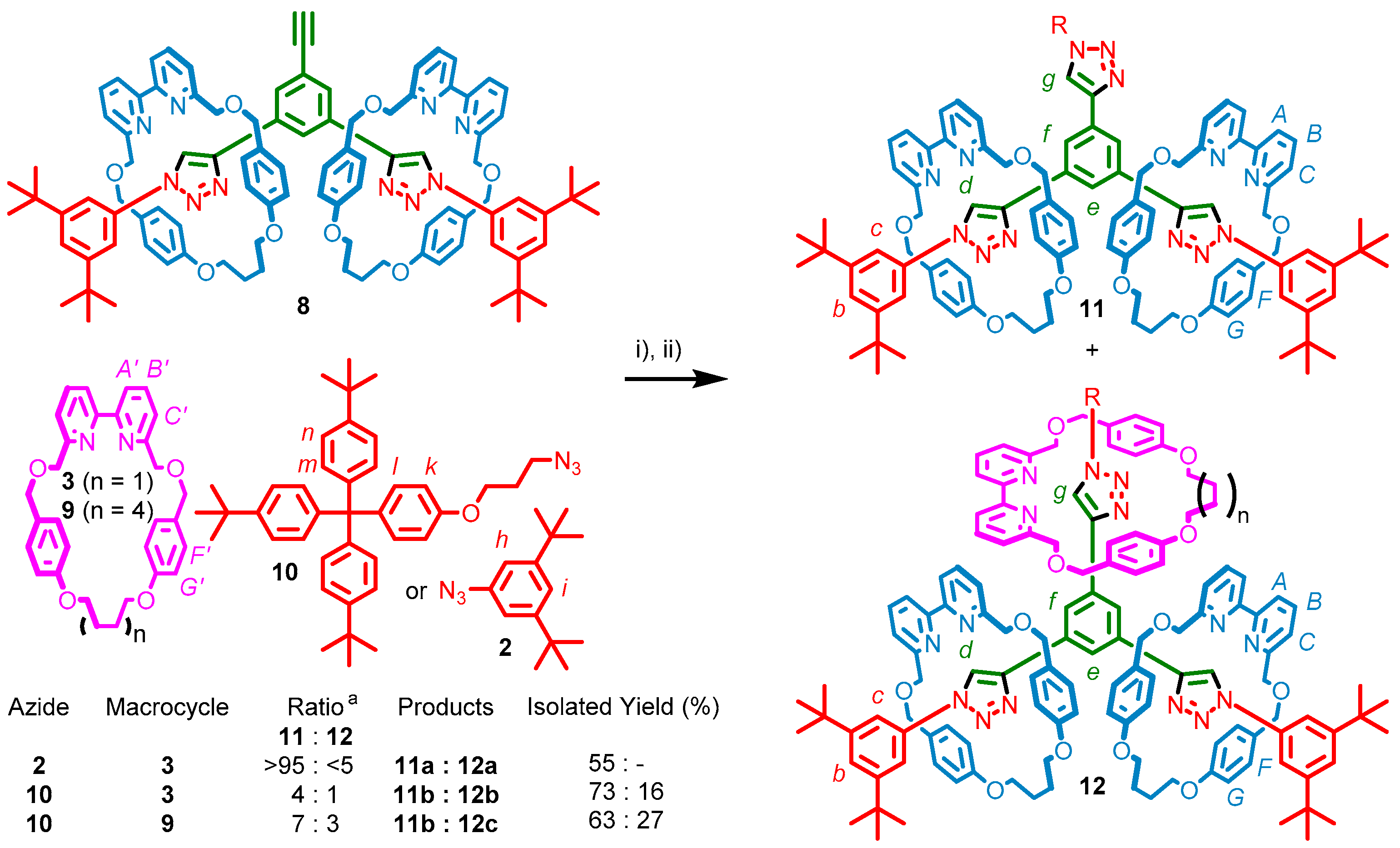
2.3. Synthesis of [4]Rotaxanes
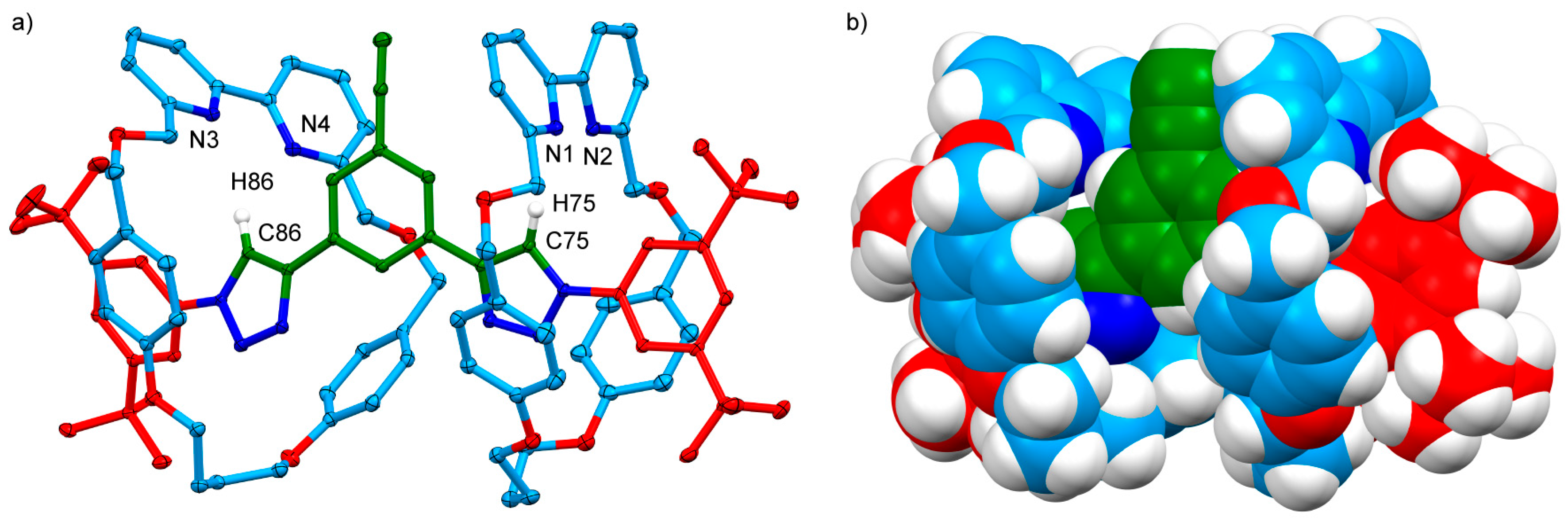
2.4. Analysis of [4]Rotaxanes 12b and 12c
3. Materials and Methods
3.1. General Information
3.2. General Procedure
3.2.1. Synthesis of [2]Rotaxane 7
3.2.2. Synthesis of [3]Rotaxane 8
3.2.3. Synthesis of [3]Rotaxane 11a
3.2.4. Synthesis of [3]Rotaxane 11b and [4]Rotaxane 12b
3.2.5. Synthesis of [3]Rotaxane 11b and [4]Rotaxane 12c
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Harrison, I.T.; Harrison, S. The Synthesis of a Stable Complex of a Macrocycle and a Threaded Chain. J. Am. Chem. Soc. 1967, 89, 5723–5724. [Google Scholar] [CrossRef]
- Ma, X.; Tian, H. Bright functional rotaxanes. Chem. Soc. Rev. 2010, 39, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.A.; Goldup, S.M. Chemical consequences of mechanical bonding in catenanes and rotaxanes: Isomerism, modification, catalysis and molecular machines for synthesis. Chem. Commun. 2014, 50, 5128. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, S.F.M.; Cantekin, S.; Elemans, J.A.A.W.; Rowan, A.E.; Nolte, R.J.M. Functional interlocked systems. Chem. Soc. Rev. 2014, 43, 99–122. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Yang, Y.; Chi, X.; Yan, X.; Huang, F. Development of Pseudorotaxanes and Rotaxanes: From Synthesis to Stimuli-Responsive Motions to Applications. Chem. Rev. 2015, 115, 7398–7501. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.E.M.; Galli, M.; Goldup, S.M. Properties and emerging applications of mechanically interlocked ligands. Chem. Commun. 2017, 53, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.J.; Davis, J.J.; Beer, P.D. Interlocked hostrotaxane and catenane structures for sensing charged guest species via optical and electrochemical methodologies. Org. Biomol. Chem. 2009, 7, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Zapata, F.; Beer, P.D. Interlocked host molecules for anion recognition and sensing. Coord. Chem. Rev. 2013, 257, 2434–2455. [Google Scholar] [CrossRef]
- Langton, M.J.; Beer, P.D. Rotaxane and Catenane Host Structures for Sensing Charged Guest Species. Acc. Chem. Res. 2014, 47, 1935–1949. [Google Scholar] [CrossRef] [PubMed]
- Leigh, D.A.; Marcos, V.; Wilson, M.R. Rotaxane catalysts. ACS Catal. 2014, 4, 4490–4497. [Google Scholar] [CrossRef]
- Hoekman, S.; Kitching, M.O.; Leigh, D.A.; Papmeyer, M.; Roke, D. Goldberg Active Template Synthesis of a [2]Rotaxane Ligand for Asymmetric Transition-Metal Catalysis. J. Am. Chem. Soc. 2015, 137, 7656–7659. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Lewis, J.E.M.; Goldup, S.M. A Stimuli-Responsive Rotaxane-Gold Catalyst: Regulation of Activity and Diastereoselectivity. Angew. Chem. Int. Ed. 2015, 54, 13545–13549. [Google Scholar] [CrossRef] [PubMed]
- Cotí, K.K.; Belowich, M.E.; Liong, M.; Ambrogio, M.W.; Lau, Y.A.; Khatib, H.A.; Zink, J.I.; Khashab, N.M.; Stoddart, J.F. Mechanised nanoparticles for drug delivery. Nanoscale 2009, 1, 16–39. [Google Scholar]
- Ambrogio, M.W.; Thomas, C.R.; Zhao, Y.-L.; Zink, J.I.; Stoddart, J.F. Mechanized Silica Nanoparticles: A New Frontier in Theranostic Nanomedicine. Acc. Chem. Res. 2011, 44, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Pairault, N.; Barat, R.; Tranoy-Opalinski, I.; Renoux, B.; Thomas, M.; Papot, S. Rotaxane-based architectures for biological applications. C. R. Chim. 2016, 19, 103–112. [Google Scholar] [CrossRef]
- Fernandes, A.; Viterisi, A.; Coutrot, F.; Potok, S.; Leigh, D.A.; Aucagne, V.; Papot, S. Rotaxane-Based Propeptides: Protection and Enzymatic Release of a Bioactive Pentapeptide. Angew. Chem. Int. Ed. 2009, 48, 6443–6447. [Google Scholar] [CrossRef] [PubMed]
- Smithrud, D.B.; Wang, X.; Tarapore, P.; Ho, S. Crown Ether Host-Rotaxanes as Cytotoxic Agents. ACS Med. Chem. Lett. 2013, 4, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Barat, R.; Legigan, T.; Tranoy-Opalinski, I.; Renoux, B.; Péraudeau, E.; Clarhaut, J.; Poinot, P.; Fernandes, A.E.; Aucagne, V.; Leigh, D.A.; et al. A mechanically interlocked molecular system programmed for the delivery of an anticancer drug. Chem. Sci. 2015, 6, 2608–2613. [Google Scholar] [CrossRef]
- Kay, E.R.; Leigh, D.A. Beyond switches: Rotaxane- and catenane-based synthetic molecular motors. Pure Appl. Chem. 2008, 80, 17–29. [Google Scholar] [CrossRef]
- Erbas-Cakmak, S.; Leigh, D.A.; McTernan, C.T.; Nussbaumer, A.L. Artificial Molecular Machines. Chem. Rev. 2015, 115, 10081–10206. [Google Scholar] [CrossRef] [PubMed]
- Coskun, A.; Banaszak, M.; Astumian, R.D.; Stoddart, J.F.; Grzybowski, B.A. Great expectations: Can artificial molecular machines deliver on their promise? Chem. Soc. Rev. 2012, 41, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Dietrich-Buchecker, C.O.; Sauvage, J.P. Une nouvelle famille de molecules: Les metallo-catenanes. Tetrahedron Lett. 1983, 24, 5095–5098. [Google Scholar] [CrossRef]
- Dietrich-Buchecker, C.O.; Sauvage, J.P.; Kern, J.M. Templated synthesis of interlocked macrocyclic ligands: The catenands. J. Am. Chem. Soc. 1984, 106, 3043–3045. [Google Scholar] [CrossRef]
- Ayme, J.-F.; Beves, J.E.; Campbell, C.J.; Leigh, D.A. Template synthesis of molecular knots. Chem. Soc. Rev. 2013, 42, 1700–1712. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.H.; Beer, P.D. Progress in the synthesis and exploitation of catenanes since the Millennium. Chem. Soc. Rev. 2014, 43, 4658–4683. [Google Scholar] [CrossRef] [PubMed]
- Crowley, J.D.; Goldup, S.M.; Lee, A.-L.; Leigh, D.A.; McBurney, R.T. Active metal template synthesis of rotaxanes, catenanes and molecular shuttles. Chem. Soc. Rev. 2009, 38, 1530–1541. [Google Scholar] [CrossRef] [PubMed]
- Beves, J.E.; Blight, B.A.; Campbell, C.J.; Leigh, D.A.; McBurney, R.T. Strategies and Tactics for the Metal-Directed Synthesis of Rotaxanes, Knots, Catenanes, and Higher Order Links. Angew. Chem. Int. Ed. 2011, 50, 9260–9327. [Google Scholar] [CrossRef] [PubMed]
- Chambron, J.-C.; Sauvage, J.-P. Topologically complex molecules obtained by transition metal templation: It is the presentation that determines the synthesis strategy. New J. Chem. 2013, 37, 49–57. [Google Scholar] [CrossRef]
- Vickers, M.S.; Beer, P.D. Anion templated assembly of mechanically interlocked structures. Chem. Soc. Rev. 2007, 36, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Mullen, K.M.; Beer, P.D. Sulfate anion templation of macrocycles, capsules, interpenetrated and interlocked structures. Chem. Soc. Rev. 2009, 38, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Clegg, J.K.; Lindoy, L.F. Template Synthesis. In Anion Coordination Chemistry; Bowman-James, K., Bianchi, A., España, G., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; pp. 289–320. [Google Scholar]
- Spence, G.T.; Beer, P.D. Expanding the Scope of the Anion Templated Synthesis of Interlocked Structures. Acc. Chem. Res. 2013, 46, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Barin, G.; Coskun, A.; Fouda, M.M.G.; Stoddart, J.F. Mechanically Interlocked Molecules Assembled by π-π Recognition. ChemPlusChem 2012, 77, 159–185. [Google Scholar] [CrossRef]
- Schalley, C.A.; Weilandt, T.; Brüggemann, J.; Vögtle, F. Hydrogen-Bond-Mediated Template Synthesis of Rotaxanes, Catenanes, and Knotanes. Top. Curr. Chem. 2005, 248, 141–200. [Google Scholar]
- Kay, E.R.; Leigh, D.A. Hydrogen Bond-Assembled Synthetic Molecular Motors and Machines. Top. Curr. Chem. 2005, 262, 133–177. [Google Scholar]
- Cheng, C.; Cheng, T.; Xiao, H.; Krzyaniak, M.D.; Wang, Y.; McGonigal, P.R.; Frasconi, M.; Barnes, J.C.; Fahrenbach, A.C.; Wasielewski, M.R.; et al. Influence of Constitution and Charge on Radical Pairing Interactions in Tris-radical Tricationic Complexes. J. Am. Chem. Soc. 2016, 138, 8288–8300. [Google Scholar] [CrossRef] [PubMed]
- Klotz, E.J.F.; Claridge, T.D.W.; Anderson, H.L. Homo- and Hetero-[3]Rotaxanes with Two π-Systems Clasped in a Single Macrocycle. J. Am. Chem. Soc. 2006, 128, 15374–15375. [Google Scholar] [CrossRef] [PubMed]
- Prikhod’ko, A.I.; Durola, F.; Sauvage, J. Iron(II)-Templated Synthesis of [3]Rotaxanes by Passing Two Threads through the Same Ring. J. Am. Chem. Soc. 2008, 130, 448–449. [Google Scholar] [CrossRef] [PubMed]
- Prikhod′ko, A.I.; Sauvage, J.-P. Passing Two Strings through the Same Ring Using an Octahedral Metal Center as Template: A New Synthesis of [3]Rotaxanes. J. Am. Chem. Soc. 2009, 131, 6794–6807. [Google Scholar] [CrossRef] [PubMed]
- Goldup, S.M.; Leigh, D.A.; McGonigal, P.R.; Ronaldson, V.E.; Slawin, A.M.Z. Two Axles Threaded Using a Single Template Site: Active Metal Template Macrobicyclic [3]Rotaxanes. J. Am. Chem. Soc. 2010, 132, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.M.; Leigh, D.A.; Maffei, F.; McGonigal, P.R.; Slawin, A.M.Z.; Wu, J. En Route to a Molecular Sheaf: Active Metal Template Synthesis of a [3]Rotaxane with Two Axles Threaded through One Ring. J. Am. Chem. Soc. 2011, 133, 12298–12303. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Mutoh, Y.; Yamasaki, R.; Kasama, T.; Saito, S. Synthesis of [3]Rotaxanes that Utilize the Catalytic Activity of a Macrocyclic Phenanthroline-Cu Complex: Remarkable Effect of the Length of the Axle Precursor. Chem. Eur. J. 2015, 21, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, R.; Mutoh, Y.; Kasama, T.; Saito, S. Synthesis of [3]Rotaxanes by the Combination of Copper-Mediated Coupling Reaction and Metal-Template Approach. J. Org. Chem. 2015, 80, 7536–7546. [Google Scholar] [CrossRef] [PubMed]
- Movsisyan, L.D.; Franz, M.; Hampel, F.; Thompson, A.L.; Tykwinski, R.R.; Anderson, H.L. Polyyne Rotaxanes: Stabilization by Encapsulation. J. Am. Chem. Soc. 2016, 138, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Danon, J.J.; Leigh, D.A.; McGonigal, P.R.; Ward, J.W.; Wu, J. Triply Threaded [4]Rotaxanes. J. Am. Chem. Soc. 2016, 138, 12643–12647. [Google Scholar] [CrossRef] [PubMed]
- Takata, T. Polyrotaxane and Polyrotaxane Network: Supramolecular Architectures Based on the Concept of Dynamic Covalent Bond Chemistry. Polym. J. 2006, 38, 1–20. [Google Scholar] [CrossRef]
- Araki, J.; Ito, K. Recent advances in the preparation of cyclodextrin-based polyrotaxanes and their applications to soft materials. Soft Matter 2007, 3, 1456–1473. [Google Scholar] [CrossRef]
- Harada, A.; Hashidzume, A.; Yamaguchi, H.; Takashima, Y. Polymeric Rotaxanes. Chem. Rev. 2009, 109, 5974–6023. [Google Scholar] [CrossRef] [PubMed]
- Arunachalam, M.; Gibson, H.W. Recent developments in polypseudorotaxanes and polyrotaxanes. Prog. Polym. Sci. 2014, 39, 1043–1073. [Google Scholar] [CrossRef]
- Wei, P.; Yan, X.; Huang, F. Supramolecular polymers constructed by orthogonal self-assembly based on host-guest and metal-ligand interactions. Chem. Soc. Rev. 2015, 44, 815–832. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Ke, C.; Fraser Stoddart, J. Cooperative capture synthesis: Yet another playground for copper-free click chemistry. Chem. Soc. Rev. 2016, 45, 3766–3780. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.A.; Vermeulen, N.A.; McGonigal, P.R.; Avestro, A.-J.; Sarjeant, A.A.; Stern, C.L.; Stoddart, J.F. Formation of a hetero[3]rotaxane by a dynamic component-swapping strategy. Chem. Commun. 2014, 50, 9665–9668. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.A.; Goldup, S.M. A Kinetic Self-Sorting Approach to Heterocircuit [3]Rotaxanes. Angew. Chem. Int. Ed. 2016, 55, 12488–12493. [Google Scholar] [CrossRef] [PubMed]
- Avestro, A.-J.; Belowich, M.E.; Stoddart, J.F. Cooperative self-assembly: Producing synthetic polymers with precise and concise primary structures. Chem. Soc. Rev. 2012, 41, 5881–5895. [Google Scholar] [CrossRef] [PubMed]
- Fuller, A.-M.L.; Leigh, D.A.; Lusby, P.J. One Template, Multiple Rings: Controlled Iterative Addition of Macrocycles onto a Single Binding Site Rotaxane Thread. Angew. Chem. Int. Ed. 2007, 46, 5015–5019. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.W.; Klotz, E.J.F.; Odell, B.; Claridge, T.D.W.; Anderson, H.L. Solid-Phase Synthesis of Oligo(phenylene ethynylene) Rotaxanes. Angew. Chem. Int. Ed. 2007, 46, 6845–6848. [Google Scholar] [CrossRef] [PubMed]
- Spruell, J.M.; Dichtel, W.R.; Heath, J.R.; Stoddart, J.F. A One-Pot Synthesis of Constitutionally Unsymmetrical Rotaxanes Using Sequential CuI-Catalyzed Azide-Alkyne Cycloadditions. Chem. Eur. J. 2008, 14, 4168–4177. [Google Scholar] [CrossRef] [PubMed]
- Fuller, A.-M.L.; Leigh, D.A.; Lusby, P.J. Sequence Isomerism in [3]Rotaxanes. J. Am. Chem. Soc. 2010, 132, 4954–4959. [Google Scholar] [CrossRef] [PubMed]
- Langton, M.J.; Matichak, J.D.; Thompson, A.L.; Anderson, H.L. Template-directed synthesis of π-conjugated porphyrin [2]rotaxanes and a [4]catenane based on a six-porphyrin nanoring. Chem. Sci. 2011, 2, 1897. [Google Scholar] [CrossRef]
- Yamada, Y.; Okada, M.; Tanaka, K. Repetitive stepwise rotaxane formation toward programmable molecular arrays. Chem. Commun. 2013, 49, 11053–11055. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, L.-J.; Wang, X.-Q.; Sun, B.; Li, X.; Zhang, Y.; Shi, J.; Yu, Y.; Zhang, L.; Liu, M.; et al. Organometallic rotaxane dendrimers with fourth-generation mechanically interlocked branches. Proc. Natl. Acad. Sci. USA 2015, 112, 5597–5601. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.E.M.; Bordoli, R.J.; Denis, M.; Fletcher, C.J.; Galli, M.; Neal, E.A.; Rochette, E.M.; Goldup, S.M. High yielding synthesis of 2,2′-bipyridine macrocycles, versatile intermediates in the synthesis of rotaxanes. Chem. Sci. 2016, 7, 3154–3161. [Google Scholar] [CrossRef]
- Aucagne, V.; Hänni, K.D.; Leigh, D.A.; Lusby, P.J.; Walker, D.B. Catalytic “Click” Rotaxanes: A Substoichiometric Metal-Template Pathway to Mechanically Interlocked Architectures. J. Am. Chem. Soc. 2006, 128, 2186–2187. [Google Scholar] [CrossRef] [PubMed]
- Lahlali, H.; Jobe, K.; Watkinson, M.; Goldup, S.M. Macrocycle Size Matters: “Small” Functionalized Rotaxanes in Excellent Yield Using the CuAAC Active Template Approach. Angew. Chem. Int. Ed. 2011, 50, 4151–4155. [Google Scholar] [CrossRef] [PubMed]
- Winn, J.; Pinczewska, A.; Goldup, S.M. Synthesis of a Rotaxane CuI Triazolide under Aqueous Conditions. J. Am. Chem. Soc. 2013, 135, 13318–13321. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.E.M.; Winn, J.; Cera, L.; Goldup, S.M. Iterative Synthesis of Oligo[n]rotaxanes in Excellent Yield. J. Am. Chem. Soc. 2016, 138, 16329–16336. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, K. Rotaxane dendrimers. Top. Curr. Chem. 2003, 228, 111–140. [Google Scholar] [PubMed]
- Ho, W.K.-W.; Lee, S.-F.; Wong, C.-H.; Zhu, X.-M.; Kwan, C.-S.; Chak, C.-P.; Mendes, P.M.; Cheng, C.H.K.; Leung, K.C.-F. Type III-B rotaxane dendrimers. Chem. Commun. 2013, 49, 10781–10783. [Google Scholar] [CrossRef] [PubMed]
- Repeating the reaction at room temperature allowed examination of each step by 1H-NMR (see Supplementary Materials), confirming that following the reaction in EtOH predominantly a single interlocked species was formed, assigned as the triazolide intermediate of the AT-CuAAC reaction, with MS supporting this conclusion. After heating in CH2Cl2 and washing with EDTA-NH3 solution to abstract Cu ions present the main species observed was the [2]rotaxane product, 7.
- Zhu, W.; Ma, D. Synthesis of aryl azides and vinyl azides via proline-promoted CuI-catalyzed coupling reactions. Chem. Commun. 2004, 888–889. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Kijima, M. 1,3,5-Tris(functionalised-phenylethynyl)benzene-metal complexes: Synthetic survey of mesoporous coordination polymers and investigation of their carbonisation. J. Mater. Chem. 2008, 18, 1037–1045. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors upon request.






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, J.E.M.; Winn, J.; Goldup, S.M. Stepwise, Protecting Group Free Synthesis of [4]Rotaxanes. Molecules 2017, 22, 89. https://doi.org/10.3390/molecules22010089
Lewis JEM, Winn J, Goldup SM. Stepwise, Protecting Group Free Synthesis of [4]Rotaxanes. Molecules. 2017; 22(1):89. https://doi.org/10.3390/molecules22010089
Chicago/Turabian StyleLewis, James E. M., Joby Winn, and Stephen M. Goldup. 2017. "Stepwise, Protecting Group Free Synthesis of [4]Rotaxanes" Molecules 22, no. 1: 89. https://doi.org/10.3390/molecules22010089
APA StyleLewis, J. E. M., Winn, J., & Goldup, S. M. (2017). Stepwise, Protecting Group Free Synthesis of [4]Rotaxanes. Molecules, 22(1), 89. https://doi.org/10.3390/molecules22010089