Abstract
Amomum kravanh is an important edible and medicinal herb, the dried fruits of which are widely used in traditional herbal medicine as cardamom. We sequenced and analyzed the complete chloroplast (cp) genome of A. kravanh with herbgenomics technologies. The size of the A. kravanh cp genome was 162,766 bp, which consisted of long (LSC; 87,728 bp) and short (SSC; 15,390 bp) single-copy regions, separated by a pair of inverted repeats (IRs; 29,824 bp). The genome encoded 114 unique genes, including 80 protein-coding genes, 30 tRNAs and four rRNAs. A total of 299 simple sequence repeats (SSRs) were identified in the A. kravanh cp genome, which provides an effective method to study species identification and population genetics of the medicinal plant. Moreover, one complement, 12 forward, 12 palindrome and two reverse repeats were detected. Comparative cp genome sequence analysis of four Zingiberaceae species indicated that their intergenic spacers are highly divergent, although the gene order, gene content and genome structure differed only minimally. In particular, there was a remarkable expansion of the IR regions in the A. kravanh cp genome. Phylogenetic analysis strongly supported a sister relationship between A. kravanh and Alpinia zerumbet. This study identified the unique characteristics of the A. kravanh cp genome and might provide valuable information for future studies aiming for Amomum identification, and provide insights into the taxonomy of the commelinids.
1. Introduction
As a major plant cell organelle, the chloroplast (cp) is crucial to the growth and development of plants through its roles in photosynthesis and secondary metabolic activities. In recent years, cp genomes have been widely used for species identification, investigation of phyletic evolution and genetic engineering because of their highly conserved nature and role in monolepsis. Development of sequencing technologies has led to a sharp increase in the number of cp genome sequences; however, much remains to be achieved, particularly for medicinal species. No Amomum cp genome sequences have yet been reported, which has led to delays in investigation of the genetics and breeding of cardamom. In addition, the lack of complete cp genomes has hindered phylogenetic analysis of the commelinids.
Amomum kravanh (family Zingiberaceae) is one of the original types of cardamom plant, which is a significant traditional Chinese medicine that had been primarily imported from Thailand, Vietnam and Cambodia since the 1960s, and is now widely cultivated in southern China. Cardamom mainly contains volatile oils, which can promote the secretion of gastric juices and have antiemetic properties. Modern pharmacological research has demonstrated that the Yishen Paizhuo decoction (traditionally recognized as a decoction to support kidney function), which is mainly composed of cardamom, can be used to treat chronic renal failure (CRF) and prolong survival of patients with this condition [1]. Not only are the dried fruits of A. kravanh medicinal, but they can also be used as a culinary spice, similar to products from many other species in the Zingiberaceae, cassia and garlic, among others [2]. The widespread use of A. kravanh has increased demand for the plant, while research related to its introduction and cultivation has been insufficient, leading to the widespread occurrence of adulterants, which represent a serious health threat to consumers.
A. kravanh is a perennial herb of the order Zingiberales, which belongs to the clade commelinids. According to Angiosperm Phylogeny Group IV, the magnolliids, monocots and eudicots comprise the core groups of angiosperms, with commelinids the basal taxon in the monocot clade; however, there is tremendous controversy regarding the phylogeny of the commelinids, basal taxa in the order Poales. Luo et al. compared monocot pollen morphological data in an attempt to catalog these disputes [3]. Further research on the phylogenetics of the commelinids will assist in determining the evolutionary relationships among the major taxonomic categories of angiosperms.
Here we report the complete cp genome sequence of A. kravanh, including description of its essential characteristics and repeat sequences and comparative analyses of the cp whole genome. Moreover, we constructed a phylogenetic tree of the commelinids. These results will assist in species identification and breeding of A. kravanh varieties, and provide insights into the taxonomy of the commelinids.
2. Results and Discussion
2.1. Characteristics of the A. kravanh cp Genome
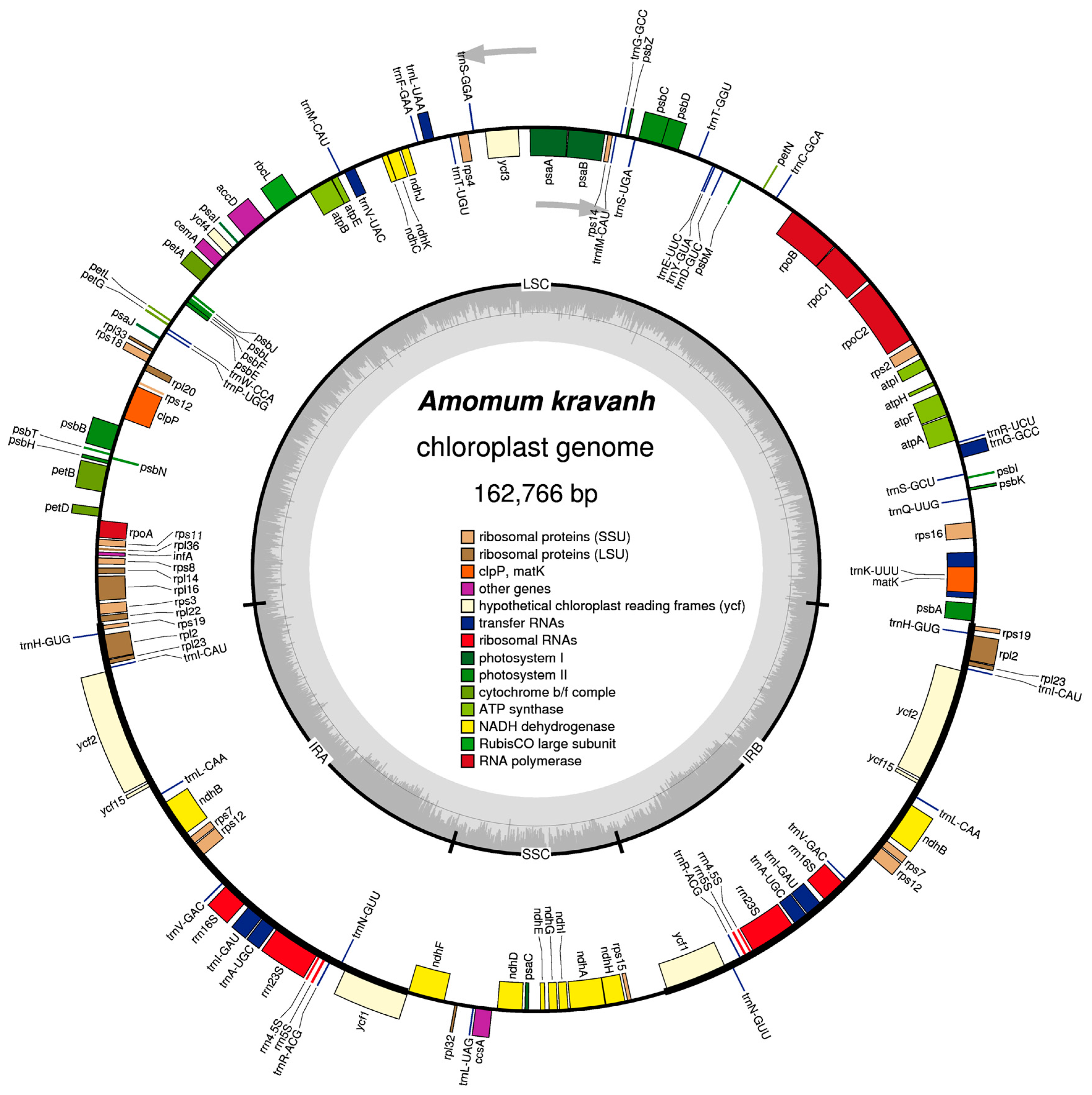
The complete cp genome of A. kravanh was revealed as 162,766 bp in size, with a typical quadripartite structure (Table 1 and Figure 1). The long (LSC; 87,728 bp) and short (SSC; 15,390 bp) single-copy regions were separated by a pair of inverted repeats (IRs; 29,824 bp each). The overall GC content of the A. kravanh cp genome was 31.3%, which is consistent with that reported for other cp genomes [4]. The IR regions had higher GC content than the LSC and SSC regions (Table 1). Within the protein-coding regions (CDS), the AT content of third-codon positions (70.7%) was higher than that of the first and second positions (Table 1), similar to other reported cp genomes. This bias can be used to discriminate cp DNA from nuclear and mitochondrial DNA [5,6,7,8]. In addition, the cp genome was almost equally split into coding and non-coding (introns, pseudogenes and intergenic spacer) regions.

Table 1.
Base composition in the Amomum kravanh chloroplast genome.
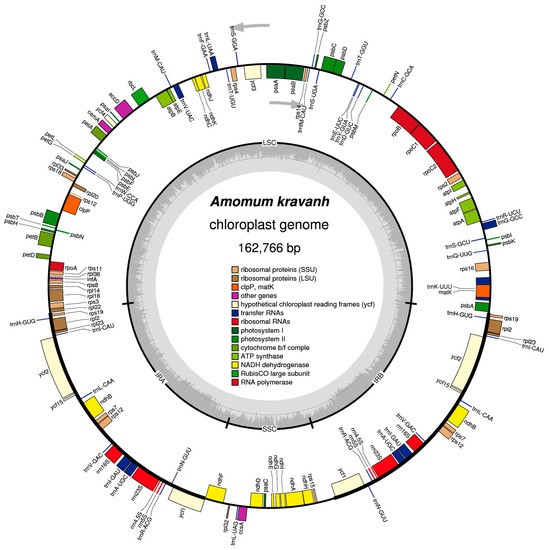
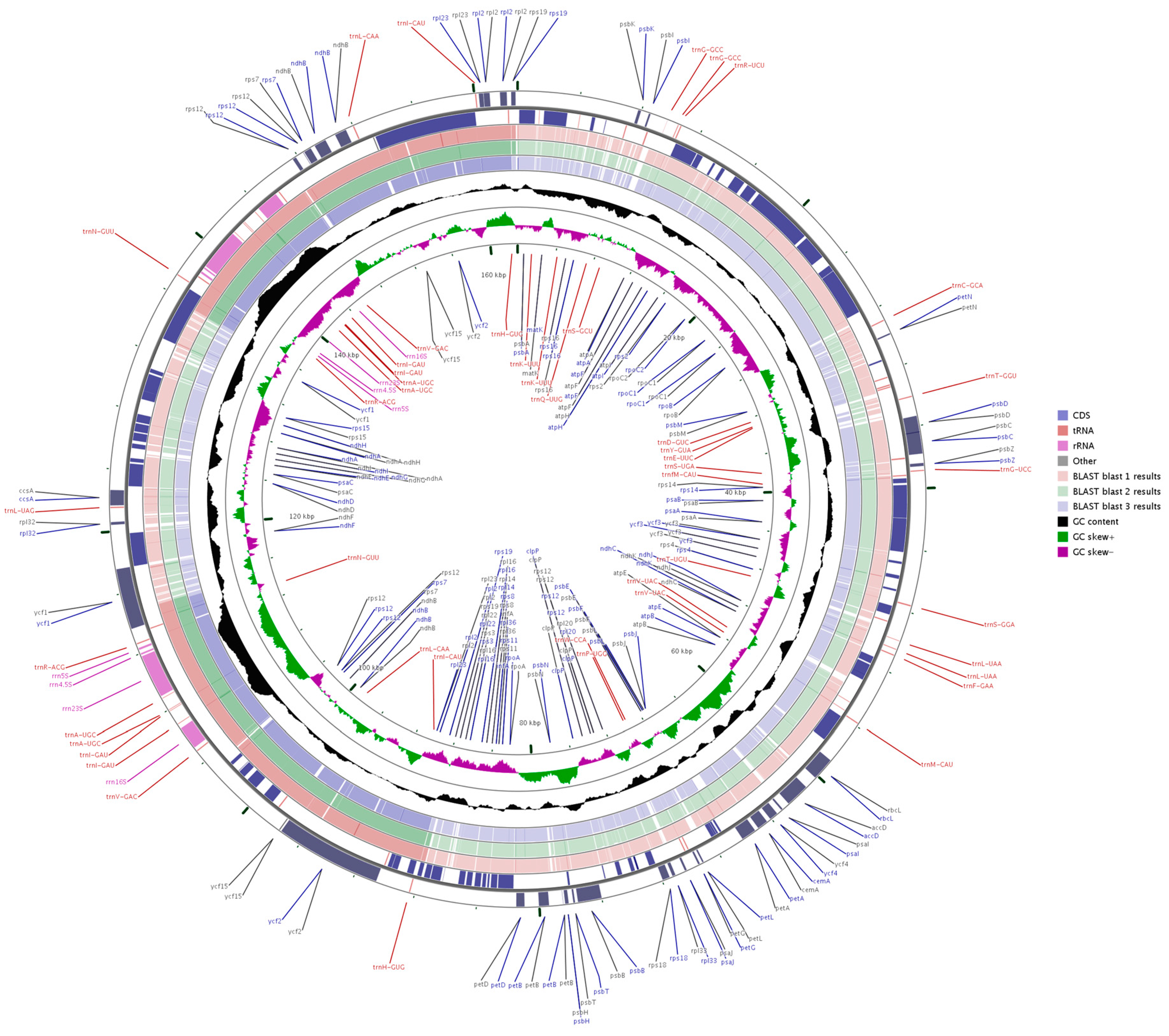
Figure 1.
Map of the A. kravanh cp genome map. Genes drawn inside the circle are transcribed clockwise, and those outside are counterclockwise. Genes belonging to different functional groups are color-coded. The darker gray in the inner circle corresponds to GC content, while the lighter gray corresponds to AT content.
In total, the A. kravanh cp genome encoded 135 functional genes, the positions of which are presented in Figure 1. There were 114 unique genes remaining after duplicates were removed, including 80 protein-coding, 30 tRNA and four rRNA genes (Table S2). Among these genes, nine protein-coding, eight tRNA and all four rRNA genes were duplicated in the IR regions. The LSC region contained 60 protein-coding and 21 tRNA genes, while 11 protein-coding genes and one tRNA gene are located in the SSC region. Introns have important roles in the regulation of gene expression, and can improve exogenous gene expression at specific locations and times; thus, introns can be useful tools for improvement of transformation efficiency [9]. Among the 114 unique genes, eight protein-coding (rps16, rpl16, rpl2, rpoC1, ndhA, ndhB, petB and atpF) and six tRNA (trnA-UGC, trnG-GCC, trnI-GAU, trnK-UUU, trnL-UAA and trnV-UAC) genes contained one intron, and three genes (clpP, rps12 and ycf3) contained two introns.
RSCU (relative synonymous codon usage) is a measure of non-uniform synonymous codon usage in coding sequences. RSCU values <1.00 indicate less-frequent use of a codon than expected, whereas codons used more frequently than excepted score >1.00. Based on the sequences of protein-coding genes (CDS), the codon usage frequency was estimated for the A. kravanh cp genome (summarized in Table 2). In total, the A. kravanh cp genome genes contain 27,214 codons. Other than Met, amino acid codons in the A. kravanh cp genome preferentially end with A or U (RSCU > 1). This codon usage pattern is similar to those reported for other cp genomes, and may be driven by a composition bias for a high proportion of A/T. In addition, codons ending in A and/or U accounted for 71.2% of all CDS codons. The majority of protein-coding genes in land-plant cp genomes employ standard ATG initiator codons. The use of the start codon (ATG) and TGG, encoding Trp, exhibited no bias (RSCU = 1) in the A. kravanh cp genome; however, two genes used alternatives to the AUG as a start codon as follows: ndhD, ATC and rpl2, ATA.
Table 2.
Codon–anticodon recognition patterns and codon usage in the A. kravanh chloroplast genome.
2.2. Analysis of SSRs and Long Repeats
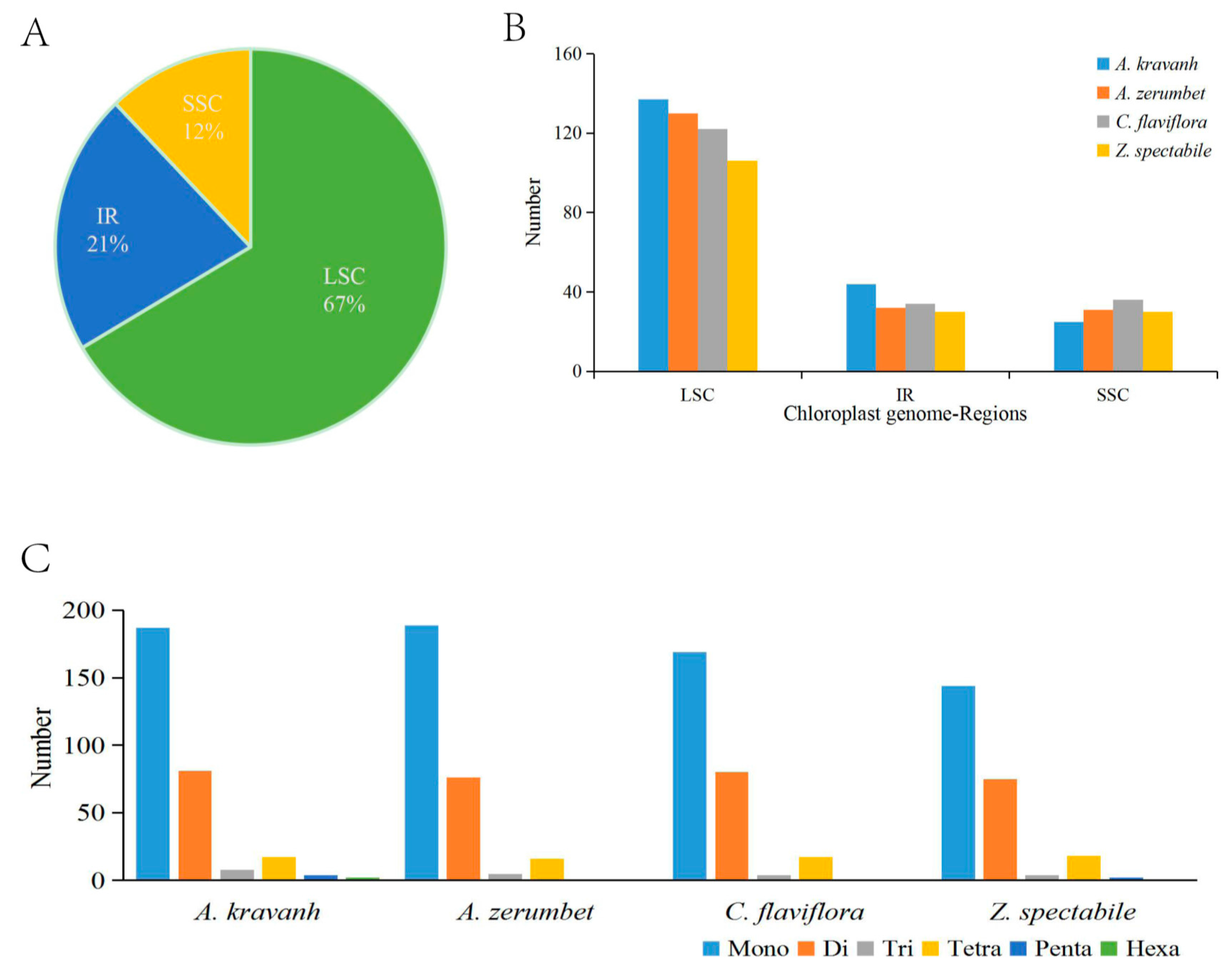
The SSRs, also known as microsatellites, are a group of tandem repeated sequences, which generally consist of 1–6 nucleotide repeat units, and are widely distributed in cp genomes. SSRs are important for plant typing and are widely used as molecular markers for species identification [10,11,12]. The distribution of SSRs in the A. kravanh cp genome was analyzed in this study, and a total of 299 SSRs were detected, including 187 mono-, 81 di-, 8 tri-, 17 tetra-, four penta- and two hexa-nucleotide SSRs, the majority of which were located in the LSC region. Our results are consistent with the opinion that SSRs in cp genomes are generally composed of short polyadenine (polyA) or polythymine (polyT) repeats, which contribute to the A/T richness of cp genomes [13]. Furthermore, we counted the number of SSRs among four Zingiberaceae cp genomes (A. kravanh, Alpinia zerumbet, Curcuma flaviflora and Zingiber spectabile) and found that there was minimal difference in the distribution pattern and number of SSRs among the four cp genomes (Figure 2), although hexa-nucleotide SSRs were only identified in the A. kravanh cp genome. These results will undoubtedly provide cp SSR marker information for the analysis of genetic diversity in A. kravanh and its related species.
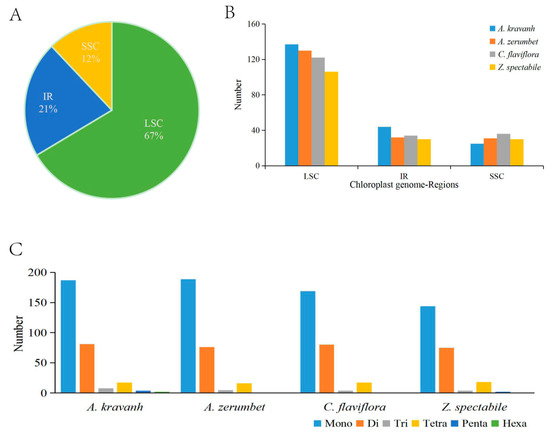
Figure 2.
SSR (simple sequence repeats) analysis of four Zingiberaceae chloroplast genomes (A. kravanh, A. zerumbet, C. flaviflora and Z. spectabile). (A) Presence of SSRs in the LSC, SSC and IR regions (A. kravanh); (B) presence of SSRs in the LSC, SSC and IR regions (A. kravanh, A. zerumbet, C. flaviflora and Z. spectabile); (C) presence of polymers in the cp genome of A. kravanh, A. zerumbet, C. flaviflora and Z. spectabile.
Repeat structure analysis revealed 27 long repeats in total: one complement, 12 forward (direct), 12 palindrome (inverted) and two reverse repeats in the A. kravanh cp genome (Table 3). More than half of the repeats were located in the intergenic or intronic regions, and the rest in protein-coding regions. The majority of these repeats were between 30 and 60 bp, while the ycf1 gene possessed the longest palindrome repeats (up to 181 bp). Two pairs of repeats are associated with tRNA genes (trnK-UUU, trnI-CAU). In addition, one complement, seven forward, six palindromic and two reverse repeats were distributed in the LSC region. Short dispersed repeats are considered to be a major factor promoting cp genome rearrangements, which may facilitate intermolecular recombination and create diversity among the cp genomes in a population. Hence, the repeats identified in this study will provide valuable information to support investigation of the phylogeny of Amomum and A. kravanh population studies.
Table 3.
Long repeat sequences in the A. kravanh chloroplast genome.
2.3. Comparative Chloroplast Genomic Analysis
Three sequences representing the Zingiberaceae (A. zerumbet, C. flaviflora and Z. spectabile) were selected for comparison with A. kravanh. Pairwise cp genome alignment between A. kravanh and the other three cp genomes revealed a high degree of synteny conservation (Figures S1–S3). The overall sequence identity among the four Zingiberaceae cp genomes was plotted using mVISTA and CGView, with the annotated A. kravanh sequence as the reference (Figure 3 and Figure S4). The comparison showed that the IR regions were less divergent than the LSC and SSC. In addition, the coding regions were more conserved than the non-coding regions, and the most highly divergent regions among the four cp genomes were in the intergenic spacers, including trnS-GCU-trnG-GCC, atpH-atpI, trnC-GCA-petN, trnE-UUC-trnT-GGU, trnT-GGU-psbD, ndhC-trnV-UAC, petA-psbJ and psbE-petL in the LSC, and ndhF-rpl32, rpl32-trnL-UAG, psaC-ndhE and rps15-ycf1 in SSC, similar to other plant cp genomes [14]. Moreover, the most divergent coding regions are the ndhA, ycf1, petB and ycf2 genes in the cp genomes. Sequence identity analysis showed that A. zerumbet had the highest sequence similarity to A. kravanh, which was consistent with the results of phylogenetic analysis. In our study, we observed that all eight rRNA genes were the most conserved. The highly divergent regions identified in this research could be used to develop markers or specific barcodes that would maximize the ability to differentiate species within the Zingiberaceae.
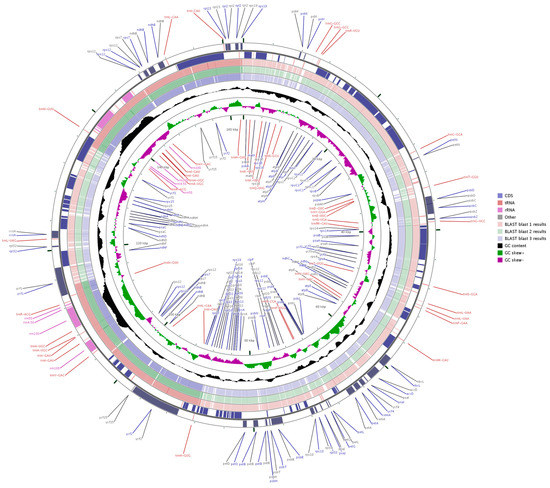
Figure 3.
Genome comparison of three commelinid cp genomes (from blast 1 to 3: A. zerumbet, C. flaviflora and Z. spectabile) with that of A. kravanh using GCView. The two outer narrow rings show gene positions based on the A. kravanh cp genome. The inner two rings indicate GC skew in A. kravanh. GC skew+ indicates G > C, GC skew- indicates G < C.
2.4. IR Contraction and Expansion
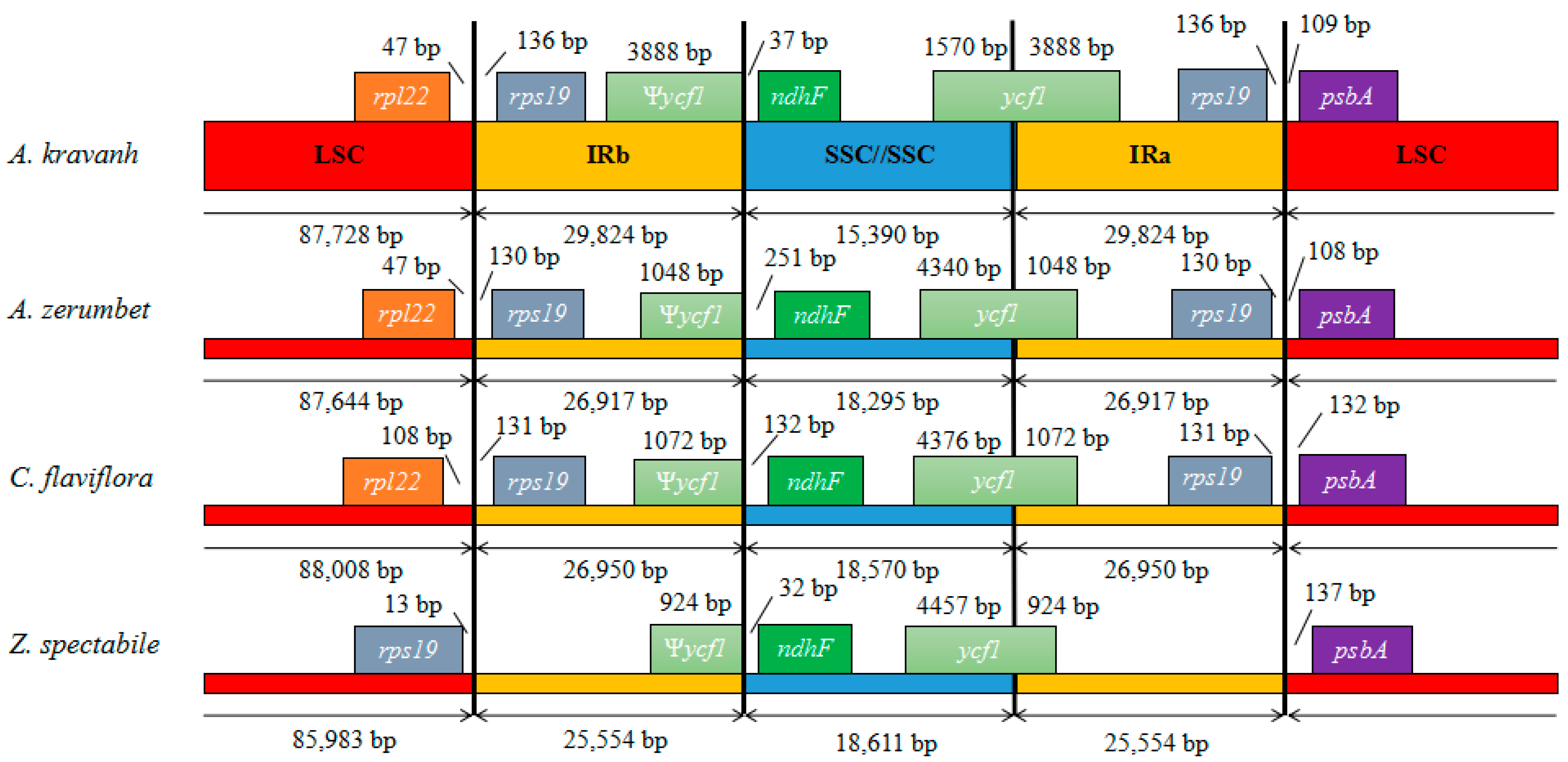
Contraction and expansion at the borders of IR regions are common evolutionary events considered the main reason for size differences among cp genomes [11,13,14,15,16]. Detailed comparison of the junctions between the IR and LSC/SSC regions among the four Zingiberaceae cp genomes (A. kravanh, A. zerumbet, C. flaviflora and Z. spectabile) is presented in Figure 4. In addition, a comparison of cp genome size among the examined Zingiberaceae species is provided in Table S3. As for other reported cp genomes, the IRa/SSC border was generally positioned in the coding region of the ycf1 gene, resulting in a deletion of the 5′ end of the gene, to generate a pseudogene at the IRb/SSC border. The ycf1 pseudogene has proven useful in analysis of cp genome variation in higher plants and algae [17], and varies in length from 924–3888 bp in the cp genomes compared. In the A. kravanh cp genome in particular, there was a remarkable expansion of the IR regions, resulting in the largest IR (29,824 bp) and smallest SSC (15,390 bp) regions relative to the other three cp genomes (Table S3). The photosynthetic gene, ndhF, was 37, 251, 132 and 32 bp from the IRb/SSC border in A. kravanh, A. zerumbet, C. flaviflora and Z. spectabile, respectively. The rps19 gene, which is one of the most abundant transcripts in the cp genome, was situated in the IR regions of all the cp genomes included in the comparison, except for Z. spectabile. In the Z. spectabile cp genome, rps19 was located in the LSC region, which may be because of the contraction of the IR regions, which makes the whole cp genome of this species shorter. Although the IR/LSC boundaries are not static among the Zingiberaceae species cp genomes analyzed, the dynamic processes appear to have been confined to conservative expansions and contractions, similar to other eukaryon plants [18].
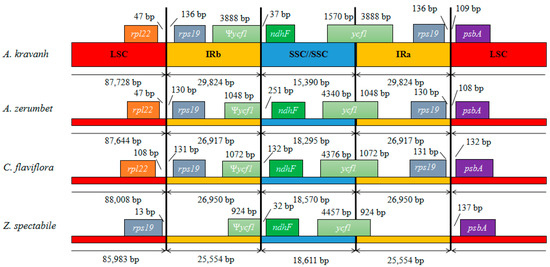
Figure 4.
Comparison of the borders of LSC, SSC and IR regions among four chloroplast genomes. Ψ, pseudogenes.
2.5. Analysis of Synonymous and Non-Synonymous Substitution Rates
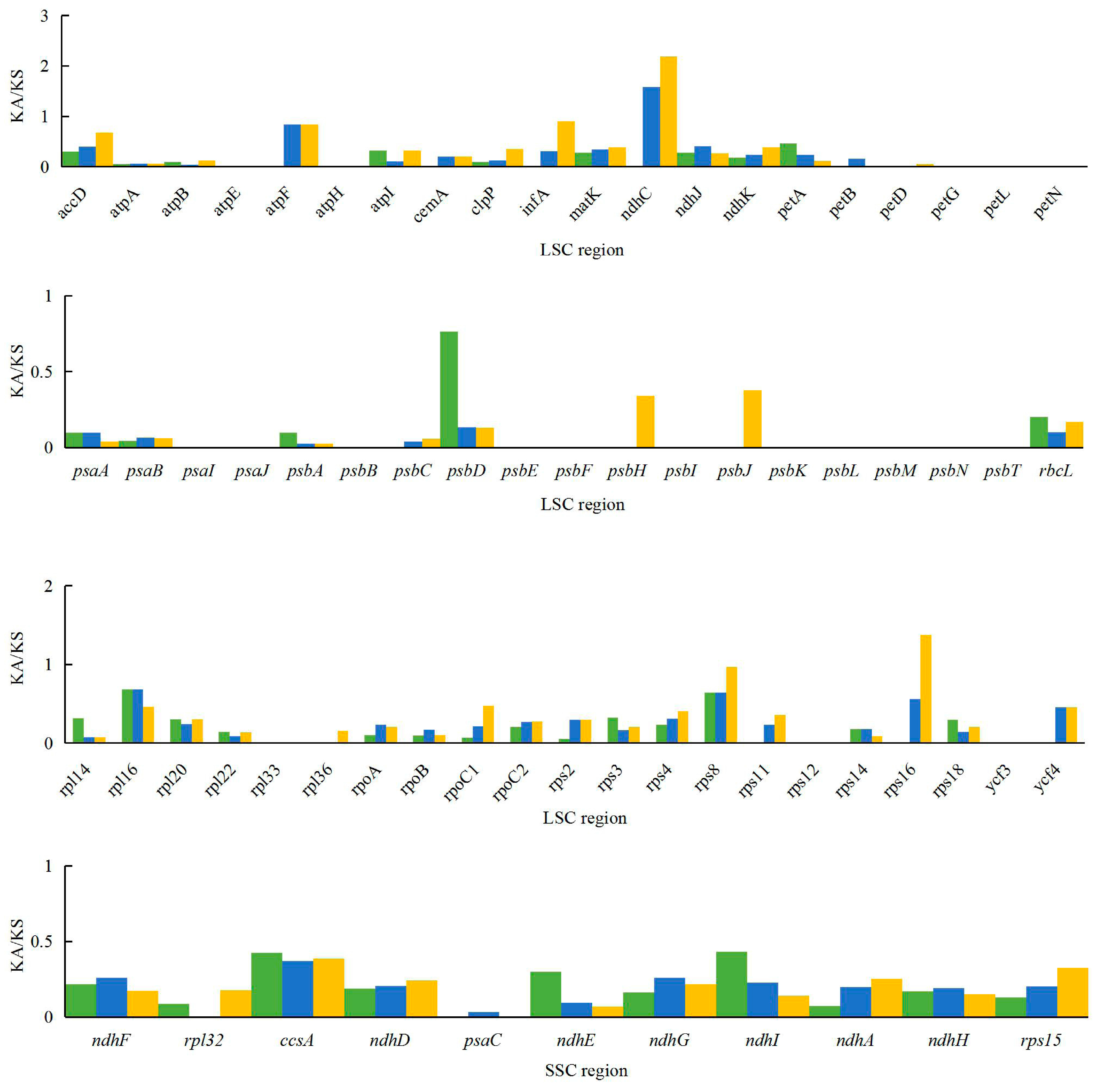
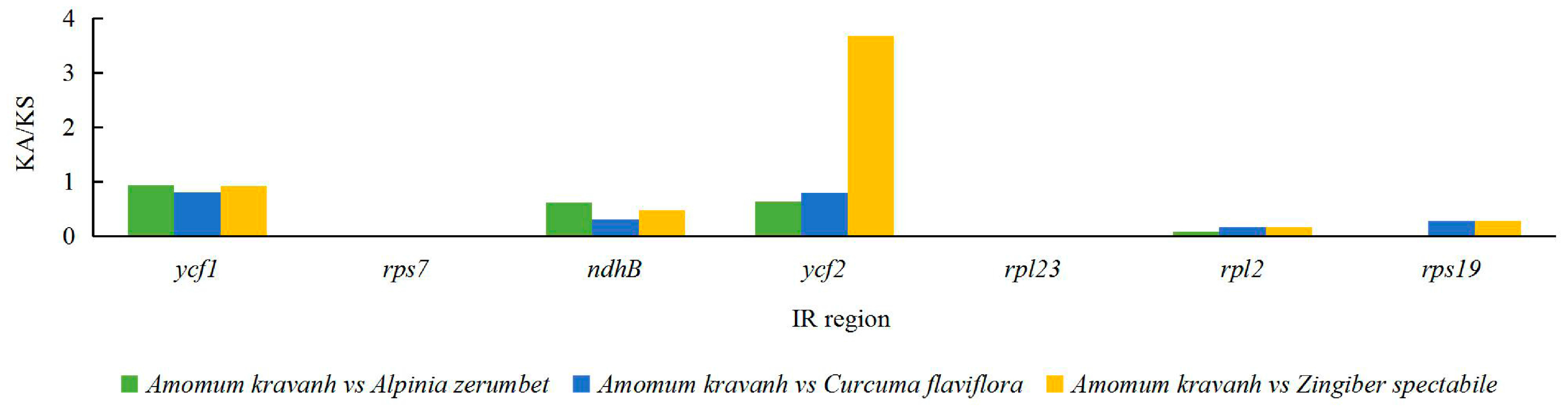
The Ka/Ks ratio is widely used to evaluate the evolutionary forces on specific groups of genes; ratios > 1 indicate positive selection, values < 1 indicate negative (purifying) selection, and values of 1 indicate neutral selection. In this study, a total of 80 protein-coding genes in the A. kravanh cp genome were included in the analysis of synonymous and non-synonymous substitutions rates, relative to A. zerumbet, C. flaviflora and Z. spectabile (Figure 5). The Ka/Ks ratios of the majority of genes were <1 compared with the three closely related species. This indicates that the majority of protein-coding regions in the A. kravanh cp genome have been under strong purifying selection during evolution. In addition to genes with high Ka/Ks values, the Ks values (≥1) of ndhC, rps16 and ycf2 indicate they have undergone positive selection, which was also found from analysis of other cp genomes [19]. However, compared with previous studies, it is clear that cp genes may be influenced by varying levels of selection pressure in different plants.
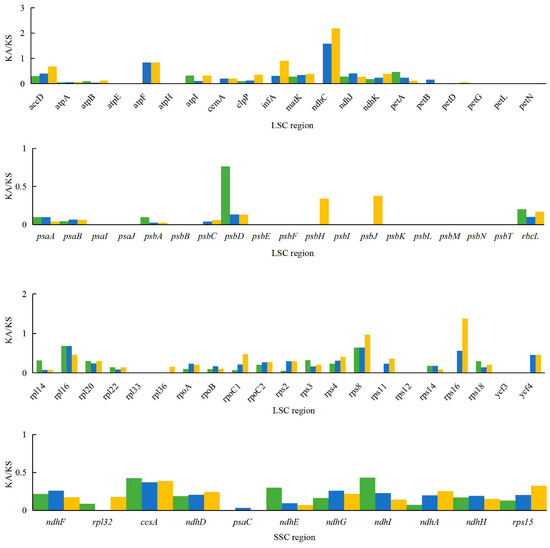
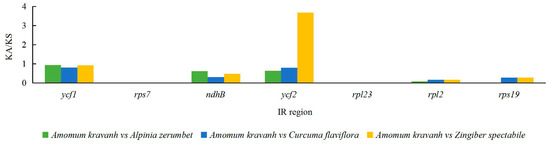
Figure 5.
Ka/Ks values of 80 protein-coding genes of A. kravanh, A. zerumbet, C. flaviflora and Z. spectabile.
2.6. Phylogenetic Analysis
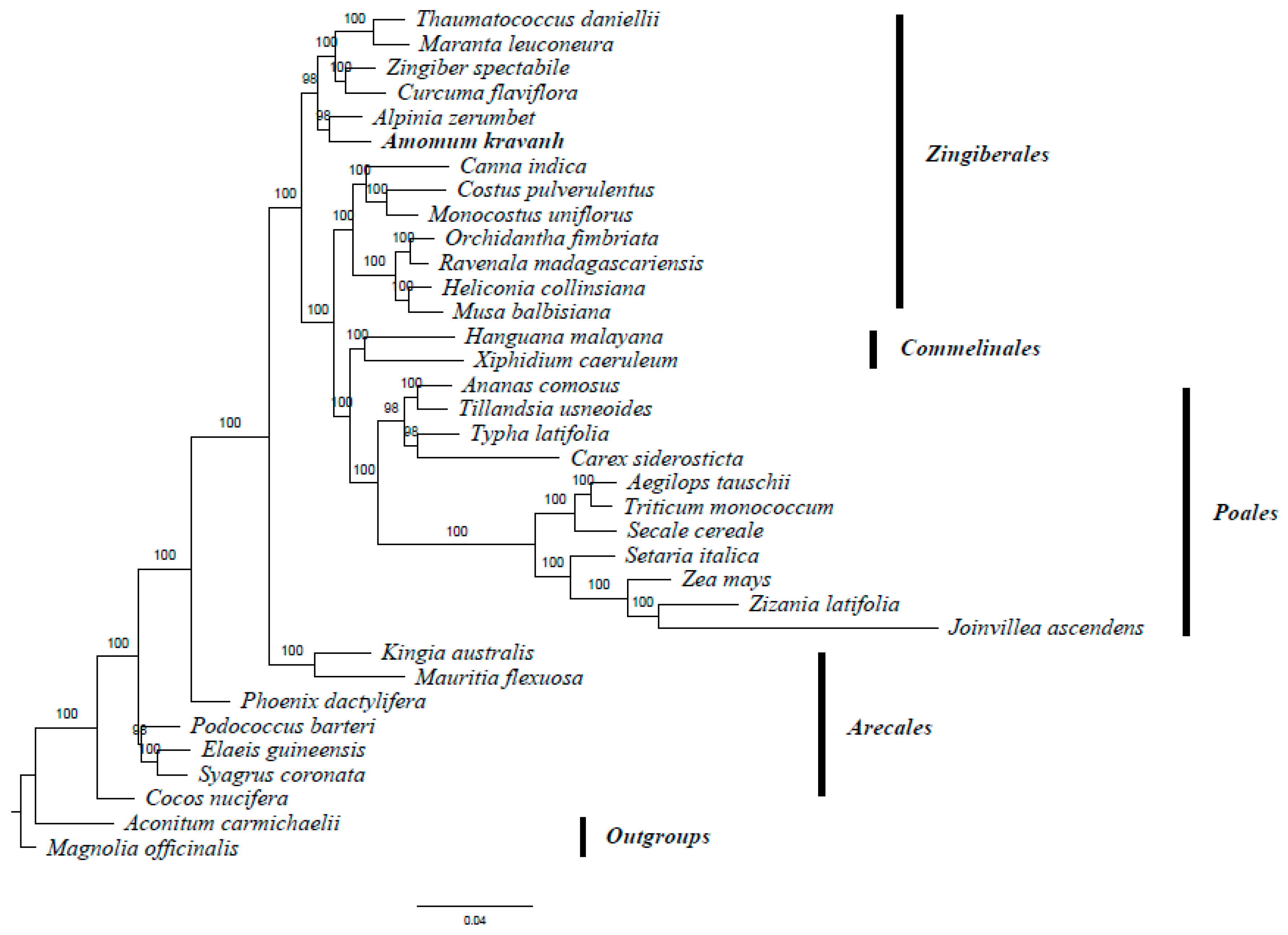
A. kravanh belongs to the Zingiberales order in the commelinid clade. Several studies have reported analysis of phylogenetic relationships within the commelinid clade based on morphological and molecular characteristics [20,21,22,23]. The availability of complete cp genome sequences of A. kravanh and other commelinid species provided us with data to investigate the molecular evolution of A. kravanh and the phylogeny of the commelinid clade. In this study, we performed multiple sequence alignments of 58 protein-coding genes commonly present in cp genomes from species belonging to 33 commelinid-clade species. The cp genomes of Magnolia officinalis and Aconitum carmichaelii were included as outgroups. On the basis of the GTR + G + I nucleotide substitution model recommended, a total of 39,750 nucleotide positions were analyzed, and the best-scoring ML (maximum likelihood) tree (final ML optimization likelihood = −281,929.050009) is presented in Figure 6. MP (maximum parsimony) analysis was also performed (Figure S5), and the resulting topology was relatively consistent with that produced by ML analysis, with a consistency index (CI) of 0.5453, a homoplasy index (HI) of 0.4547 and a retention index (RI) of 0.6772. The relationships among major basal commelinid lineages in our analyses are basically consistent with APG IV. ML and MP phylogenetic results both strongly supported the position in the Zingiberaceae of A. kravanh as a sister of the closely related species A. zerumbet, which was also used as cardamom in ancient China [24]. The results of this study should be validated in future analyses with more complete cp genomes and sophisticated analytical approaches.
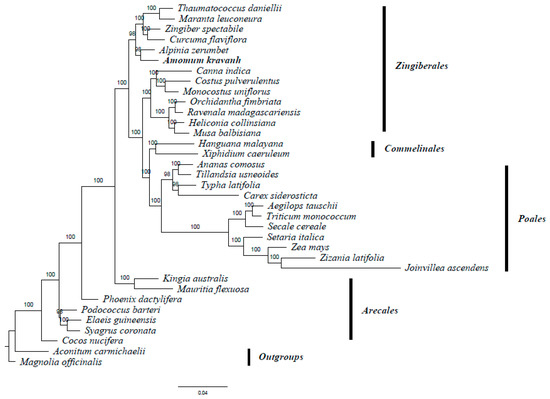
Figure 6.
ML phylogenetic tree of 33 taxa in the commelinid clade based on the concatenated sequences of 58 shared chloroplast protein-coding genes. Numbers above each node are ML bootstrap values >50%. Magnolia officinalis and Aconitum carmichaelii were used as outgroups.
3. Materials and Methods
3.1. DNA Sequencing and Genome Assembly
Fresh A. kravanh leaves were collected from cultivated fields in Hainan Province, China. Total chloroplast DNA (cpDNA) was extracted from approximately 100 g of leaves using the sucrose gradient centrifugation method, as improved by Li et al. (2012) [25]. The concentration of cpDNA was estimated by measuring A260 and A280 using an ND-2000 spectrometer (Nanodrop Technologies, Wilmington, DE, USA); samples were also visually examined by gel electrophoresis. Pure cpDNA was used to construct shotgun libraries, which were sequenced by Herbgenomics Technologies on the 454 GS FLX Titanium platform (Roche Diagnostics, Basel, Switzerland), according to the manufacturer’s instructions. The resulting sff-file was pre-processed by trimming low-quality (Q < 20) and short (L < 50 bp) reads. This resulted in about 60× coverage of this cp genome. Cleaned reads were used for sequence assembly using GS FLX De Novo Assembler Software (Newbler V2.6). To verify the assembly, the four junctions between the inverted repeat (IR) and long single-copy/short single-copy (LSC/SSC) regions were confirmed by PCR amplification and Sanger sequencing.
3.2. Genome Annotation
The initial gene annotation of the A. kravanh cp genome was performed using CPGAVAS (http://www.herbalgenomics.org/cpgavas) [26] and confirmed with BLAST and DOGMA (http://dogma.ccbb.utexas.edu/) [27]. tRNA genes were identified using tRNAscanSE (http://lowelab.ucsc.edu/tRNAscan-SE/) [28] with default settings. Then, the position of each gene was manually corrected using Apollo [29], according to the positions of start and stop codons. The circular cp genome map was drawn using OGDRAW (http://ogdraw.mpimp-golm.mpg.de/) [30]. The final cp genome of A. kravanh was submitted to GenBank (Accession Number: MF991963).
3.3. Sequence Analyses
To analyze the characteristics of synonymous codon usage, relative synonymous codon usage values (RSCU), codon usage and GC content were determined using MEGA7 [31]. Simple sequence repeats (SSRs) were detected with MISA [32], using the following thresholds: 8, 4, 4, 3, 3 and 3 repeat units for mono-, di-, tri-, tetra-, penta- and hexa-nucleotide SSRs, respectively. We performed the same SSR analysis for three other Zingiberaceae cp genomes, and conducted analysis with mVISTA to assess the relative number of SSRs in the four different cp genomes. For analysis of repeat structures, REPuter (http://bibiserv.tech--fak.uni-bielefeld.de/reputer/) [33] was used to visualize forward, reverse, palindrome and complement sequences of size ≥30 bp and identity ≥90% in the cp genome. All repeats recognized were manually verified, and redundant results removed.
3.4. Genome Comparison
MUMmer [34] was used to perform pairwise cp genomic alignment, and dotplots were drawn using a Perl script. The mVISTA program was employed in Shuffle-LAGAN mode [35,36] to compare the cp genome of A. kravanh with those of Alpinia zerumbet (JX088668), Curcuma flaviflora (KR967361) and Zingiber spectabile (JX088661). Additionally, the A. kravanh cp genome was compared with three available commelinid cp genomes (A. zerumbet, JX088668; Musa balbisiana, KT595228; Kingia australis, JX051651) using CGView (http://stothard.afns.ualberta.ca/cgview_server/index.html) [37]. GC distributions were measured based on GC skew, using the following equation: GC skew = (G − C)/(G + C).
3.5. Analysis of Synonymous and Non-Synonymous Substitution Rates
The A. kravanh cp genome sequence was compared with those of A. zerumbet, C. flaviflora and Z. spectabile. To evaluate synonymous (Ks) and non-synonymous (Ka) substitution ratios, shared individual protein-coding exons were extracted using a Python script and separately aligned with MEGA7. Ks and Ka substitution rates for each protein-coding gene were estimated in DnaSP [38].
3.6. Phylogenetic Analysis
For phylogenetic analysis, 33 complete cp genome sequences were downloaded from NCBI (Table S1), and a dataset of 58 protein-coding genes commonly present in the 35 cp genomes included in the analysis was used to construct the phylogenetic tree. All shared genes were aligned separately using ClustalW2 [39]. Jmodeltest [40] was employed to determine the most appropriate model for maximum likelihood (ML), based on Akaike information criterion (AIC), then ML analysis was performed using the tool RAxML-HPC 2.7.6.3 [41] in XSEDE at the CIPRES Science Gateway (http://www.phylo.org/) [42] with 1000 bootstrap replicates. Similarly, maximum parsimony (MP) analysis was performed using PAUP in XSEDE. Magnolia officinalis and Aconitum carmichaelii were included as outgroups.
4. Conclusions
The complete cp genome sequence of A. kravanh was assembled, annotated and analyzed in this study. Detailed comparisons with the cp genomes of three other Zingiberaceae species demonstrated that the A. kravanh cp genome has clearly undergone expansion of the IR regions, while the gene content, gene order, genome structure and SSRs were broadly similar. Repeated sequences, together with the aforementioned SSRs, are informative sources for the development of new molecular markers. Phylogenetic relationships among 33 commelinid species strongly supported a sister relationship between A. kravanh and A. zerumbet. The comprehensive data presented in this study provide insights into the characteristics of the entire A. kravanh cp genome and the phylogenetic relationships within the commelinid clade, which will facilitate a breeding program, genetic engineering and evolutionary studies among the major angiosperm taxonomic categories.
Supplementary Materials
Supplementary materials are available online.
Acknowledgments
This work is supported by the grants from the National Key Technology Support Program (2015BAI05B02) and from the China Academy of Chinese Medical Sciences Special Fund for Thirteen-five key research (ZZ10-007).
Author Contributions
S.C. and X.L. conceived and designed the research framework; Q.L. prepared the sample and performed the experiments; M.W. analyzed the data and wrote the paper. X.L. and Z.H. made revisions to the final manuscript. All authors have read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shu, Y.M.; Zhou, M.Q.; Zhang, Y.J.; Yue, W.Q. Clinical efficacy study of Yishen\Jiangzhuo granule on chronic renal failure. Chin. Tradit. Pat. Med. 1993, 2, 20–21. [Google Scholar]
- Pu, Q.; Qin, H.Z.; Huang, Y.Q.; Xie, P.; Luo, J.; Wong, M.Z. Advances in studies on the correlation between medicinal properties and genetic relationship of five genera in Zingiberaceae. China Pharm. 2016, 27, 3301–3303. [Google Scholar]
- Luo, Y.; Lu, L.; Wortley, A.H.; Li, D.Z.; Wang, H.; Blackmore, S. Evolution of Angiosperm Pollen. 3. Monocots1. Ann. Mo. Bot. Gard. 2015, 101, 406–455. [Google Scholar] [CrossRef]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.H.; Wang, L.; Biradar, S.S. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Qian, J.; Sun, Z.Y.; Xu, X.L.; Chen, S.L. Complete chloroplast genome of medicinal plant Lonicera japonica: Genome rearrangement, intron gain and loss, and implications for phylogenetic studies. Molecules 2017, 22, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Xiang, B.; Li, X.; Qian, J.; Wang, L.; Ma, L.; Tian, X. The complete chloroplast genome sequence of the medicinal plant Swertia mussotii using the PacBio RS II platform. Molecules 2016, 21, 1029–1031. [Google Scholar] [CrossRef] [PubMed]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.K.; Lee, H.L.; Sun, B.Y.; Chung, M.Y.; Kim, K.J. The complete chloroplast DNA sequence of Eleutherococcus senticosus (Araliaceae); comparative evolutionary analyses with other three asterids. Mol. Ceflls. 2012, 33, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Bryan, G.J.; McNicol, J.W.; Meyer, R.C.; Ramsay, G.; De Jong, W.S. Polymorphic simple sequence repeat markers in chloroplast genomes of Solanaceous plants. Theor. Appl. Genet. 1999, 99, 859–867. [Google Scholar] [CrossRef]
- Provan, J. Novel chloroplast microsatellites reveal cytoplasmic variation in Arabidopsis thaliana. Mol. Ecol. 2000, 9, 2183–2185. [Google Scholar] [CrossRef] [PubMed]
- Flannery, M.L.; Mitchell, F.J.; Coyne, S.; Kavanagh, T.A.; Burke, J.I.; Salamin, N. Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs. Theor. Appl. Genet. 2006, 113, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.H.; Zhao, Z.L.; Xu, H.X.; Chen, S.L.; Dorje, G. The complete chloroplast genome of Gentiana straminea (Gentianaceae), an endemic species to the Sino-Himalayan subregion. Gene 2016, 577, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evolut. Biol. 2008, 8, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Ye, Y.; Bai, T.; Xu, M.; Xu, L.-A. Complete chloroplast genome of Pinus massoniana (Pinaceae): Gene rearrangements, loss of ndh genes, and short inverted repeats contraction, expansion. Molecules 2017, 22, 1528. [Google Scholar] [CrossRef] [PubMed]
- De Cambiaire, J.C.; Otis, C.; Lemieux, C.; Turmel, M. The complete chloroplast genome sequence of the chlorophycean green alga Scenedesmus obliquus reveals a compact gene organization and a biased distribution of genes on the two DNA strands. BMC Evolut. Biol. 2006, 6, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yang, B.; Zhu, W.; Sun, L.; Tian, J.; Wang, X. The complete chloroplast genome sequence of Mahonia bealei (Berberidaceae) reveals a significant expansion of the inverted repeat and phylogenetic relationship with other angiosperms. Gene 2013, 528, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Drescher, A.; Ruf, S.; Calsa, T.J.; Carrer, H.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plant are essential genes. Plant J. 2000, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Johansen, L.B.; Hoot, S.B. Phylogeny of Orchidantha (Lowiaceae) and the Zingiberales based on six DNA regions. Syst. Botany 2005, 30, 106–117. [Google Scholar] [CrossRef]
- Givnish, T.J.; Evans, T.M.; Pires, J.C.; Systma, K.J. Polyphyly and convergent morphological evolution in Commelinales and Commelinidae: Evidence from rbcL sequence data. Mol. Phylogenet. Evolut. 1999, 12, 360–385. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.B. Phylogenetic analysis of the subfamily Alpinioideae (Zingiberaceae), particularly Etlingera giseke, based on nuclear and plastid DNA. Plant Syst. Evolut. 2004, 245, 239–258. [Google Scholar] [CrossRef]
- Smith, J.F.; Kress, W.J.; Zimmer, E.A. Phylogenetic analysis of the Zingiberales based on rbcL sequences. Ann. Mo. Bot. Gard. 1993, 80, 620–630. [Google Scholar] [CrossRef]
- Dong, H.; Mei, Q.C.; Xu, G.J.; Xu, L.S. The herbal textual research of Alpinia katsumadai and cardamom. J. Chin. Mater. Med. 1992, 17, 451–453. [Google Scholar]
- Li, X.W.; Hu, Z.G.; Lin, X.H.; Li, Q.; Gao, H.H.; Luo, G.A. High-throughput pyrosequencing of the complete chloroplast genome of Magnolia officinalis and its application in species identification. Acta Pharm. Sin. 2012, 47, 124–130. [Google Scholar]
- Liu, C.; Shi, Y.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.E.; Searle, S.M.; Harris, N.; Gibson, M.; Iyer, V.; Richter, J. Apollo: A sequence annotation editor. Genome Biol. 2002, 3, RESEARCH0082. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. Organellar Genome DRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evolut. 2016, 33, 1870. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.M.; Sun, J.T.; Xue, X.F.; Zhu, W.C.; Hong, X.Y. Development and characterization of 18 Novel EST-SSRs from the western flower Thrips, Frankliniella occidentalis (Pergande). Int. J. Mol. Sci. 2012, 13, 2863–2876. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evolut. 2008, 25, 1253. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. The CIPRES science gateway: A community resource for phylogenetic analyses. In Proceedings of the Teragrid Conference: Extreme Digital Discovery, Salt Lake City, UT, USA, 18–21 July 2011. [Google Scholar]
Sample Availability: Sequence data of Amomum kravanh are available from the authors. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).