New 2-Phenylthiazoles as Potential Sortase A Inhibitors: Synthesis, Biological Evaluation and Molecular Docking

 ,
,  , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Assays
2.2.1. Antimicrobial Activity—Initial In Vitro Qualitative Screening Study
2.2.2. Antimicrobial Activity—In Vitro Quantitative Assay
2.2.3. Anti-Biofilm Activity Assay
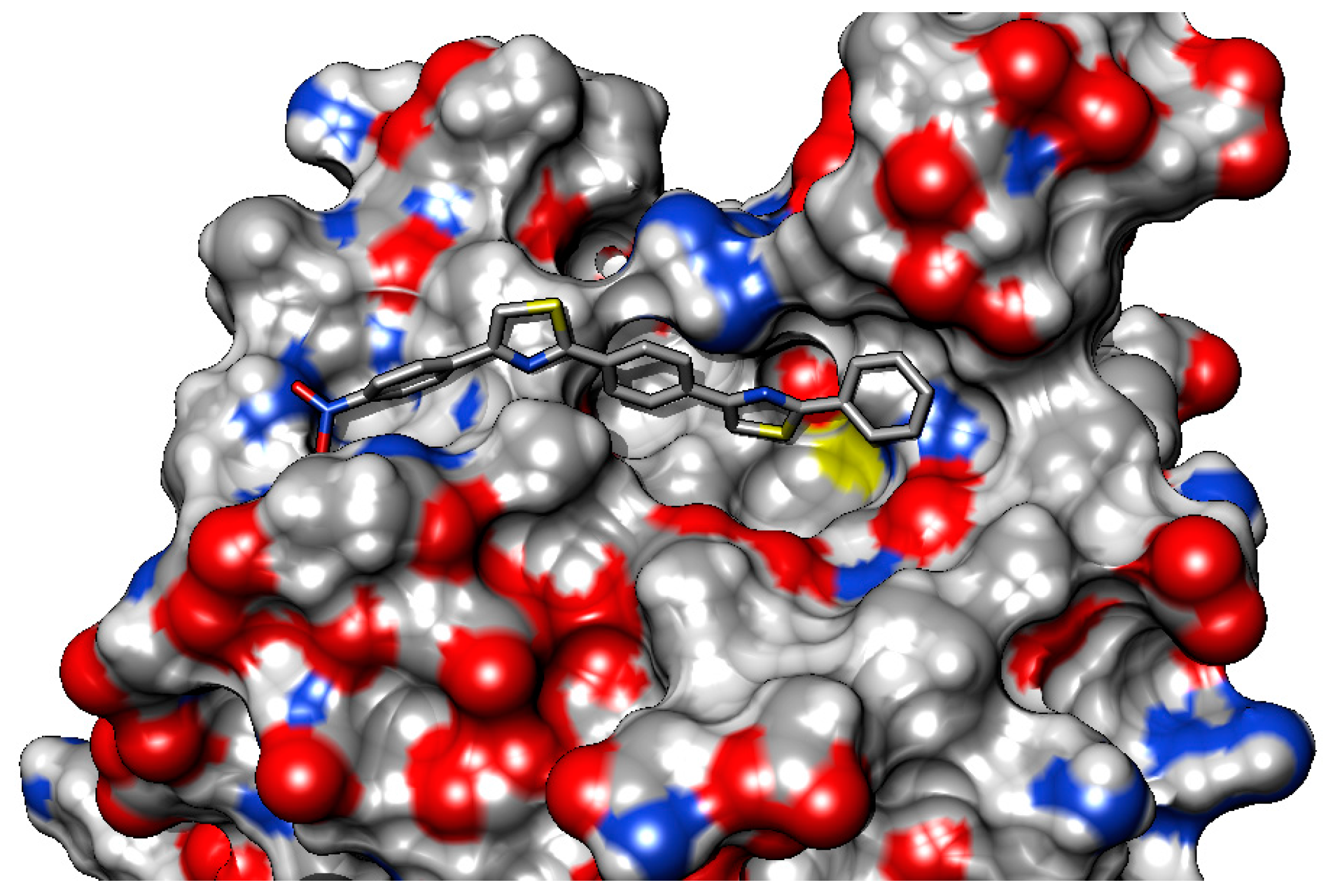
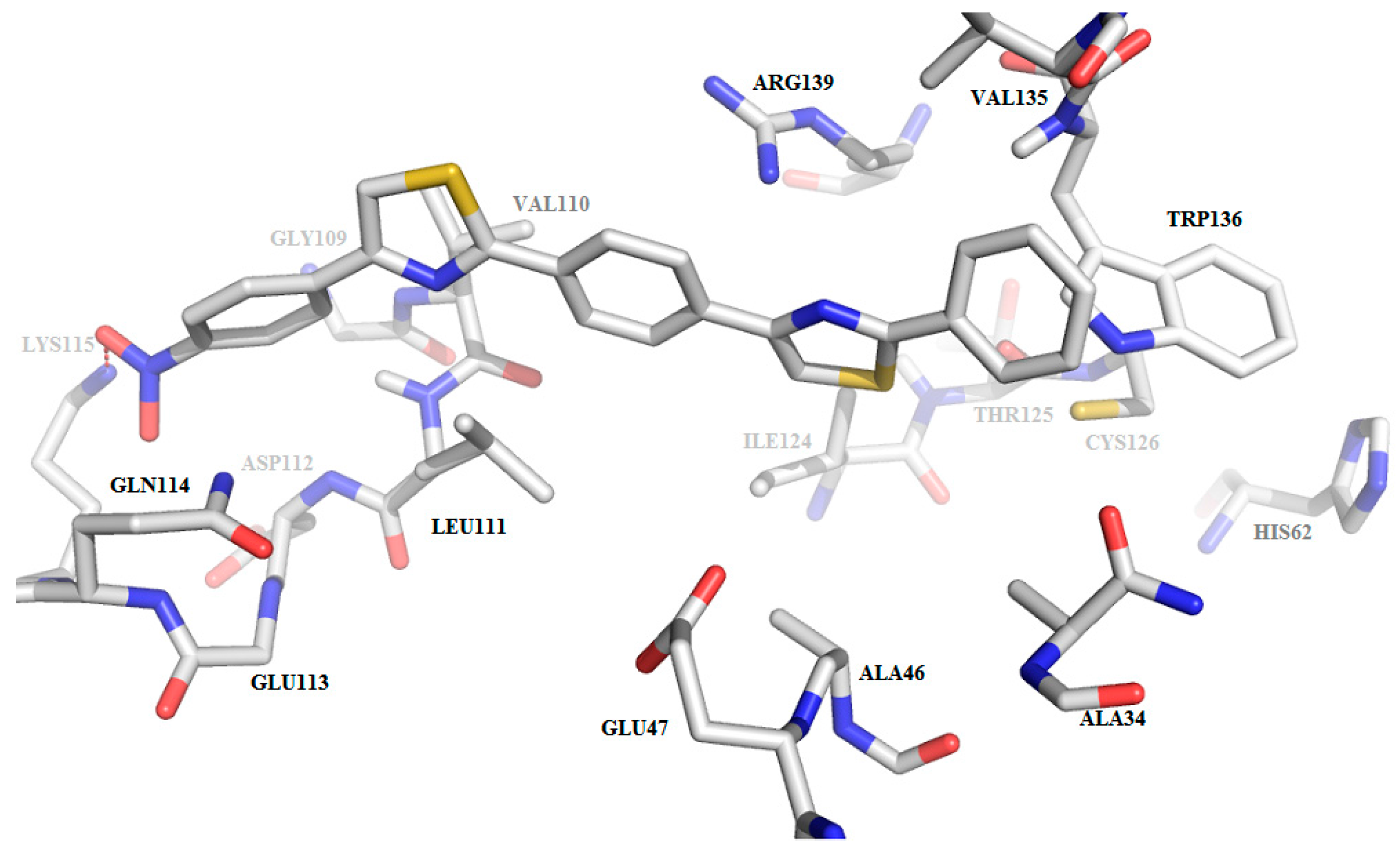
2.3. Molecular Docking
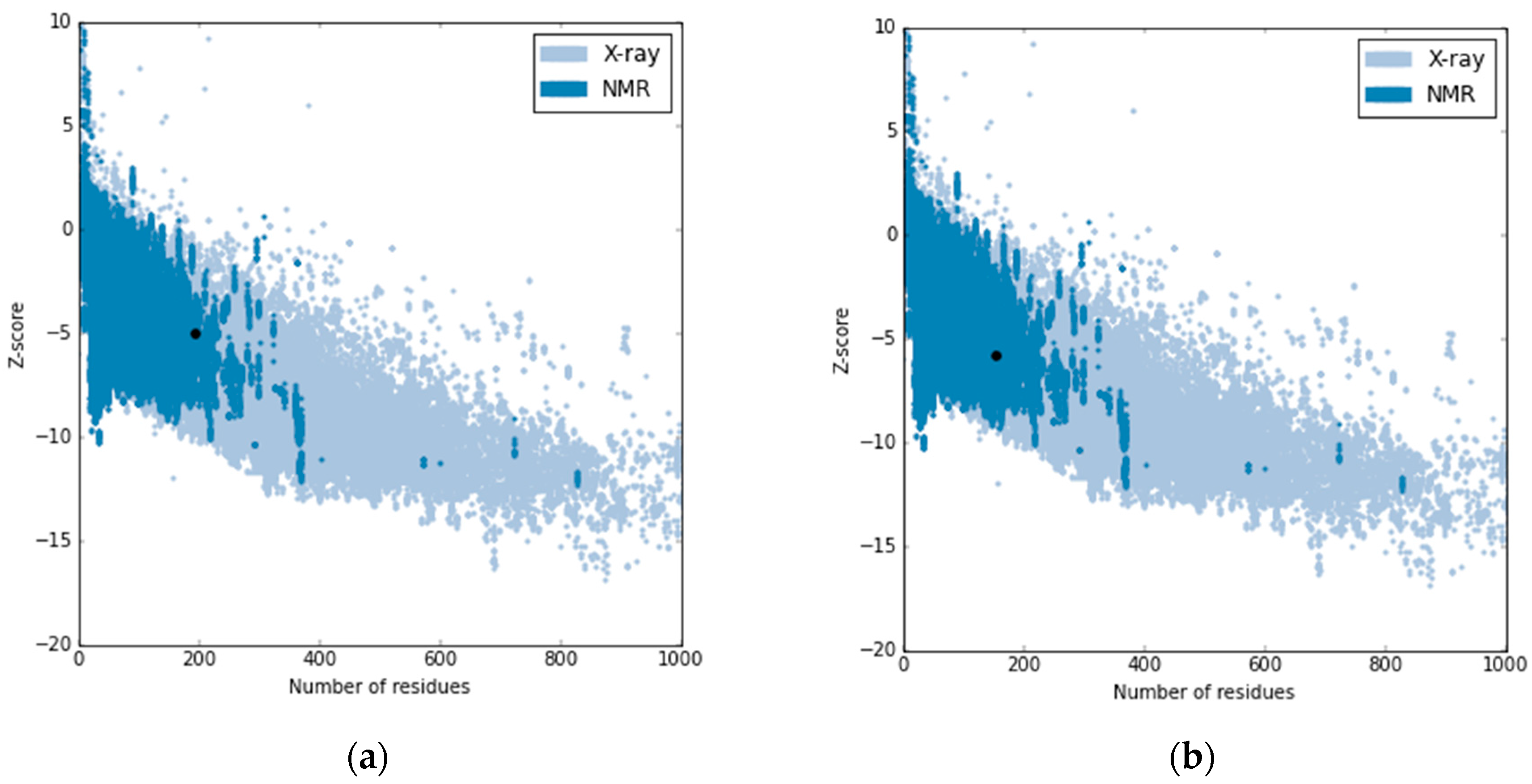
2.3.1. Sequence Alignment and Validation of the Generated Model for E. faecalis ATCC 29212 Sortase A
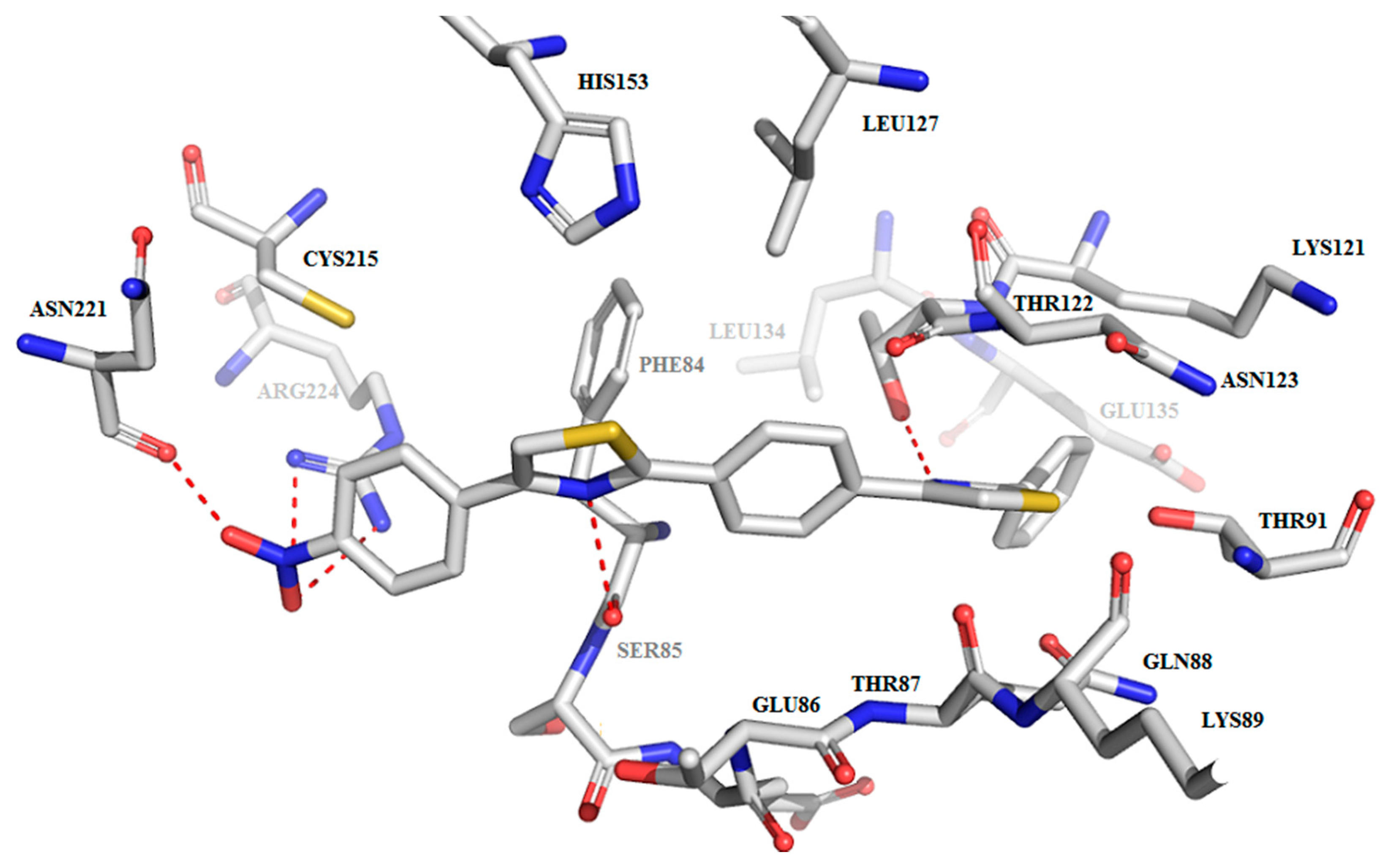
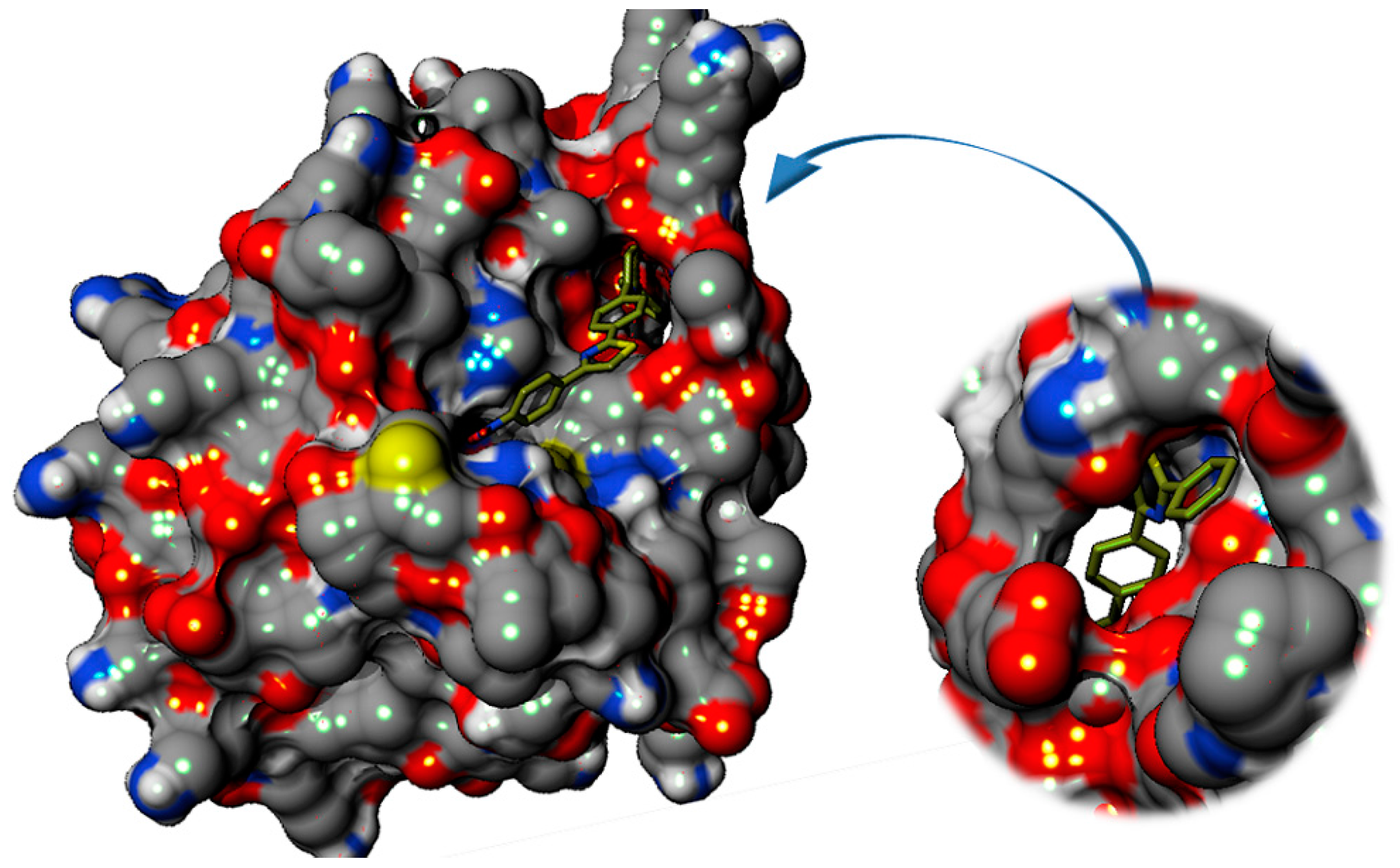
2.3.2. Binding Mode of the C1–8 2-Phenylthiazoles to the Catalytic Site of Sortases
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.3. Biological Assays
3.3.1. Antimicrobial Activity—Initial In Vitro Qualitative Screening Study
3.3.2. Antimicrobial Activity—In Vitro Quantitative Assay
3.3.3. Anti-Biofilm Activity Assay
3.4. Molecular Docking Study
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organisation. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Available online: http://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/ (accessed on 15 August 2017).
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Rice, L.B. Antimicrobial resistance in gram-positive bacteria. Am. J. Infect. Control 2006, 34, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Schneewind, O.; Fowler, A.; Faull, K.F. Structure of the cell wall anchor of surface proteins in Staphylococcus aureus. Science 1995, 268, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Schneewind, O.; Missiakas, D. Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Spirig, T.; Weiner, E.M.; Clubb, R.T. Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 2011, 82, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Tsompanidou, E.; Denham, E.L.; Sibbald, M.J.J.B.; Yang, X.; Seinen, J.; Friedrich, A.W.; Buist, G.; Van Dijl, J.M. The Sortase A Substrates FnbpA, FnbpB, ClfA and ClfB Antagonize Colony Spreading of Staphylococcus aureus. PLoS ONE 2012, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.; Pozzi, C.; Houston, P.; Humphreys, H.; Robinson, D.A.; Loughman, A.; Foster, T.J.; O’Gara, J.P. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J. Bacteriol. 2008, 190, 3835–3850. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, H.; Rudkin, J.K.; Black, N.S.; Gallagher, L.; O’Neill, E.; O’Gara, J.P. Methicillin resistance and the biofilm phenotype in Staphylococcus aureus. Front. Cell. Infect. Microbiol. 2015, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Dunny, G.; Hancock, L.; Shankar, N. Enterococcal Biofilm Structure and Role in Colonization and Disease. In Enterococci: from Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M., Clewell, D., Ike, Y., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014. [Google Scholar]
- Guiton, P.S.; Hung, C.S.; Kline, K.A.; Roth, R.; Kau, A.L.; Hayes, E.; Heuser, J.; Dodson, K.W.; Caparon, M.G.; Hultgren, S.J. Contribution of autolysin and sortase A during Enterococcus faecalis DNA-dependent biofilm development. Infect. Immun. 2009, 77, 3626–3638. [Google Scholar] [CrossRef] [PubMed]
- Pericàs, J.M.; Corredoira, J.; Moreno, A.; García-País, M.J.; Falces, C.; Rabuñal, R.; Mestres, C.A.; Alonso, M.P.; Marco, F.; Quintana, E.; et al. Relationship Between Enterococcus faecalis Infective Endocarditis and Colorectal Neoplasm: Preliminary Results From a Cohort n Rabun. Rev. Española Cardiol. 2017. [Google Scholar] [CrossRef]
- Nallapareddy, S.R.; Singh, K.V.; Sillanpää, J.; Garsin, D.A.; Höök, M.; Erlandsen, S.L.; Murray, B.E. Endocarditis and biofilm-associated pili of Enterococcus faecalis. J. Clin. Investig. 2006, 116, 2799–2807. [Google Scholar] [CrossRef] [PubMed]
- Van Harten, R.M.; Willems, R.J.L.; Martin, N.I.; Hendrickx, A.P.A. Multidrug-Resistant Enterococcal Infections: New Compounds, Novel Antimicrobial Therapies? Trends Microbiol. 2017, 25, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, A.; Rasmussen, M. Mature biofilms of Enterococcus faecalis and Enterococcus faecium are highly resistant to antibiotics. Diagn. Microbiol. Infect. Dis. 2016, 84, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Totsika, M.; Schillaci, D. Sortase A: An ideal target for anti-virulence drug development. Microb. Pathog. 2014, 77, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Li, P.; Liu, X.; Luo, L. Chinese herb medicine against Sortase A catalyzed transformations, a key role in gram-positive bacterial infection progress. J. Enzym. Inhib. Med. Chem. 2016, 31, 184–196. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Zhang, Y.; Bao, J.; Deng, X.; Batara, J.; Casey, S.; Guo, Q.; Jiang, F.; Fu, L. Synthesis, biological evaluation and molecular docking analysis of 2-phenyl-benzofuran-3-carboxamide derivatives as potential inhibitors of Staphylococcus aureus Sortase A. Bioorg. Med. Chem. 2017, 25, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bao, J.; Deng, X.-X.; He, W.; Fan, J.-J.; Jiang, F.-Q.; Fu, L. Synthesis, biological evaluation and molecular docking of 2-phenyl-benzo[d]oxazole-7-carboxamide derivatives as potential Staphylococcus aureus Sortase A inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4081–4085. [Google Scholar] [CrossRef] [PubMed]
- Maggio, B.; Raffa, D.; Raimondi, M.V.; Cascioferro, S.; Plescia, F.; Schillaci, D.; Cusimano, M.G.; Leonchiks, A.; Zhulenkovs, D.; Basile, L.; et al. Discovery of a new class of sortase a transpeptidase inhibitors to tackle gram-positive pathogens: 2-(2-phenylhydrazinylidene)alkanoic acids and related derivatives. Molecules 2016, 21, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhulenkovs, D.; Rudevica, Z.; Jaudzems, K.; Turks, M.; Leonchiks, A. Discovery and structure-activity relationship studies of irreversible benzisothiazolinone-based inhibitors against Staphylococcus aureus sortase A transpeptidase. Bioorg. Med. Chem. 2014, 22, 5988–6003. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.K.; Ganesh Kumar, V.; Leela Madhuri, C.; Pardhasaradhi, M.; Durga Lakshmi, R.; Ravi, M.; Sri Ramudu, B.; Venkata Rao, S.V.; Ramachandran, D. Designing of potential inhibitors against Staphylococcus aureus sortase A: Combined analogue and structure based approach with in vitro validation. J. Mol. Graph. Model. 2015, 60, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Uddin, R.; Lodhi, M.U.; Ul-Haq, Z. Combined Pharmacophore and 3D-QSAR Study on A Series of Staphylococcus aureus Sortase A inhibitors. Chem. Biol. Drug Des. 2012, 80, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, H.; Zhu, K.; Gong, S.; Dramsi, S.; Wang, Y.-T.; Li, J.; Chen, F.; Zhang, R.; Zhou, L.; et al. Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc. Natl. Acad. Sci. USA 2014, 111, 13517–13522. [Google Scholar] [CrossRef] [PubMed]
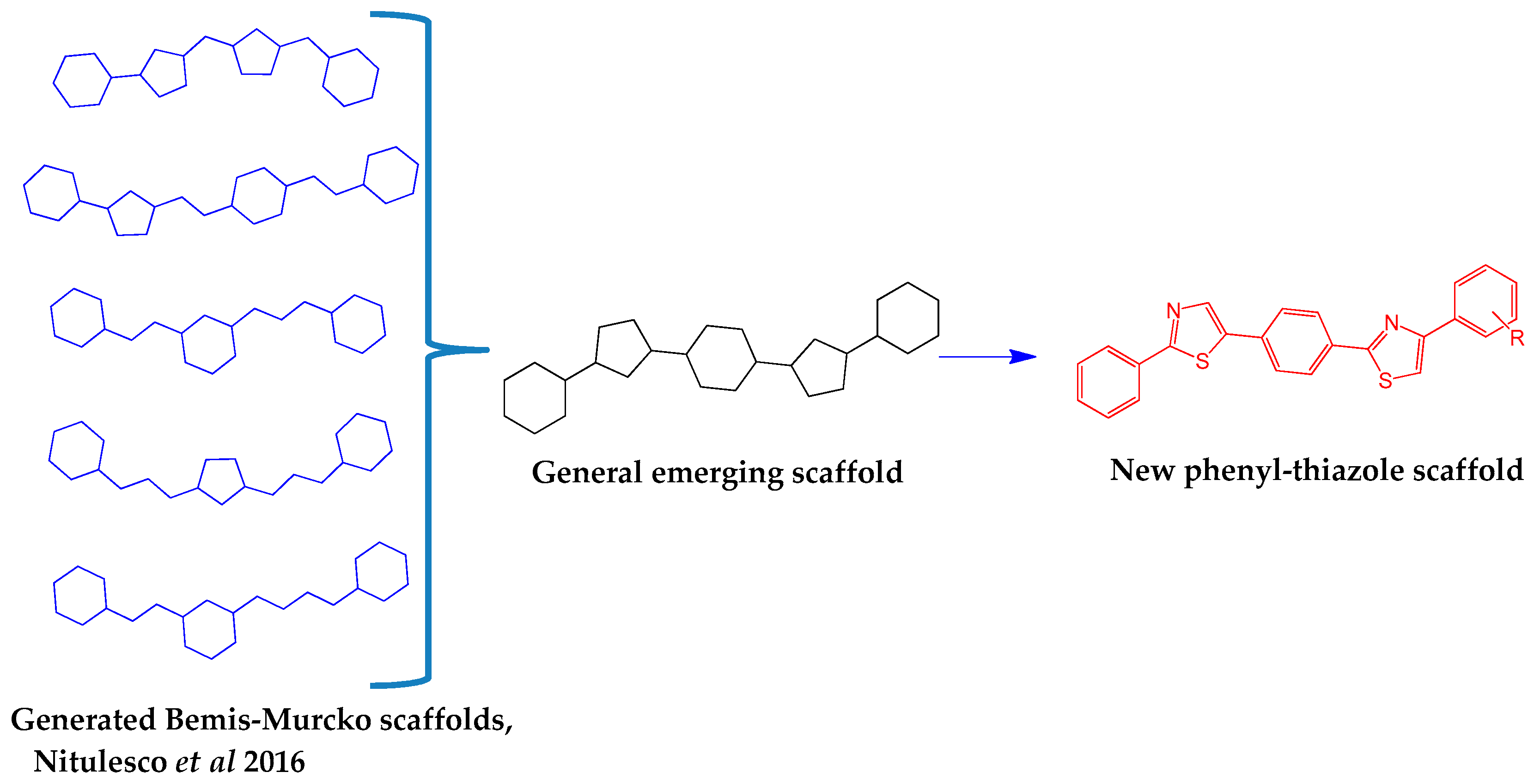
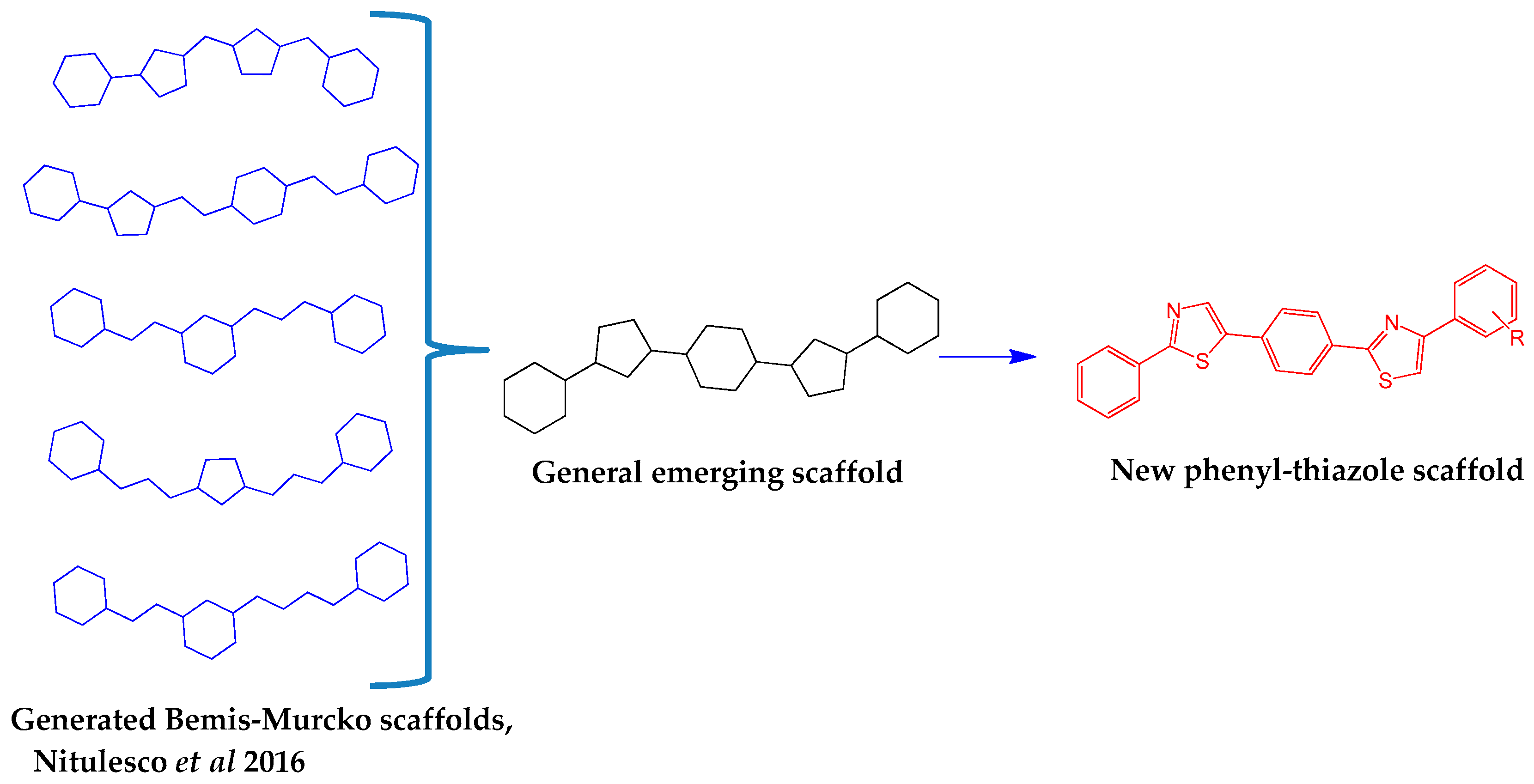
- Nitulescu, G.; Zanfirescu, A.; Olaru, O.T.; Nicorescu, I.M.; Nitulescu, G.M.; Margina, D. Structural analysis of sortase A inhibitors. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Raffa, D.; Maggio, B.; Raimondi, M.V.; Schillaci, D.; Daidone, G. Sortase A Inhibitors: Recent Advances and Future Perspectives. J. Med. Chem. 2015, 58, 9108–9123. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Uzelac, I.; Gottfries, J.; Eriksson, L.A. Exploration of multiple Sortase A protein conformations in virtual screening. Sci. Rep. 2016, 6, 20413. [Google Scholar] [CrossRef] [PubMed]
- Araniciu, C.; Maruţescu, L.; Oniga, S.; Oniga, O.; Chifiriuc, M.C.; Palage, M. Evaluation of the antimicrobial and anti-biofilm activity of some 4,2 and 5,2 bisthiazoles derivatives. Dig. J. Nanomater. Biostruct. 2014, 9, 123–131. [Google Scholar]
- Araniciu, C.; Oniga, S.; Oniga, O.; Palage, M.; Chifiriuc, M.-C.; Marutescu, L. Antimicrobial and Anti-Pathogenic Activity Evaluation of Some 2-(Trimethoxyphenyl)-4-Ar 1-5-R 2-Thiazoles. Farmacia 2015, 63, 40–45. [Google Scholar]
- Oniga, S.; Duma, S.; Oniga, O.; Tiperciuc, B.; Pirnau, A.; Araniciu, C.; Palage, M. Synthesis of some new 4-methyl-2-(4-pyridyl)-thiazole-5-yl-azoles as potential antimicrobial agents. Farmacia 2015, 63, 171–178. [Google Scholar]
- Oniga, S.; Araniciu, C.; Palage, M.; Stoica, C.; Chifiriuc, M.C.; Marutescu, L. Synthesis and bioevaluation of the antimicrobial features of some new thiazolyl-Azoles. Rev. Chim. 2016, 67, 426–429. [Google Scholar]
- Nitulescu, G.; Olaru, O.; Nitulescu, G.M.; Ungurianu, A.; Margina, D. Toxicity assessment of sortases inhibitors. Toxicol. Lett. 2016, 258, S75. [Google Scholar] [CrossRef]
- Suree, N.; Yi, S.W.; Thieu, W.; Marohn, M.; Damoiseaux, R.; Chan, A.; Jung, M.E.; Clubb, R.T. Discovery and structure-activity relationship analysis of Staphylococcus aureus sortase A inhibitors. Bioorg. Med. Chem. 2009, 17, 7174–7185. [Google Scholar] [CrossRef] [PubMed]
- Dramsi, S.; Trieu-Cuot, P.; Bierne, H. Sorting sortases: A nomenclature proposal for the various sortases of Gram-positive bacteria. Res. Microbiol. 2005, 156, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Okamiya, J. The Preparation and the Rates of the Reaction of Heterocyclic (Thiophen and Thiazole) Bromoketones with Thioamides. Nippon Kagaku Zasshi 1966, 87, 594–600. [Google Scholar] [CrossRef]
- Balaure, P.C.; Andronescu, E.; Grumezescu, A.M.; Ficai, A.; Huang, K.S.; Yang, C.H.; Chifiriuc, C.M.; Lin, Y.S. Fabrication, characterization and in vitro profile based interaction with eukaryotic and prokaryotic cells of alginate-chitosan-silica biocomposite. Int. J. Pharm. 2013, 441, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Limban, C.; Chifiriuc, M.C. Antibacterial activity of new dibenzoxepinone oximes with fluorine and trifluoromethyl group substituents. Int. J. Mol. Sci. 2011, 12, 6432–6444. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Sippl, M.J. Recognition of errors in three-dimensional structures of proteins. Proteins Struct. Funct. Genet. 1993, 17, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining Global and Local Measures for Structure-Based Druggability Predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; De Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Jain, H.; Mulay, S.; Mullany, P. Persistence of endodontic infection and Enterococcus faecalis: Role of horizontal gene transfer. Gene Rep. 2016, 5, 112–116. [Google Scholar] [CrossRef]
Sample Availability: Samples of all the compounds are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Enterococcus faecalis ATCC 29212 | Staphylococcus aureus ATCC 6538 | Staphylococcus aureus BAA 1026 | Staphylococcus saprophyticus ATCC 15305 | Bacillus subtilis ATCC 6633 | Escherichia coli ATCC 8739 | Pseudomonas aeruginosa ATCC 27853 |
|---|---|---|---|---|---|---|---|
| C1 | 14 | 9 | 10 | 10 | 0 | 0 | 0 |
| C2 | 13 | 8 | 8 | 11 | 0 | 0 | 0 |
| C3 | 14 | 8 | 7 | 13 | 0 | 0 | 0 |
| C4 | 12 | 8 | 8 | 10 | 0 | 0 | 0 |
| C5 | 13 | 9 | 8 | 12 | 0 | 0 | 0 |
| C6 | 14 | 10 | 0 | 0 | 0 | 0 | 0 |
| C7 | 10 | 0 | 0 | 0 | 0 | 0 | 0 |
| C8 | 12 | 9 | 10 | 10 | 0 | 0 | 0 |
| Ciprofloxacin | 15 | 14 | 15 | 16 | 14 | 14 | 16 |
| DMSO | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Compound | Enterococcus faecalis ATCC 29212 | Staphylococcus aureus ATCC 6538 | Staphylococcus aureus BAA 1026 | Staphylococcus saprophyticus ATCC 15305 |
|---|---|---|---|---|
| C1 | 0.25 | >1 | >1 | >1 |
| C2 | 0.125 | >1 | 0.125 | 0.032 |
| C3 | 0.062 | >1 | >1 | 0.032 |
| C4 | 0.062 | >1 | 0.032 | 0.125 |
| C5 | 0.062 | >1 | >1 | 0.016 |
| C6 | 0.016 | >1 | - | - |
| C7 | 0.25 | - | - | - |
| C8 | 0.032 | >1 | >1 | 0.002 |
| Ciprofloxacin | 0.002 | 0.0005 | 0.001 | 0.0005 |
| Compound | Enterococcus faecalis ATCC 29212 | Staphylococcus aureus ATCC 6538 | Staphylococcus aureus BAA 1026 | Staphylococcus saprophyticus ATCC 15305 | Bacillus subtilis ATCC 6633 | Escherichia coli ATCC 8739 | Pseudomonas aeruginosa ATCC 27853 |
|---|---|---|---|---|---|---|---|
| C1 | 0.004 | 1 | 1 | >1 | 1 | 1 | 1 |
| C2 | 0.002 | 1 | 1 | >1 | 1 | 1 | 1 |
| C3 | 0.002 | >1 | >1 | >1 | 1 | >1 | 1 |
| C4 | 0.004 | >1 | 0.062 | >1 | 1 | >1 | 1 |
| C5 | 0.002 | 1 | 0.25 | >1 | 1 | 1 | >1 |
| C6 | 0.004 | 1 | 0.25 | >1 | >1 | 1 | >1 |
| C7 | 0.008 | 1 | 0.25 | >1 | >1 | 1 | 1 |
| C8 | 0.016 | >1 | >1 | >1 | >1 | 1 | 1 |
| Compound | E. faecalis Sortase A | S. aureus Sortase A | ||
|---|---|---|---|---|
| Binding Energy (kcal/mol) | Inhibition Constant (nM) | Binding Energy (kcal/mol) | Inhibition Constant (nM) | |
| C1 | −9.54 | 101.64 | −8.16 | 1043.84 |
| C2 | −9.46 | 116.34 | −7.25 | 4849.25 |
| C3 | −10.54 | 18.80 | −9.00 | 252.88 |
| C4 | −9.94 | 51.75 | −7.28 | 4609.83 |
| C5 | −10.64 | 15.88 | −8.50 | 588.05 |
| C6 | −10.56 | 18.17 | −7.90 | 1618.89 |
| C7 | −9.67 | 81.62 | −8.37 | 732.32 |
| C8 | −10.01 | 45.98 | −8.61 | 488.41 |

| Compound | Ligand Atom ID | Interacting AA Residue | Bond Length (Å) |
|---|---|---|---|
| C1 | N1 | Thr122-OH | 2.9 |
| N1 | Thr122-C=O | 3.1 | |
| C2 | N1 | Thr122-OH | 3.1 |
| C3 | N1 | Thr122-OH | 2.8 |
| N2 | Ser85-C=O | 3.0 | |
| O-nitro | Arg224-C=NH | 2.8 | |
| O-nitro | Arg224-C-NH2 | 3.5 | |
| O-nitro | Asn221-C-NH2 | 2.5 | |
| C4 | N1 | Thr122-OH | 2.9 |
| N2 | Ser85-C=O | 2.9 | |
| O-methoxy | Arg224-C=NH | 3.0 | |
| O-methoxy | Arg224-C-NH2 | 3.3 | |
| C5 | N1 | Thr122-OH | 2.8 |
| N-cyano | Asn221-C=O | 3.5 | |
| C6 | N1 | Thr122-OH | 2.9 |
| N1 | Thr122-C=O | 3.1 | |
| C7 | N2 | Thr122-OH | 2.9 |
| N2 | Thr122-C=O | 3.1 | |
| OH | Glu135-COOH | 2.8 | |
| NH2 | Asp82-COOH | 3.0 | |
| C8 | N1 | Thr122-OH | 2.9 |
| N1 | Thr122-OH | 3.0 |
| Parameter | E. faecalis | S. aureus |
|---|---|---|
| Volume | 580.03 Ǻ3 | 387.65 Ǻ3 |
| Internal surface | 1022.95 Ǻ2 | 654.68 Ǻ2 |
| H bond donor AA | 23 | 9 |
| H bond acceptor AA | 56 | 24 |
| Non polar AA | 29% | 52% |
| Polar non-ionic AA | 34% | 14% |
| Cationic AA | 21% | 24% |
| Anionic AA | 16% | 10% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oniga, S.D.; Araniciu, C.; Palage, M.D.; Popa, M.; Chifiriuc, M.-C.; Marc, G.; Pirnau, A.; Stoica, C.I.; Lagoudis, I.; Dragoumis, T.; et al. New 2-Phenylthiazoles as Potential Sortase A Inhibitors: Synthesis, Biological Evaluation and Molecular Docking. Molecules 2017, 22, 1827. https://doi.org/10.3390/molecules22111827
Oniga SD, Araniciu C, Palage MD, Popa M, Chifiriuc M-C, Marc G, Pirnau A, Stoica CI, Lagoudis I, Dragoumis T, et al. New 2-Phenylthiazoles as Potential Sortase A Inhibitors: Synthesis, Biological Evaluation and Molecular Docking. Molecules. 2017; 22(11):1827. https://doi.org/10.3390/molecules22111827
Chicago/Turabian StyleOniga, Smaranda Dafina, Cătălin Araniciu, Mariana Doina Palage, Marcela Popa, Mariana-Carmen Chifiriuc, Gabriel Marc, Adrian Pirnau, Cristina Ioana Stoica, Ioannis Lagoudis, Theodoros Dragoumis, and et al. 2017. "New 2-Phenylthiazoles as Potential Sortase A Inhibitors: Synthesis, Biological Evaluation and Molecular Docking" Molecules 22, no. 11: 1827. https://doi.org/10.3390/molecules22111827
APA StyleOniga, S. D., Araniciu, C., Palage, M. D., Popa, M., Chifiriuc, M.-C., Marc, G., Pirnau, A., Stoica, C. I., Lagoudis, I., Dragoumis, T., & Oniga, O. (2017). New 2-Phenylthiazoles as Potential Sortase A Inhibitors: Synthesis, Biological Evaluation and Molecular Docking. Molecules, 22(11), 1827. https://doi.org/10.3390/molecules22111827