Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity


Abstract
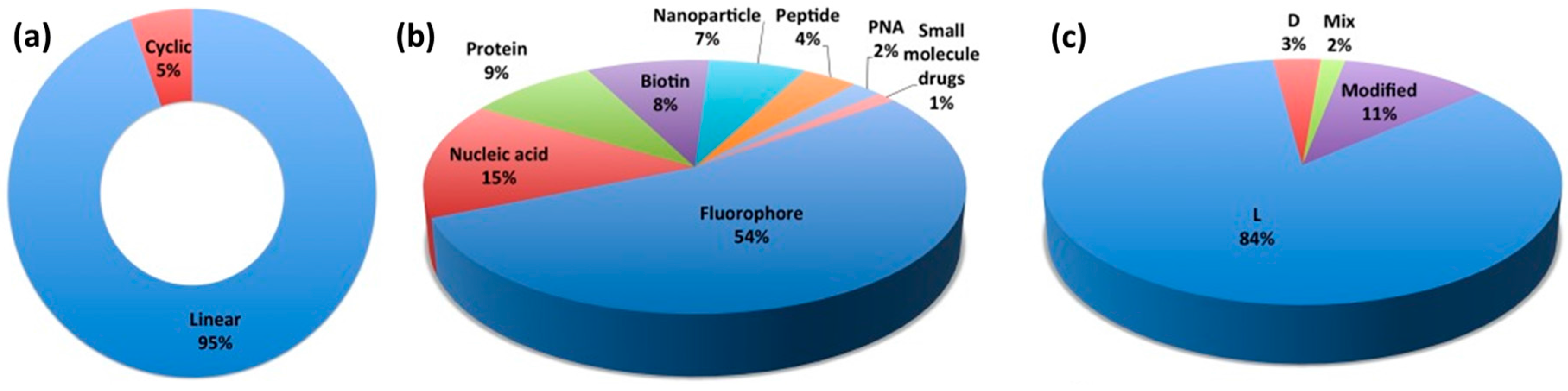
1. Introduction
2. From Protein Domains to the Design of Peptides with Cell Penetration Properties
2.1. Natural and Fusion Sequences
2.2. Structural and Functional Plasticity
3. Mechanistic Challenges
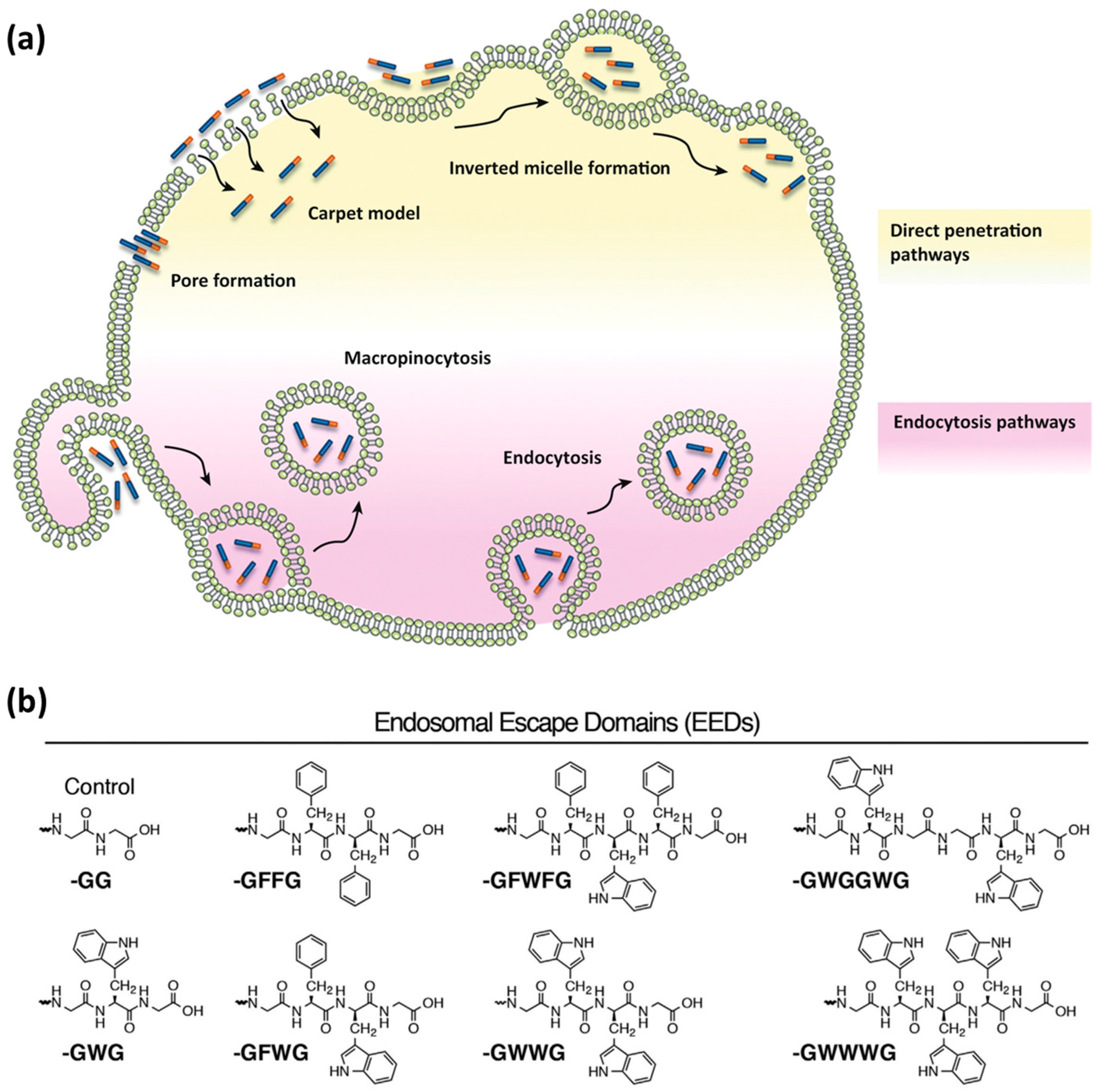
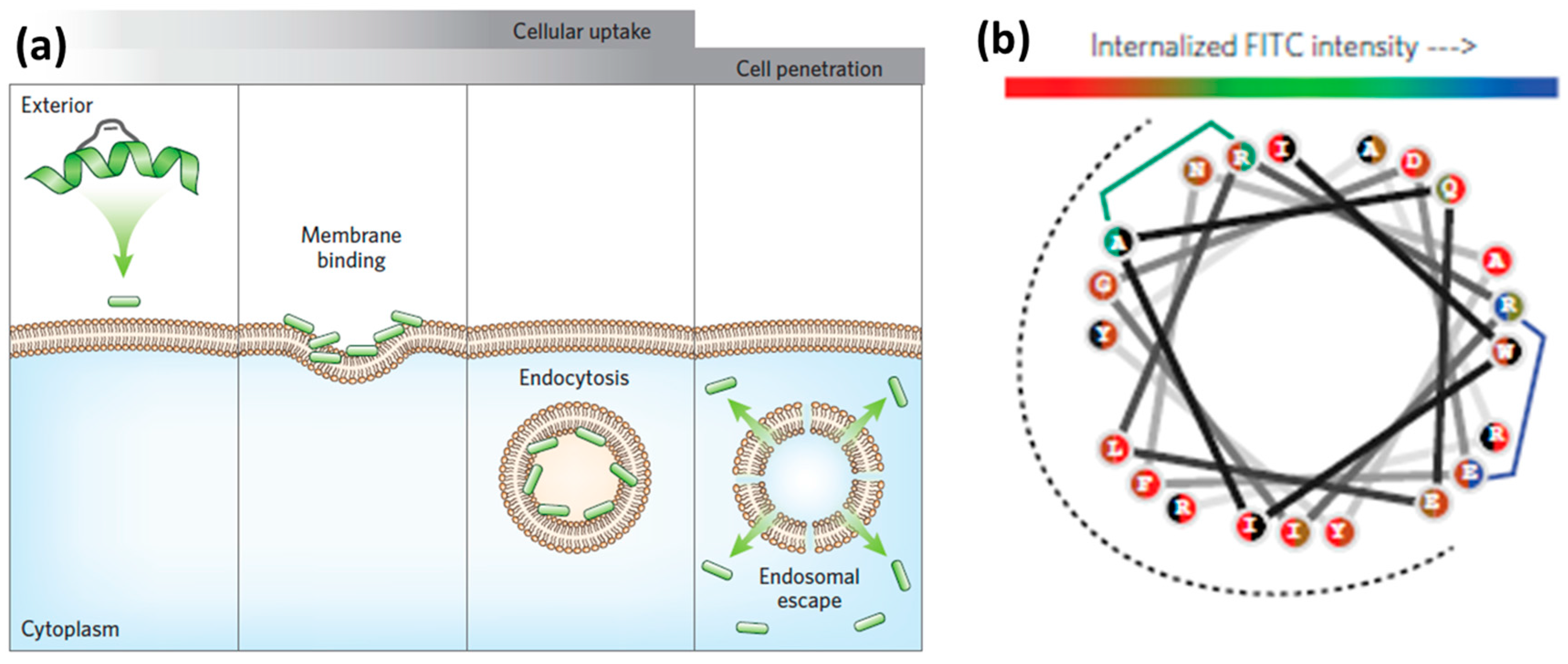
3.1. Internalization Mechanisms
3.2. Membrane Composition
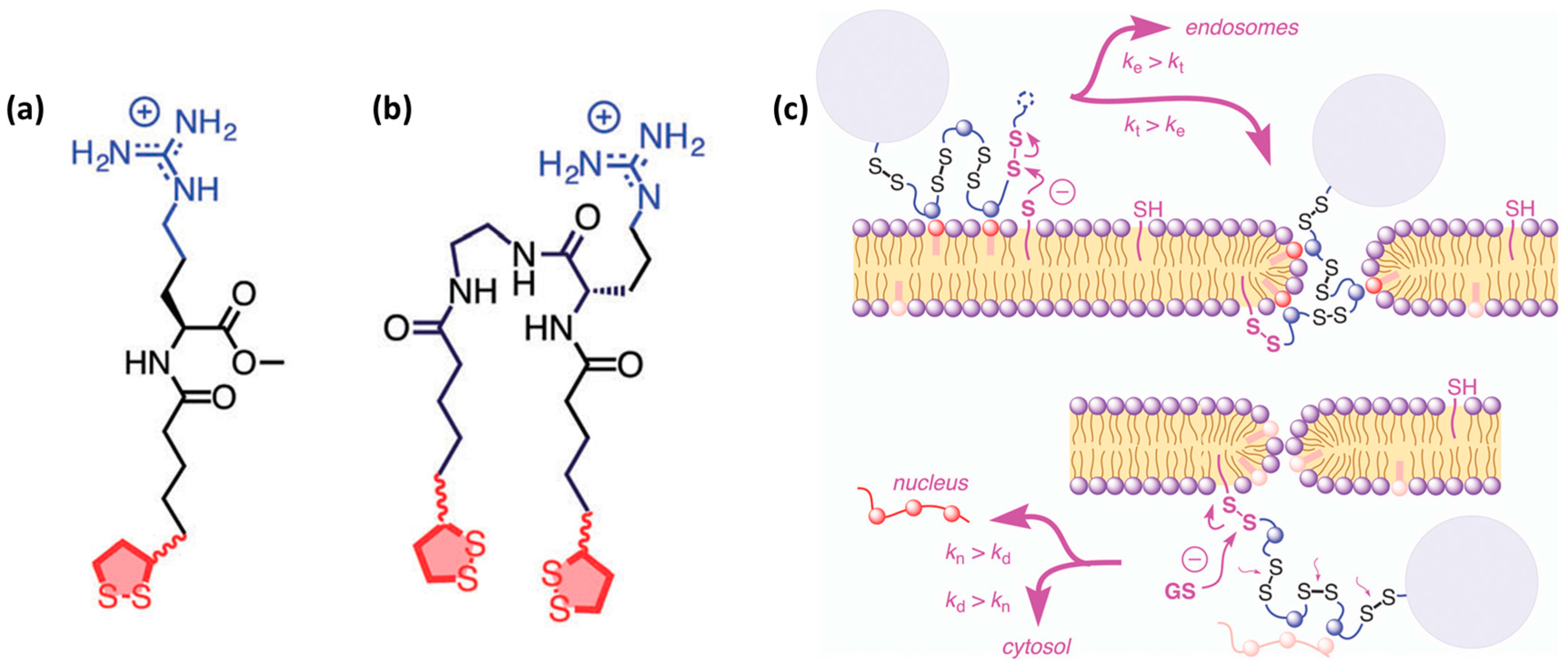
3.3. Endosomal Escape
3.4. Influence of Cargo
4. Design Strategies
4.1. The Importance of the Primary Sequence
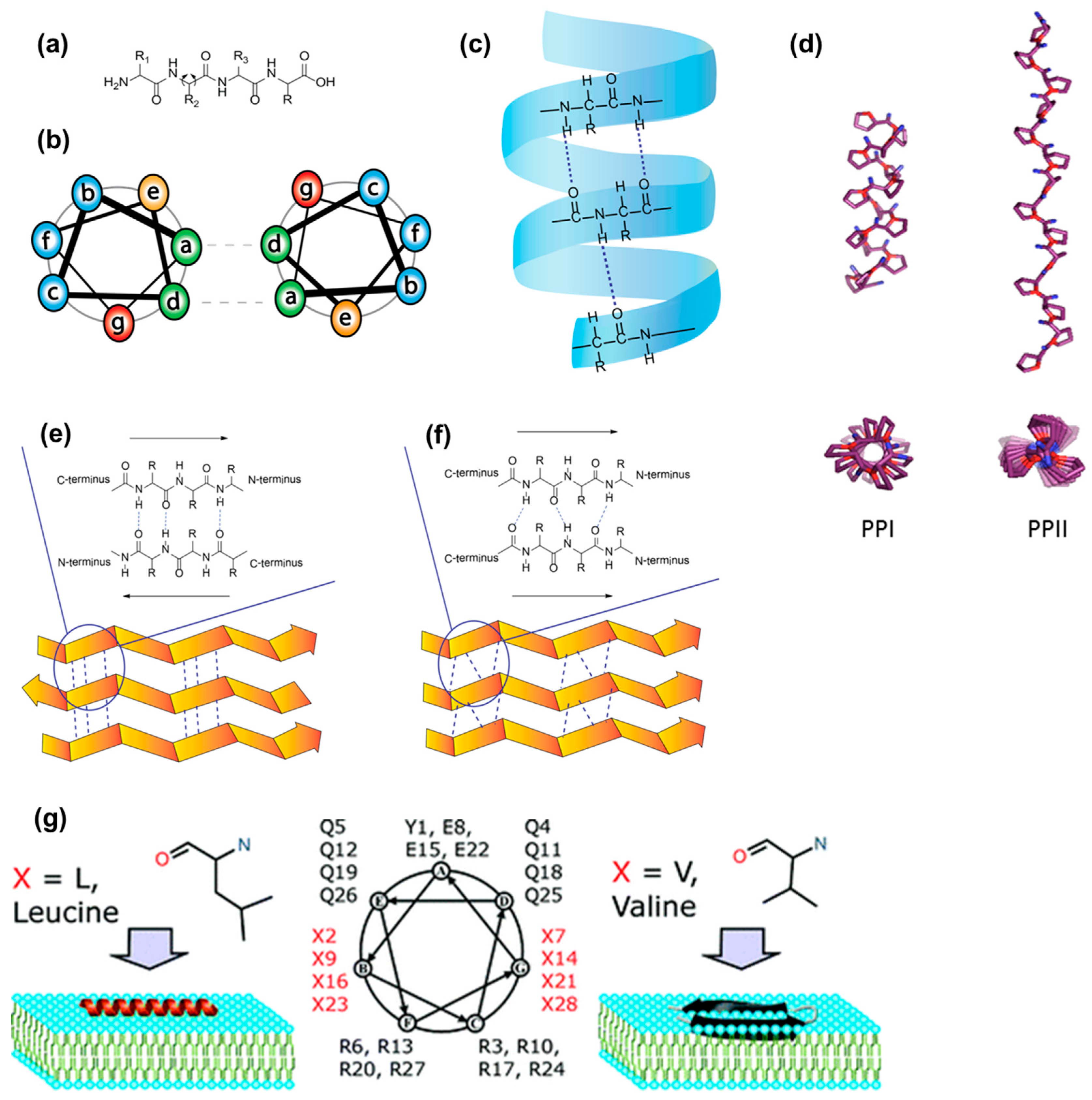
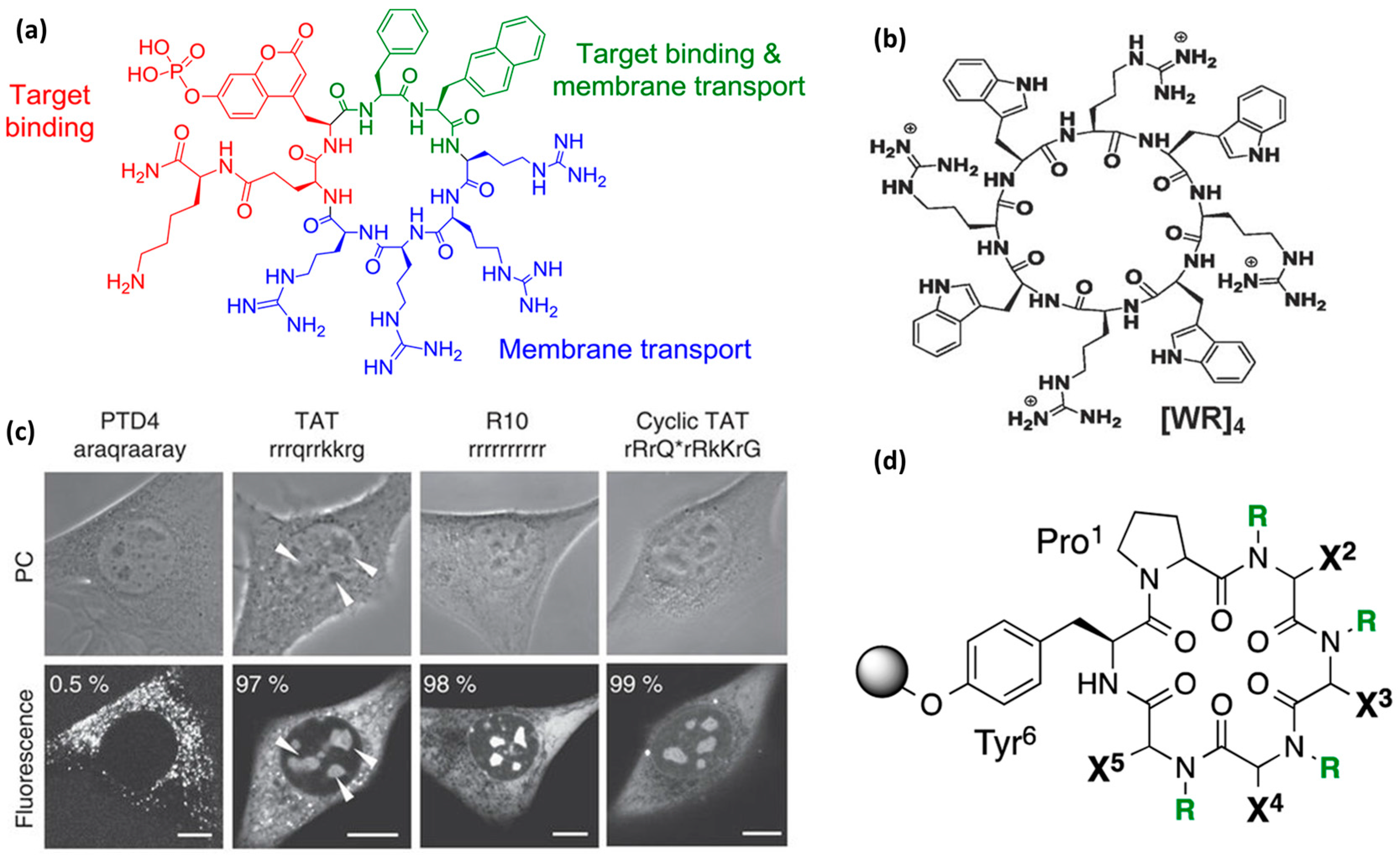
4.2. Flexible Amphipathic CPPs
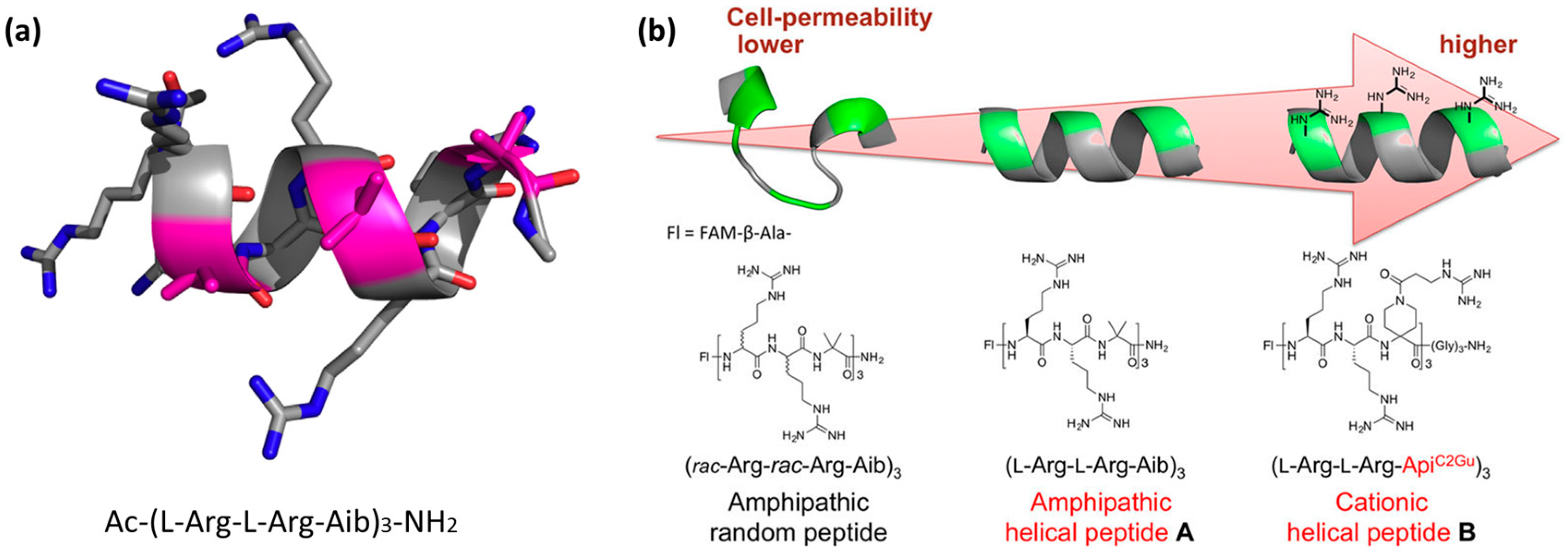
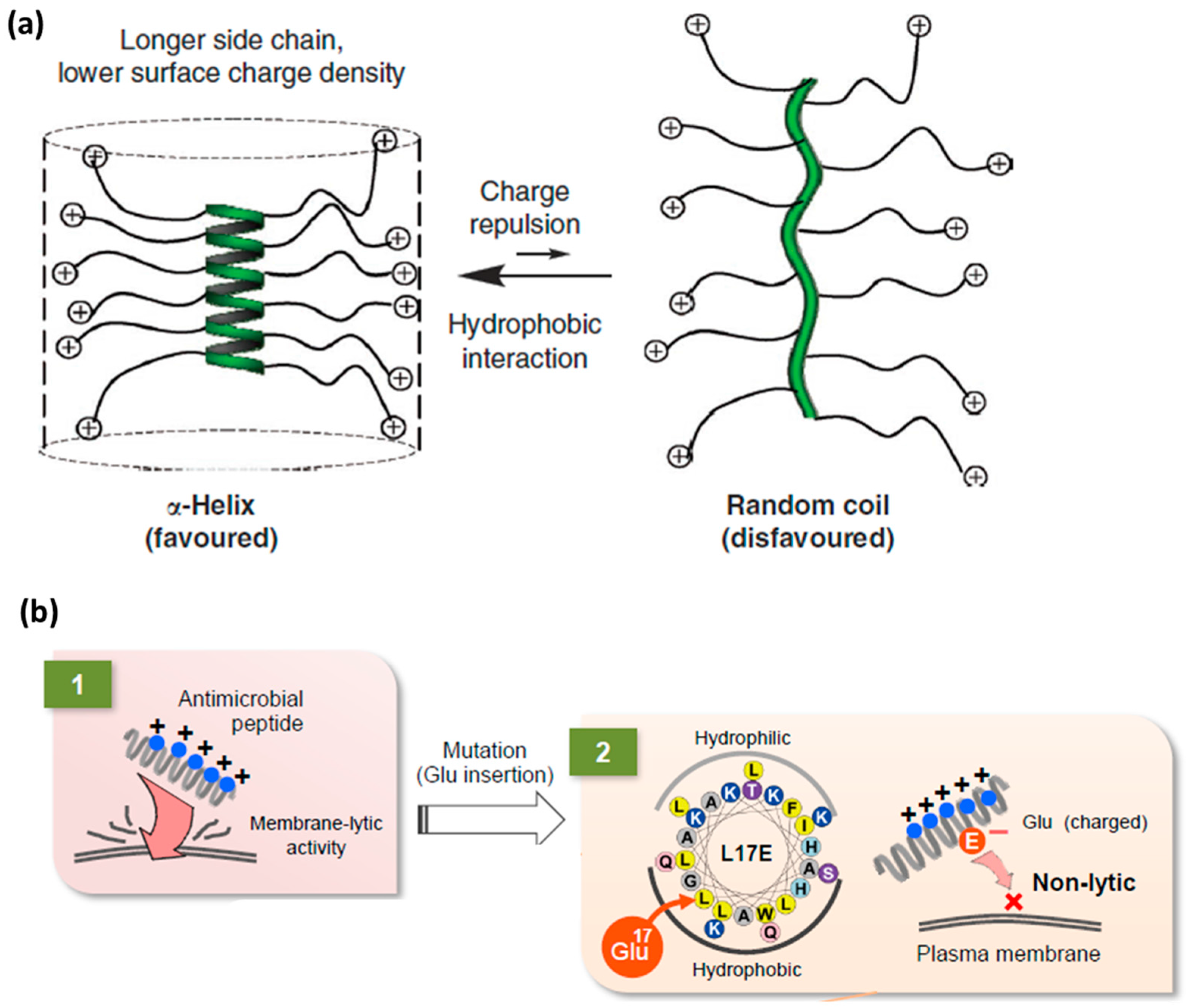
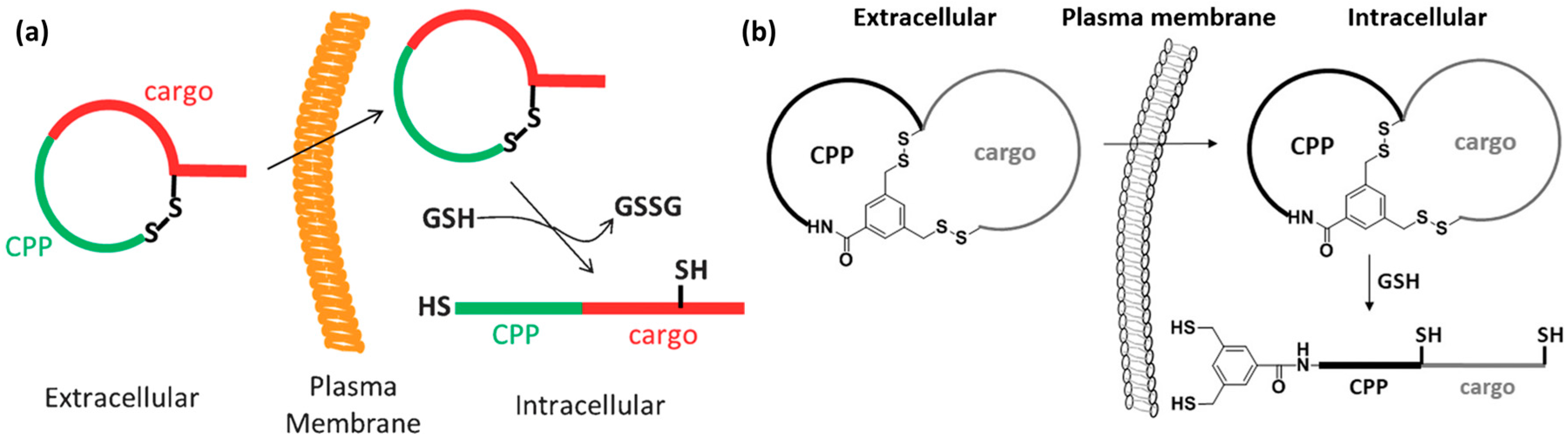
4.3. Rigidity as an Emerging Concept in CPP Design
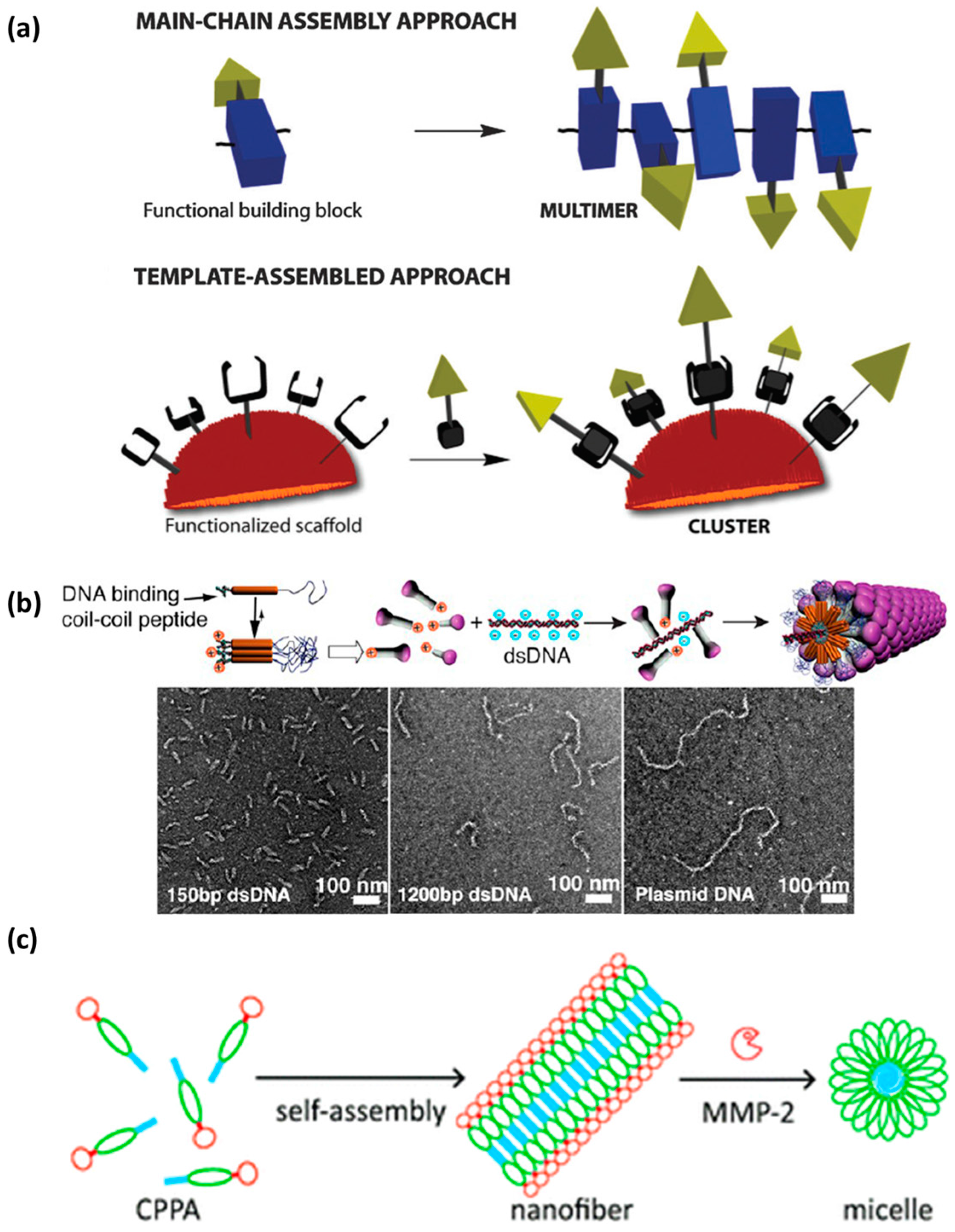
4.4. Controlled Self-Assembly
5. Challenges and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [PubMed]
- Lindgren, M.; Langel, Ü. Classes and Prediction of Cell-Penetrating Peptides. In Cell-Penetrating Peptides; Humana Press: New York, NY, USA, 2011; Volume 683, pp. 3–19. [Google Scholar]
- Sani, M.A.; Separovic, F. How Membrane-Active Peptides Get into Lipid Membranes. Acc. Chem. Res. 2016, 49, 1130–1138. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, W.B.; Fuselier, T.; He, J.; Wimley, W.C. Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci. 2015, 40, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Di Pisa, M.; Chassaing, G.; Swiecicki, J.M. Translocation mechanism(s) of cell-penetrating peptides: Biophysical studies using artificial membrane bilayers. Biochemistry 2015, 54, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Carneado, J.; Kogan, M.J.; Pujals, S.; Giralt, E. Amphipathic Peptides and Drug Delivery. Biopolymers 2004, 76, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Singh, H.; Tyagi, A.; Chaudhary, K.; Kumar, R.; Kapoor, P.; Raghava, G.P.S. Database tool CPPsite: A curated database of cell penetrating peptides. Database 2012, bas015. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.S.; Gautam, A. CPPsite 2.0 : A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 2016, 44, 1098–1103. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, G.; Bang, E.-K.; Montenegro, J.; Matile, S. Cellular uptake: Lessons from supramolecular organic chemistry. Chem. Commun. 2015, 51, 10389–10402. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.; Feher, K.M.; Hu, C.; Slowinska, K. Peptide internalization enabled by folding: Triple helical cell-penetrating peptides. J. Pept. Sci. 2015, 21, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Eiríksdóttir, E.; Konate, K.; Langel, Ü.; Divita, G.; Deshayes, S. Secondary structure of cell-penetrating peptides controls membrane interaction and insertion. Biochim. Biophys. Acta 2010, 1798, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Veiman, K.-L.; Künnapuu, K.; Lehto, T.; Kiisholts, K.; Pärn, K.; Langel, Ü.; Kurrikoff, K. PEG shielded MMP sensitive CPPs for efficient and tumor specific gene delivery in vivo. J. Control. Release 2015, 209, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.T.; Sayers, E.J. Cell entry of cell penetrating peptides: Tales of tails wagging dogs. J. Control. Release 2012, 161, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Nakase, I.; Tadokoro, A.; Takeuchi, T.; Jones, A.T. Arginine-rich peptides and their internalization mechanisms. Biochem. Soc. Trans. 2007, 35, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed]
- Lättig-tünnemann, G.; Prinz, M.; Hoffmann, D.; Behlke, J.; Palm-apergi, C.; Morano, I.; Herce, H.D.; Cardoso, M.C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, 2, 453. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001, 19, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997, 25, 2730–2736. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Jha, D.; Mishra, R.; Gottschalk, S.; Wiesm, K.; Ugurbil, K.; Maier, M.E. CyLoP-1: A Novel Cysteine-Rich Cell-Penetrating Peptide for Cytosolic Delivery of Cargoes. Bioconj. Chem. 2011, 22, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, S.; Falanga, A.; Vitiello, M.; Browne, H.; Pedone, C.; Galdiero, M. Fusogenic domains in herpes simplex virus type 1 glycoprotein H. J. Biol. Chem. 2005, 280, 28632–28643. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, S.; Falanga, A.; Morelli, G.; Galdiero, M. GH625: A milestone in understanding the many roles of membranotropic peptides. Biochim. Biophys. Acta Biomembr. 2015, 1848, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, R.; Accardo, A.; Falanga, A.; Guarnieri, D.; Vitiello, G.; Netti, P.; D’Errico, G.; Morelli, G.; Galdiero, S. Clickable functionalization of liposomes with the gH625 peptide from herpes simplex virus type i for intracellular drug delivery. Chem. A Eur. J. 2011, 17, 12659–12668. [Google Scholar] [CrossRef] [PubMed]
- Perillo, E.; Allard-Vannier, E.; Falanga, A.; Stiuso, P.; Vitiello, M.T.; Galdiero, M.; Galdiero, S.; Chourpa, I. Quantitative and qualitative effect of gH625 on the nanoliposome-mediated delivery of mitoxantrone anticancer drug to HeLa cells. Int. J. Pharm. 2015, 488, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Vitiello, M.T.; Cantisani, M.; Tarallo, R.; Guarnieri, D.; Mignogna, E.; Netti, P.; Pedone, C.; Galdiero, M.; Galdiero, S. A peptide derived from herpes simplex virus type 1 glycoprotein H: Membrane translocation and applications to the delivery of quantum dots. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Carberry, T.P.; Tarallo, R.; Falanga, A.; Finamore, E.; Galdiero, M.; Weck, M.; Galdiero, S. Dendrimer functionalization with a membrane-interacting domain of herpes simplex virus type 1: Towards intracellular delivery. Chem. A Eur. J. 2012, 18, 13678–13685. [Google Scholar] [CrossRef] [PubMed]
- Smaldone, G.; Falanga, A.; Capasso, D.; Guarnieri, D.; Correale, S.; Galdiero, M.; Netti, P.A.; Zollo, M.; Galdiero, S.; Di Gaetano, S.; et al. gH625 is a viral derived peptide for effective delivery of intrinsically disordered proteins. Int. J. Nanomed. 2013, 8, 2555–2565. [Google Scholar]
- Perillo, E.; Hervé-Aubert, K.; Allard-Vannier, E.; Falanga, A.; Galdiero, S.; Chourpa, I. Synthesis and in vitro evaluation of fluorescent and magnetic nanoparticles functionalized with a cell penetrating peptide for cancer theranosis. J. Colloid Interface Sci. 2017, 499, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Takeuchi, T.; Kuwata, K.; Chiba, J.; Hatanaka, Y.; Nakase, I.; Futaki, S. Syndecan—4 Is a Receptor for Clathrin-Mediated Endocytosis of Arginine-Rich Cell-Penetrating Peptides. Bioconj. Chem. 2016, 27, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Sun, Y.; Huang, H.W. Membrane-mediated peptide conformation change from B-monomers to B-aggregates. Biophys. J. 2010, 98, 2236–2245. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Dustin, D.; Jiang, L.; Samways, D.S.K.; Dong, H. Designed filamentous cell penetrating peptides: Probing supramolecular structure-dependent membrane activity and transfection efficiency. Chem. Commun. 2015, 51, 11757–11760. [Google Scholar] [CrossRef] [PubMed]
- Frederix, P.W.J.M.; Scott, G.G.; Abul-Haija, Y.M.; Kalafatovic, D.; Pappas, C.G.; Javid, N.; Hunt, N.T.; Ulijn, R.V.; Tuttle, T. Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels. Nat. Chem. 2015, 7, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, U. Cell-penetrating peptides: Design, synthesis, and applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Carneado, J.; Kogan, M.J.; Van Mau, N.; Pujals, S.; López-Iglesias, C.; Heitz, F.; Giralt, E. Fatty acyl moieties: Improving Pro-rich peptide uptake inside HeLa cells. J. Pept. Res. 2005, 65, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Regberg, J.; Srimanee, A.; Langel, Ü. Applications of Cell-Penetrating Peptides for Tumor Targeting. Pharmaceuticals 2012, 5, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Martín, I.; Teixidó, M.; Giralt, E. Building cell selectivity into CPP-mediated strategies. Pharmaceuticals 2010, 3, 1456–1490. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huang, P.; Chen, X. Stimuli-Responsive Programmed Specific Targeting in Nanomedicine. ACS Nano 2016, 10, 2991–2994. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hu, Y.; Ochbaum, G.; Lin, R.; Bitton, R.; Cui, H.; Azevedo, H.S. Enzymatic activation of cell-penetrating peptides in self-assembled nanostructures triggers fibre-to-micelle morphological transition. Chem. Commun. 2017, 53, 7037–7040. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Sun, L.; Ye, J.; Liu, E.; Chen, S.; Liang, Q.; Shin, M.C.; Yang, V.C. Enzyme-triggered, cell penetrating peptide-mediated delivery of anti-tumor agents. J. Control. Release 2015, 240, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Kalafatovic, D.; Nobis, M.; Son, J.; Anderson, K.I.; Ulijn, R.V. MMP-9 triggered self-assembly of doxorubicin nanofiber depots halts tumor growth. Biomaterials 2016, 98, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating Peptides. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Daniels, D.S.; Coplin, A.E.; Jordan, G.E.; McGregor, L.M.; Schepartz, A. Minimally cationic cell-permeable miniature proteins via α-helical arginine display. J. Am. Chem. Soc. 2008, 130, 2948–2949. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wang, J.; Bai, Y.; Lang, J.W.; Liu, S.; Lin, Y.; Cheng, J. Ionic polypeptides with unusual helical stability. Nat. Commun. 2011, 2, 206. [Google Scholar] [CrossRef] [PubMed]
- Horn, M.; Reichart, F.; Natividad-Tietz, S.; Diaz, D.; Neundorf, I. Tuning the properties of a novel short cell-penetrating peptide by intramolecular cyclization with a triazole bridge. Chem. Commun. 2016, 52, 2261–2264. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Adjobo-Hermans, M.J.W.; Kohze, R.; Enderle, T.; Brock, R.; Milletti, F. Identification of short hydrophobic CPPs for cytosolic peptide delivery by rational design. Bioconj. Chem. 2017, 28, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Fernández-carneado, J.; López-iglesias, C.; Kogan, M.J.; Giralt, E. Mechanistic aspects of CPP-mediated intracellular drug delivery : Relevance of CPP self-assembly. Biochim. Biophys. Acta 2006, 1758, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Alves, I.D.; Goasdoué, N.; Correia, I.; Aubry, S.; Galanth, C.; Sagan, S.; Lavielle, S.; Chassaing, G. Membrane interaction and perturbation mechanisms induced by two cationic cell penetrating peptides with distinct charge distribution. Biochim. Biophys. Acta Gen. Subj. 2008, 1780, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Mg, S.; Mayor, S. Endocytosis unplugged: Multiple ways to enter the cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef] [PubMed]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Nakase, I.; Kawabata, N.; Yu, H.H.; Pujals, S.; Imanishi, M.; Giralt, E.; Futaki, S. Cytosolic targeting of macromolecules using a pH-dependent fusogenic peptide in combination with cationic liposomes. Bioconj. Chem. 2009, 20, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, G.; Nakase, I.; Fukuda, Y.; Masuda, R.; Oishi, S.; Shimura, K.; Kawaguchi, Y.; Takatani-Nakase, T.; Langel, Ü.; Gräslund, A.; et al. CXCR4 stimulates macropinocytosis: Implications for cellular uptake of arginine-rich cell-penetrating peptides and HIV. Chem. Biol. 2012, 19, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, A.; Dowdy, S.F. Protein transduction domain delivery of therapeutic macromolecules. Curr. Opin. Biotechnol. 2011, 22, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Lönn, P.; Kacsinta, A.D.; Cui, X.; Hamil, A.S.; Kaulich, M.; Gogoi, K.; Dowdy, S.F. Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci. Rep. 2016, 6, 32301. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of Macromolecules Using Arginine-Rich Cell-Penetrating Peptides: Ways to Overcome Endosomal Entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Bushweller, J.H.; Cafiso, D.S.; Tamm, L.K. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 2001, 8, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Hirose, H.; Tanaka, G.; Pujals, S.; Katayama, S.; Nakase, I.; Futaki, S. Effect of the attachment of a penetration accelerating sequence and the influence of hydrophobicity on octaarginine-mediated intracellular delivery. Mol. Pharm. 2012, 9, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Nakase, I.; Michiue, H.; Takeuchi, T.; Tomizawa, K.; Matsui, H.; Futaki, S. Enhanced intracellular delivery using arginine-rich peptides by the addition of penetration accelerating sequences (Pas). J. Control. Release 2009, 138, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Mäe, M.; EL Andaloussi, S.; Lundin, P.; Oskolkov, N.; Johansson, H.J.; Guterstam, P.; Langel, Ü. A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy. J. Control. Release 2009, 134, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nicol, F.; Szoka, F.C. GALA: A designed synthetic pH-responsive amphipathic peptide with applications in drug and gene delivery. Adv. Drug Deliv. Rev. 2004, 56, 967–985. [Google Scholar] [CrossRef] [PubMed]
- Santiwarangkool, S.; Akita, H.; Nakatani, T.; Kusumoto, K.; Kimura, H.; Suzuki, M.; Nishimura, M.; Sato, Y.; Harashima, H. PEGylation of the GALA Peptide Enhances the Lung-Targeting Activity of Nanocarriers That Contain Encapsulated siRNA. J. Pharm. Sci. 2017, 106, 2420–2427. [Google Scholar] [CrossRef] [PubMed]
- Akishiba, M.; Takeuchi, T.; Kawaguchi, Y.; Sakamoto, K.; Yu, H.-H.; Nakase, I.; Takatani-Nakase, T.; Madani, F.; Gräslund, A.; Futaki, S. Cytosolic antibody delivery by lipid-sensitive endosomolytic peptide. Nat. Chem. 2017, 9, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.T.; Castanho, M.A.R.B. Environmental factors that enhance the action of the cell penetrating peptide pep-1: A spectroscopic study using lipidic vesicles. Biochim. Biophys. Acta Biomembr. 2005, 1669, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.M.; Horton, K.L.; Kelley, S.O. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org. Biomol. Chem. 2008, 6, 2242–2255. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Cell-penetrating peptides: From molecular mechanisms to therapeutics. Biol. Cell 2008, 100, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, M.; Birch, D.; Nielsen, H.M. Applications and challenges for use of cell-penetrating peptides as delivery vectors for peptide and protein cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Freire, J.M.; Veiga, S.; de la Torre, B.G.; Andreu, D.; Castanho, M.A.R.B. Quantifying molecular partition of cell- penetrating peptide—Cargo supramolecular complexes into lipid membranes: Optimizing peptide-based drug delivery systems. J. Pept. Sci. 2013, 19, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Svensen, N.; Walton, J.G.A.; Bradley, M. Peptides for cell-selective drug delivery. Trends Pharmacol. Sci. 2012, 33, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Zorko, M.; Langel, U. Cell-penetrating peptides: Mechanism and kinetics of cargo delivery. Adv. Drug Deliv. Rev. 2005, 57, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Martin, I.; Teixido, M.; Giralt, E. Intracellular Fate of Peptide-Mediated Delivered Cargoes. Curr. Pharm. Des. 2013, 19, 2924–2942. [Google Scholar] [CrossRef] [PubMed]
- Reches, M.; Gazit, E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science 2003, 300, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Fernández-Carneado, J.; Kogan, M.J.; Martinez, J.; Cavelier, F.; Giralt, E. Replacement of a proline with silaproline causes a 20-fold increase in the cellular uptake of a pro-rich peptide. J. Am. Chem. Soc. 2006, 128, 8479–8483. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Yin, L.; Kim, K.H.; Cheng, J. Helical poly(arginine) mimics with superior cell-penetrating and molecular transporting properties. Chem. Sci. 2013, 4, 3839. [Google Scholar] [CrossRef] [PubMed]
- DeRonde, B.M.; Birke, A.; Tew, G.N. Design of Aromatic-containing Cell Penetrating Peptide Mimics with Structurally Modified π-electronics. Chem. A Eur. J. 2015, 21, 3013–3019. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Ohashi, W.; Suzuki, T.; Niwa, M.; Tanaka, S.; Ueda, K.; Harashima, H.; Sugiura, Y. Stearylated arginine-rich peptides: A new class of transfection systems. Bioconj. Chem. 2001, 12, 1005–1011. [Google Scholar] [CrossRef]
- Liu, Y.; Mei, L.; Xu, C.; Yu, Q.; Shi, K.; Zhang, L.; Wang, Y.; Zhang, Q.; Gao, H.; Zhang, Z.; He, Q. Dual receptor recognizing cell penetrating peptide for selective targeting, efficient intratumoral diffusion and synthesized anti-glioma therapy. Theranostics 2016, 6, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef] [PubMed]
- Tünnemann, G.; Ter-Avetisyan, G.; Martin, R.; Stöckl, M.; Herrmann, A.; Cardoso, M.C. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J. Pept. Sci. 2008, 14, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Rothbard, J.B.; Jessop, T.C.; Lewis, R.S.; Murray, B.A.; Wendner, P.A. Role of Membrane Potential and Hydrogen Bonding in the Mechanism of Translocation of Guanidium-Rich Peptides into Cells. J. Am. Chem. Soc. 2004, 126, 9506–9507. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Lai, G.H.; Schmidt, N.W.; Sun, V.Z.; Rodriguez, A.R.; Tong, R.; Tang, L.; Cheng, J.; Deming, T.J.; Kamei, D.T.; et al. Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 16883–16888. [Google Scholar] [CrossRef] [PubMed]
- Lein, M.; Deronde, B.M.; Sgolastra, F.; Tew, G.N.; Holden, M.A. Protein transport across membranes: Comparison between lysine and guanidinium-rich carriers. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2980–2984. [Google Scholar] [CrossRef] [PubMed]
- Kölmel, D.K.; Hörner, A.; Rönicke, F.; Nieger, M.; Schepers, U.; Bräse, S. Cell-penetrating peptoids: Introduction of novel cationic side chains. Eur. J. Med. Chem. 2014, 79, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Kölmel, D.K.; Fürniss, D.; Susanto, S.; Lauer, A.; Grabher, C.; Bräse, S.; Schepers, U. Cell penetrating peptoids (CPPos): Synthesis of a small combinatorial library by using IRORI MiniKans. Pharmaceuticals 2012, 5, 1265–1281. [Google Scholar] [CrossRef] [PubMed]
- Horton, K.L.; Stewart, K.M.; Fonseca, S.B.; Guo, Q.; Kelley, S.O. Mitochondria-Penetrating Peptides. Chem. Biol. 2008, 15, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.R.; Ahmed, M.; Lei, E.K.; Wisnovsky, S.P.; Kelley, S.O. Peptide-Mediated Delivery of Chemical Probes and Therapeutics to Mitochondria. Acc. Chem. Res. 2016, 49, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Bang, E.-K.; Ward, S.; Gasparini, G.; Sakai, N.; Matile, S. Cell-penetrating poly(disulfide)s: Focus on substrate-initiated co-polymerization. Polym. Chem. 2014, 5, 2433–2441. [Google Scholar] [CrossRef]
- Morelli, P.; Martin-Benlloch, X.; Tessier, R.; Waser, J.; Sakai, N.; Matile, S. Ethynyl benziodoxolones: Functional terminators for cell-penetrating poly(disulfide)s. Polym. Chem. 2016, 7, 3465–3470. [Google Scholar] [CrossRef]
- Gasparini, G.; Matile, S. Protein delivery with cell-penetrating poly(disulfide)s. Chem. Commun. 2015, 51, 17160–17162. [Google Scholar] [CrossRef] [PubMed]
- Chuard, N.; Gasparini, G.; Roux, A.; Sakai, N.; Matile, S. Cell-penetrating poly(disulfide)s: The dependence of activity, depolymerization kinetics and intracellular localization on their length. Org. Biomol. Chem. 2015, 13, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Som, A.; Reuter, A.; Tew, G.N. Protein transduction domain mimics: The role of aromatic functionality. Angew. Chem. Int. Ed. 2012, 51, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Som, A.; Tezgel, A.O.; Gabriel, G.J.; Tew, G.N. Self-activation in de novo designed mimics of cell-penetrating peptides. Angew. Chem. Int. Ed. 2011, 50, 6147–6150. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nature 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Sakai, N.; Matile, S. Anion-Mediated Transfer of Polyarginine across Liquid and Bilayer Membranes. J. Am. Chem. Soc. 2003, 125, 14348–14356. [Google Scholar] [CrossRef] [PubMed]
- Perret, F.; Nishihara, M.; Takeuchi, T.; Futaki, S.; Lazar, A.N.; Coleman, A.W.; Sakai, N.; Matile, S. Anionic fullerenes, calixarenes, coronenes, and pyrenes as activators of oligo/polyarginines in model membranes and live cells. J. Am. Chem. Soc. 2005, 127, 1114–1115. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Gong, C.; Ma, Y.; Fan, F.; Luo, M.; Yang, F.; Zhang, Y.H. Direct cytosolic delivery of cargoes in vivo by a chimera consisting of d- and l-arginine residues. J. Control. Release 2012, 162, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Sabidó, E.; Tarragó, T.; Giralt, E. all-D proline-rich cell-penetrating peptides: A preliminary in vivo internalization study. Biochem. Soc. Trans. 2007, 35, 794–796. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Signorelli, S.; Cannistraro, S.; Beattie, C.W.; Bizzarri, A.R. Chirality switching within an anionic cell-penetrating peptide inhibits translocation without affecting preferential entry. Mol. Pharm. 2015, 12, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.S.; Hoang, H.N.; Lohman, R.J.; Hill, T.A.; Lucke, A.J.; Craik, D.J.; Edmonds, D.J.; Griffith, D.A.; Rotter, C.J.; Ruggeri, R.B.; et al. Improving on Nature: Making a Cyclic Heptapeptide Orally Bioavailable. Angew. Chem. Int. Ed. 2014, 53, 12059–12063. [Google Scholar] [CrossRef] [PubMed]
- Verdurmen, W.P. R.; Bovee-Geurts, P.H.; Wadhwani, P.; Ulrich, A.S.; Hällbrink, M.; Van Kuppevelt, T.H.; Brock, R. Preferential uptake of L-versus D-amino acid cell-penetrating peptides in a cell type-dependent manner. Chem. Biol. 2011, 18, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. USA 1951, 37, 251. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H. C. The packing of α-helices: Simple coiled-coils. Acta Crystallogr. 1953, 6, 689–697. [Google Scholar] [CrossRef]
- Woolfson, D.N.; Mahmoud, Z.N. More than just bare scaffolds: Towards multi-component and decorated fibrous biomaterials. Chem. Soc. Rev. 2010, 39, 3464–3479. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.H.; Rudra, J.S.; Gasiorowski, J.Z.; Jung, J.P. Multi-component extracellular matrices based on peptide self-assembly. Chem. Soc. Rev. 2010, 39, 3413–3424. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Shai, Y. New lytic peptides based on the d,l-amphipathic helix motif preferentially kill tumor cells compared to normal cells. Biochemistry 2003, 42, 9346–9354. [Google Scholar] [CrossRef] [PubMed]
- Nygren, P.; Lundqvist, M.; Liedberg, B.; Jonsson, B.H.; Ederth, T. Secondary structure in de novo designed peptides induced by electrostatic interaction with a lipid bilayer membrane. Langmuir 2010, 26, 6437–6448. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Navarro, M.; Teixidó, M.; Giralt, E. Jumping Hurdles: Peptides Able To Overcome Biological Barriers. Acc. Chem. Res. 2017, 50, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Crespo, L.; Montaner, B.; Pe, R.; Royo, M.; Pons, M.; Albericio, F.; Giralt, E. Peptide Dendrimers Based on Polyproline Helices. J. Am. Chem. Soc. 2002, 124, 8876–8883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotechnol. 2003, 21, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gelain, F.; Zhao, X. Designer self-assembling peptide nanofiber scaffolds for 3D tissue cell cultures. Semin. Cancer Biol. 2005, 15, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, X.; Spirio, L. Scaffolding in Tissue Engineering. In PuraMatrix: Self-assembling Peptide Nanofiber Scaffolds; CRC Press: Boca Raton, FL, USA, 2005; p. 217. [Google Scholar]
- Sinthuvanich, C.; Veiga, A.S.; Gupta, K.; Gaspar, D.; Blumenthal, R.; Schneider, J.P. Anticancer β-hairpin peptides: Membrane-induced folding triggers activity. J. Am. Chem. Soc. 2012, 134, 6210–6217. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; McMaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A New Potent Secondary Amphipathic Cell–penetrating Peptide for siRNA Delivery Into Mammalian Cells. Mol. Ther. 2009, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Magzoub, M.; Eriksson, L.E. G.; Gräslund, A. Comparison of the interaction, positioning, structure induction and membrane perturbation of cell-penetrating peptides and non-translocating variants with phospholipid vesicles. Biophys. Chem. 2003, 103, 271–288. [Google Scholar] [CrossRef]
- Deshayes, S.; Decaffmeyer, M.; Brasseur, R.; Thomas, A. Structural polymorphism of two CPP: An important parameter of activity. Biochim. Biophys. Acta Biomembr. 2008, 1778, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Kogan, M.J.; Dalcol, I.; Gorostiza, P.; Lopez-Iglesias, C.; Pons, M.; Sanz, F.; Ludevid, D.; Giralt, E. Self-assembly of the amphipathic helix (VHLPPP)(8). A mechanism for zein protein body formation. J. Mol. Biol. 2001, 312, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Carneado, J.; Kogan, M.J.; Castel, S.; Giralt, E. Potential peptide carriers: Amphipathic proline-rich peptides derived from the N-terminal domain of γ-zein. Angew. Chemie Int. Ed. 2004, 43, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Martín, I.; Teixidó, M.; Giralt, E. Design, Synthesis and Characterization of a New Anionic Cell-Penetrating Peptide: SAP(E). ChemBioChem 2011, 12, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Franz, J.; Lelle, M.; Peneva, K.; Bonn, M.; Weidner, T. SAP(E)—A cell-penetrating polyproline helix at lipid interfaces. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2028–2034. [Google Scholar] [CrossRef] [PubMed]
- Farrera-Sinfreu, J.; Giralt, E.; Castel, S.; Albericio, F.; Royo, M. Cell-penetrating cis-gamma-amino-l-proline-derived peptides. J. Am. Chem. Soc. 2005, 127, 9459–9468. [Google Scholar] [CrossRef] [PubMed]
- Farrera-Sinfreu, J.; Zaccaro, L.; Vidal, D.; Salvatella, X.; Giralt, E.; Pons, M.; Albericio, F.; Royo, M. A New Class of Foldamers Based on cis-γ-Amino-l-proline. J. Am. Chem. Soc. 2004, 126, 6048–6057. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Varenik, M.; Bloch, D.N.; Atsmon-Raz, Y.; Jacoby, G.; Adler-Abramovich, L.; Shimon, L.J.W.; Beck, R.; Miller, Y.; Regev, O.; et al. A minimal length rigid helical peptide motif allows rational design of modular surfactants. Nat. Commun. 2017, 8, 14018. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Demizu, Y.; Shoda, T.; Sato, Y.; Oba, M.; Tanaka, M.; Kurihara, M. Amphipathic short helix-stabilized peptides with cell-membrane penetrating ability. Bioorg. Med. Chem. 2014, 22, 2403–2408. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Kato, T.; Oba, M.; Misawa, T.; Hattori, T.; Ohoka, N.; Tanaka, M.; Naito, M.; Kurihara, M.; Demizu, Y. Development of a Cell-penetrating Peptide that Exhibits Responsive Changes in its Secondary Structure in the Cellular Environment. Sci. Rep. 2016, 6, 33003. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.; Schepartz, A. Intrinsically cell-permeable miniature proteins based on a minimal cationic PPII motif. J. Am. Chem. Soc. 2007, 129, 14578–14579. [Google Scholar] [CrossRef] [PubMed]
- Bartolami, E.; Bouillon, C.; Dumy, P. Bioactive clusters promoting cell penetration and nucleic acid complexation for drug and gene delivery applications: From designed to self-assembled and responsive systems. Chem. Commun. 2016, 52, 4257–4273. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Xu, X.; Amacher, J.F.; Madden, D.R.; Cormet-Boyaka, E.; Pei, D. Intracellular delivery of peptidyl ligands by reversible cyclization: Discovery of a PDZ domain inhibitor that rescues CFTR activity. Angew. Chem. Int. Ed. 2015, 54, 5874–5878. [Google Scholar] [CrossRef] [PubMed]
- Trinh, T.B.; Upadhyaya, P.; Qian, Z.; Pei, D. Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides. ACS Comb. Sci. 2016, 18, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Adams, E.V.; Wayman, G.A.; Aguilar, C.; Saludes, J.P. Branched dimerization of Tat peptide improves permeability to HeLa and hippocampal neuronal. Chem. Commun. 2015, 51, 5463–5466. [Google Scholar]
- Mandal, D.; Nasrolahi Shirazi, A.; Parang, K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chemie Int. Ed. 2011, 50, 9633–9637. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, W.M.; Leung, S.S.F.; Pye, C.R.; Ponkey, A.R.; Bednarek, M.; Jacobson, M.P.; Lokey, R.S. Cell-permeable cyclic peptides from synthetic libraries inspired by natural products. J. Am. Chem. Soc. 2015, 137, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Rhodes, C.A.; McCroskey, L.C.; Wen, J.; Appiah-Kubi, G.; Wang, D.J.; Guttridge, D.C.; Pei, D. Enhancing the Cell Permeability and Metabolic Stability of Peptidyl Drugs by Reversible Bicyclization. Angew. Chem. Int. Ed. 2017, 56, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Rothbard, J.B.; Kreider, E.; VanDeusen, C.L.; Wright, L.; Wylie, B.L.; Wender, P.A. Arginine-rich molecular transporters for drug delivery: Role of backbone spacing in cellular uptake. J. Med. Chem. 2002, 45, 3612–3618. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Liu, T.; Liu, Y.Y.; Briesewitz, R.; Barrios, A.M.; Jhiang, S.M.; Pei, D. Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem. Biol. 2013, 8, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Bode, S.A.; Wallbrecher, R.; Brock, R.; van Hest, J.C.M.; Löwik, D.W.P.M. Activation of cell-penetrating peptides by disulfide bridge formation of truncated precursors. Chem. Commun. 2014, 50, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Walensky, L.D.; Bird, G.H. Hydrocarbon-stapled peptides: Principles, practice, and progress. J. Med. Chem. 2014, 57, 6275–6288. [Google Scholar] [CrossRef] [PubMed]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An All-Hydrocarbon Cross-Linking System for Enhancing the Helicity and Metabolic Stability of Peptides. J. Am. Chem. Soc. 2000, 122, 12364–12365. [Google Scholar] [CrossRef]
- Bernal, F.; Tyler, A.F.; Korsmeyer, S.J.; Walensky, L.D.; Verdine, G.L.; Hughes, H.; Har, V. Reactivation of the p53 Tumor Suppressor Pathway by a Stapled p53 Peptide. J. Am. Chem. Soc. 2007, 129, 2456–2457. [Google Scholar] [CrossRef] [PubMed]
- Walensky, L.D.; Kung, A.L.; Escher, I.; Malia, T.J.; Wright, R.D.; Wagner, G.; Verdine, G.L.; Stanley, J. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 2004, 305, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Bird, G.H.; Mazzola, E.; Opoku-Nsiah, K.; Lammert, M.A.; Godes, M.; Neuberg, D.S.; Walensky, L.D. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat. Chem. Biol. 2016, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.J.; Schmidt, S.; Wadhwani, P.; Bürck, J.; Reichert, J.; Afonin, S.; Berditsch, M.; Schober, T.; Brock, R.; Kansy, M.; et al. Lactam-Stapled Cell-Penetrating Peptides: Cell Uptake and Membrane Binding Properties. J. Med. Chem. 2017, 60, 8071–8082. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, J.A. Stapled peptides: How to be quick on the uptake. Nat. Chem. Biol. 2016, 12, 764–765. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Jiang, Y.; Tian, Y.; Yang, D.; Qin, X. Improving cell penetration of helical peptides stabilized by N-terminal crosslinked aspartic acids. Org. Biomol. 2017, 15, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, Q.S.; Geng, H.; Tian, Y.; Cheng, M.; Jiang, Y.H.; Xie, M.S.; Niu, X.G.; Jiang, F.; Zhang, Y.O.; et al. Crosslinked Aspartic Acids as Helix-Nucleating Templates. Angew. Chem. Int. Ed. 2016, 55, 12088–12093. [Google Scholar] [CrossRef] [PubMed]
- Ghadiri, M.R.; Granja, J.R.; Milligan, R.A.; McRee, D.E.; Khazanovich, N. Self-assembling organic nanotubes based on a cyclic peptide architecture. Nature 1993, 366, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Holmes, T.; Lockshin, C.; Rich, A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc. Natl. Acad. Sci. USA 1993, 90, 3334–3338. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Webber, M.J.; Stupp, S.I. Self-assembly of peptide amphiphiles: From molecules to nanostructures to biomaterials. Biopolymers 2010, 94, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Raeburn, J.; Zamith Cardoso, A.; Adams, D.J. The importance of the self-assembly process to control mechanical properties of low molecular weight hydrogels. Chem. Soc. Rev. 2013, 42, 5143–5156. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.A.; Czeisler, C.; Niece, K.L.; Beniash, E.; Harrington, D.A.; Kessler, J.A.; Stupp, S.I. Selective differentiation of neural progenitor cells by high-epitope density nanofibers. Science 2004, 303, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Lampel, A.; Mcphee, S.A.; Park, H.; Scott, G.G.; Humagain, S.; Hekstra, D.R.; Yoo, B.; Frederix, P.W.J.M.; Li, T.-D.; Bettinger, C.J.; et al. Polymeric peptide pigments with sequence-encoded properties. Science 2017, 356, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Ruff, Y.; Moyer, T.; Newcomb, C.J.; Demeler, B.; Stupp, S.I. Precision templating with DNA of a virus-like particle with peptide nanostructures. J. Am. Chem. Soc. 2013, 135, 6211–6219. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, C.J.; Sur, S.; Ortony, J.H.; Lee, O.-S.; Matson, J.B.; Boekhoven, J.; Yu, J.M.; Schatz, G.C.; Stupp, S.I. Cell death versus cell survival instructed by supramolecular cohesion of nanostructures. Nat. Commun. 2014, 5, 3321. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jiang, L.; Deridder, L.; Elmore, B.; Bukhari, M.; Wei, Q.; Samways, D.S.K.; Dong, H. Membrane activity of a supramolecular peptide-based chemotherapeutic enhancer. Mol. Biosyst. 2016, 12, 2695–2699. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, I.; Jha, D.; Admane, P.; Panda, A.K.; Haridas, V. Self-assembling tryptophan-based designer peptides as intracellular delivery vehicles. Bioorg. Med. Chem. Lett. 2016, 26, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Mosquera, J.; Couceiro, J.R.; Nitschke, J.; Vázquez, M.E.; Mascareñas, J.L. Anion Recognition as a Supramolecular Switch of Cell Internalization. J. Am. Chem. Soc. 2017, 139, 55–58. [Google Scholar] [CrossRef] [PubMed]
- García-López, V.; Chen, F.; Nilewski, L.G.; Aliyan, A.; Wang, G.; Pal, R.; Tour, J.M. Molecular machines open cell membranes. Nature 2017, 548, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, J.; Xu, D. Cell-penetrating peptides as noninvasive transmembrane vectors for the development of novel multifunctional drug-delivery systems. J. Control. Release 2016, 229, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Jiang, K.; Tai, L.; Liu, Y.; Wei, G.; Lu, W.; Pan, W. Facile Noninvasive Retinal Gene Delivery Enabled by Penetratin. ACS Appl. Mater. Interfaces 2016, 8, 19256–19267. [Google Scholar] [CrossRef] [PubMed]
- Dutot, L.; Lécorché, P.; Burlina, F.; Marquant, R.; Point, V.; Sagan, S.; Chassaing, G.; Mallet, J.M.; Lavielle, S. Glycosylated cell-penetrating peptides and their conjugates to a proapoptotic peptide: Preparation by click chemistry and cell viability studies. J. Chem. Biol. 2010, 3, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, S.; Russo, L.; Falanga, A.; Cantisani, M.; Vitiello, M.; Fattorusso, R.; Malgieri, G.; Galdiero, M.; Isernia, C. Structure and orientation of the gH625-644 membrane interacting region of herpes simplex virus type 1 in a membrane mimetic system. Biochemistry 2012, 51, 3121–3128. [Google Scholar] [CrossRef] [PubMed]
- Pooga, M.; Soomets, U.; Hällbrink, M.; Valkna, A.; Saar, K.; Rezaei, K.; Kahl, U.; Hao, J.X.; Xu, X.J.; Wiesenfeld-Hallin, Z.; et al. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat. Biotechnol. 1998, 16, 857–861. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | CPP Name | Sequence | α-Carbon Configuration | Source | Design Approach | Structural Features | Development Stage | Ref. |
|---|---|---|---|---|---|---|---|---|
| Linear Peptides | ||||||||
| 1 | Tat | GRKKRRQRRRPQ | all-l | Natural (transcription protein of HIV-1-positions 48–60) | Virus derived material | Unstructured in buffer solutions; random coil | In clinical trials | [2,3,8,9,15] |
| 2 | Penetratin | RQIKIWFQNRRMKWKK | all-l | Natural (Drosophila Antennapedia homeodomain) | Derived from natural Antennapedia homeoprotein | Secondary amphipathic; forms helices or β-sheets depending on the environment | In vivo data | [4,9,15,76,120,163,164] |
| 3 | Pep-1 | KETWWETWWTEWSQP-KKKRKV | all-l | Fusion CPP: Trp-rich segment; Lys-rich segment; and NLS derived from a virus (SV-40 T-antigen) | Combination of designed and natural segments | Primary amphipathic; forms helices in the presence of phospholipids | In vivo data | [8,9,15,22] |
| 4 | MPG | GALFLGFLGAAGSTMGAWSQP-KKKRKV | all-l | Fusion CPP: NLS derived from a virus (SV-40 T-antigen) and viral hydrophobic domain from HIV-gp-41 segment | Combination of natural segments | Primary amphipathic; forms helices in the presence of phospholipids | In vivo data | [8,23] |
| 5 | Polyarginine (R9, R8) | RRRRRRRRR | all-l | Design inspired by entry 1 and 2 | Designed to be R-rich | Flexible; unstructured; random coil | In clinical trials | [8,9,15,18,19,20,34,56] |
| 6 | R6/W3 | RRWWRRWRR | all-l | Design inspired by entry 2 | Designed to be R-rich and hydrophobic | Secondary amphipathic, forms helices in the presence of phospholipids | Cell assays | [9,15,165] |
| 7 | SAP | (VRLPPP)3 (vrlppp)3 | all-l all-d | Design inspired by a natural protein of maize, γ-zein VHL(PPP)8 | Derived from natural γ-zein protein | Polyproline II helical structure | Cell assays; In vivo data | [24,39,79,103,122,123] |
| 8 | SAP(E) | Ac-CGGW(VELPPP)3 | all-l | Design inspired by SAP; Arg residue replaced by Glu | Designed to be negatively charged | Polyproline II helical structure | Biophysical data | [124,125] |
| 9 | CyLoP-1 | CRWRWKCCKK | all-l | Derived from crotamine toxin found in snake venom, crot(27–39) | Rationally designed, by substitution/ deletion of crot(27–39) sequence | Disulfide-containing form effective; oxidation status of the cysteines important for the uptake | Cell assays | [25] |
| 10 | gH 625 | HGLASTLTRWAHYNALIRAF | all-l | Natural; based on the 625–644 residues of the glycoprotein HSV 1 | Derived from Herpes Simplex virus type I (HSV 1) | Amphipathic; α-helical conformation in contact with model membranes | In vivo data | [26,27,28,29,30,32,33,166] |
| 11 | GALA | WEAALAEALAEALAEHLAEALAEALEALAA | all-l | Glu-rich an containing His (imidazole group) in order to be pH responsive (endosomes) | Designed to efficiently escape endosomes | Flexible but assumes helical structure able to lyse endosomal membranes | In vivo data | [55,67,68] |
| 12 | TP10 | AGYLLGKINLKALAALAKKIL | all-l | Fusion CPP: N-terminal amino acids from galanin (AGYLLGKINLK) linked to matoparan (ALAALAKKIL) | Derived from the neuropeptide galanin linked to a toxin from the wasp venom | Primary amphipathic; forms helices in the presence of phospholipids | In vivo data | [8,9,167] |
| 13 | CADY | Ac-GLWRALWRLLRSLWRLLWRA-cysteamide | all-l | Designed; based on chimeric peptide carrier PPTG1 derived from the fusion peptide JTS1 | Designed by combining aromatic (W) and cationic (R) residues into a secondary amphipathic CPP | Secondary amphipathic; helical conformation | Cell assays | [5,102,119] |
| 14 | L17E | IWLTALKFLGKHAAKHEAKQQLSKL | all-l | Natural; inspired by the spider venom M-lycotoxin | Designed to contain E residues in the hydrophobic part of the amphipathic helix | Secondary amphipathic | Cell assays | [69] |
| 15 | MPPs | Mitochondria-penetrating peptides (example: FXrFXKFXrFXK) | Combination of l- and d- | Designed to contain un-natural, cyclohexylalanine (Fx) residues | Designed to have differential intracellular localization | Flexible | In vivo data | [91,92] |
| 16 | Ac-1 * Ent Ac-1 Ac-2 | Ac-(RR-Aib)3 Ac-(rr-Aib)3 Ac-(Rr-Aib)3 | all-l, all-d or combination of l- and d-Arg | Designed to contain Aib, natural non-coded amino acid | Helix stabilization is introduced at the primary sequence level | α-Helical structure | Cell assays | [129] |
| 17 | Peptide 3 | FAM-β-Ala-(RRPGu)3G3 | all-l | Designed to contain -L proline or guanidinyl -L proline | Pro residue introduced in order to sense hydrophobic and amphipathic environments | Helical structure in contact with membranes | Cell assays | [130] |
| 18 | RR5-App RR4-App * RR3-aPP | RRPRRPRRPRRPGRR-APVEDLIRFYNDLQQYLNVVTRHRYC RRPRRPRRPGRR-APVEDLIRFYNDLQQYLNVVTRHRYC GPRRPRRPGRR-APVEDLIRFYNDLQQYLNVVTRHRYC | all-l | Small proteins (36-residue polypeptides) | Arg residues were located on the solvent-exposed side of PPII helices | PPII-type helix | Cell assays | [47,131] |
| 19 | TATp-D |  | all-l | Designed as analogue of Tat | Covalent dimeric branched peptide; dimerization obtained through bis-Fmoc protected lysine near the C-terminus | Branched peptide | Cell assays | [135] |
| 20 | R4–R4 * R5–R5 | RRRRC-CRRRR RRRRRC-CRRRRR | all-l | Extended peptides obtained through disulfide bridge formation of truncated oligoarginines | Designed as small inactive oligoarginine fragments (R4) activated by linkage through C-terminal cysteines | Branched peptide | Cell assays | [141] |
| Cyclic Peptides | ||||||||
| 21 | [WR]4 * ([FK]4, [AK]4, [EL]4, [RFEF]2, [EK]4, [ER]4, [FR]4, [RFE]3, [WR]3, [WR]5) | c[WRWRWRWR]c | all-l | Designed to contain combinations of hydrophobic (W, F, L) and charged (R, K, E) residues | Designed to obtain optimal amphipathic CPP resistant to proteolysis | Cyclic | Cell assays | [136] |
| 22 | Cyclic Tat | c[K-rRrQrRkKrG-E]c | Combination of l- and d- | Lys- and Glu- amino acids added to the linear Tat sequence to obtain a ring with the same overall charge as the native form | Designed to introduce structural rigidity and controlled spatial distribution of guanidinium groups | Cyclic | Cell assays | [21] |
| 23 | cFΦR4 | c[FΦRRRRQ]c | all-l | Designed to contain Φ (L-2-naphthylalanine) | Varying design parameters used: sequence lengths; stereochemistry; or combination of the two | Cyclic | In vivo data | [60,133] |
| 24 | Danamide D | c-[I(Thz)-tBuGly-FPIP] | all-l | Design based on cyclic heptapeptide sanguinamide A | Rigid scaffold obtained through formation of a heterocycle; rigidity reinforced through bulky hydrophobic tertbutyl glycine side chains | Cyclic | In vivo data | [105] |
| 25 | Pro-(Xaa)4-Tyr | c[d-Pro−l-MeLeu−d-MeLeu−d/l-MeLeu−d/l-Leu−l-Tyr]c * | all-l, all-d or combination of l- and d- | Inspired by cyclic natural products Guangomide A and baceridin | Stereochemistry and N-methylation used to obtain backbone geometries with different conformational preferences | Cyclic | Biophysical data | [137] |
| 26 | Cyclic sC18 | c[GLRKRLRKFRNK]c-IKEK * | all-l | Inspired by the CAP-18 antimicrobial peptide; cyclization obtained by connecting residues at positions 1 + 4, 1 + 8 and 1 + 12 of the linear peptide | Gly and Lys residues replaced by propargylglycine and ε-azidolysine to allow cyclization through triazole bridge formation by click chemistry | Cyclic | Cell assays | [49] |
| 27 | RRRRΦF * |  | all-l | Bicyclic compounds; cyclization through cysteine side chains and 3,5-bis(mercaptomethyl) benzoic acid (BMB) | Designed to be conformationally constrained with the goal to induce the uptake of generally impermeable peptidyl drugs | Cyclic | Cell assays | [134,138] |
| 28 | BIM SAHB9 SAH–SOS1 | IWIAQELRXIGDXFNAYYARR * ZFGIYLTXILKTEEGN | all-l | Designed to have i + 4 staples (between X pairs; X is S-pentenylalanine) or i + 7 staples (between Z and X; Z is R-octenyl alanine | Hydrophobic staple introduced for α-helical stabilization | Stapled | Cell assays | [146] |
| 29 | 4-R 4-W | FITC-β-A (iso-DRRX)WRRW FITC-β-A (iso-DWWX)RWWR | l- or d-stereoisomers of Asp used | Peptide bond formation between the side chain of the terminal Asp and the -NH2 of Dap (X is Dap) to obtain a crosslink | Introduction of crosslinks (at similar distance to that introduced through stapling) to stabilize helical conformations | N-terminal crosslinking | Cell assays | [149] |
| Supramolecular CPPs | ||||||||
| 30 | Sp-CC-PEG2000 | Sp-REGVAKALRAVANALHYNASALEEVADALQKVKM-PEG | all-l | Obtained by the self-assembly of coiled-coil peptides decorated with cationic segments (Sp = spermine) and PEG placed at opposite termini | Supramolecular structures form due to the interaction of the peptide derivative with a DNA segment | Supramolecular filamentous virus-like nanostructures | Biophysical data | [157] |
| 31 | K10(QW)6 | KKKKKKKKKKQWQWQWQWQWQW | all-l | The design was based on combining W and K at the primary structure level to obtain self-assembly into a variety of nanostructures | Supramolecular structures form due to the interplay of electrostatic and hydrophobic interactions alongside with hydrogen bonding among QL/QW repeating units | Supramolecular fibers (based on β-sheets) | Cell assays | [36,159] |
| 32 | YTA4 | C16-IAWVKAFIRKLRKGPLG-GPLGIAGQ-RGDS | all-l | CPP amphiphile system based on the self-assembly of multi-domain peptide sequences | Design based on four main structural and functional parameters: palmitic acid (C16) tail to drive self-assembly into nanofibers; a CPP moiety; an enzyme-sensitive moiety; and an RGD-based targeting moiety | Self-assembly into nanofibers | Biophysical data | [43] |
| 33 | V2 | FITC–AβGG-POGPOGPOGPOGPOGPOGPOGPOGRRRRRR | all-l | Designed to contain (POG)n collagen like motif to induce triple helix formation (O is hydroxyproline) | Stable helix obtained inspired by collagen decorated with R6 | Triple helix | Cell assays | [14] |
| 34 | W3 | WWW | all-l | The design comprised linear, tripodal and dendrimeric W rich structures | Design based on self-assembly info spherical aggregates | Spherical aggregates | Cell assays | [160] |
| Peptido mimetics | ||||||||
| 35 | CPPMs | Synthetic mimics of CPPs | Polypeptides obtained by ring opening polymerization | Designed to mimic Tat | Designed to have a polymeric backbone with guanidines at specific positions, aromatic or aliphatic functionality | Flexible | Biophysical data | [81,88,97,98] |
| 36 | Peptoids (CPPos) | Peptidomimetic molecules | Not applicable | Have side chains on the nitrogen atom rather than on α-carbon | Specific organelle localization | Flexible | Cell assays | [89,90] |
| 37 | CPDs | Cell-penetrating disulfides | Not applicable | Designed to have a polydisulfide backbone; guanidinium rich | Designed to be responsive to reductive environments | Flexible/disulfide bridges | Cell assays | [13,93,94,95,96] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. https://doi.org/10.3390/molecules22111929
Kalafatovic D, Giralt E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules. 2017; 22(11):1929. https://doi.org/10.3390/molecules22111929
Chicago/Turabian StyleKalafatovic, Daniela, and Ernest Giralt. 2017. "Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity" Molecules 22, no. 11: 1929. https://doi.org/10.3390/molecules22111929
APA StyleKalafatovic, D., & Giralt, E. (2017). Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules, 22(11), 1929. https://doi.org/10.3390/molecules22111929