Predict the Relationship between Gene and Large Yellow Croaker’s Economic Traits


Abstract
:1. Introduction
2. Results
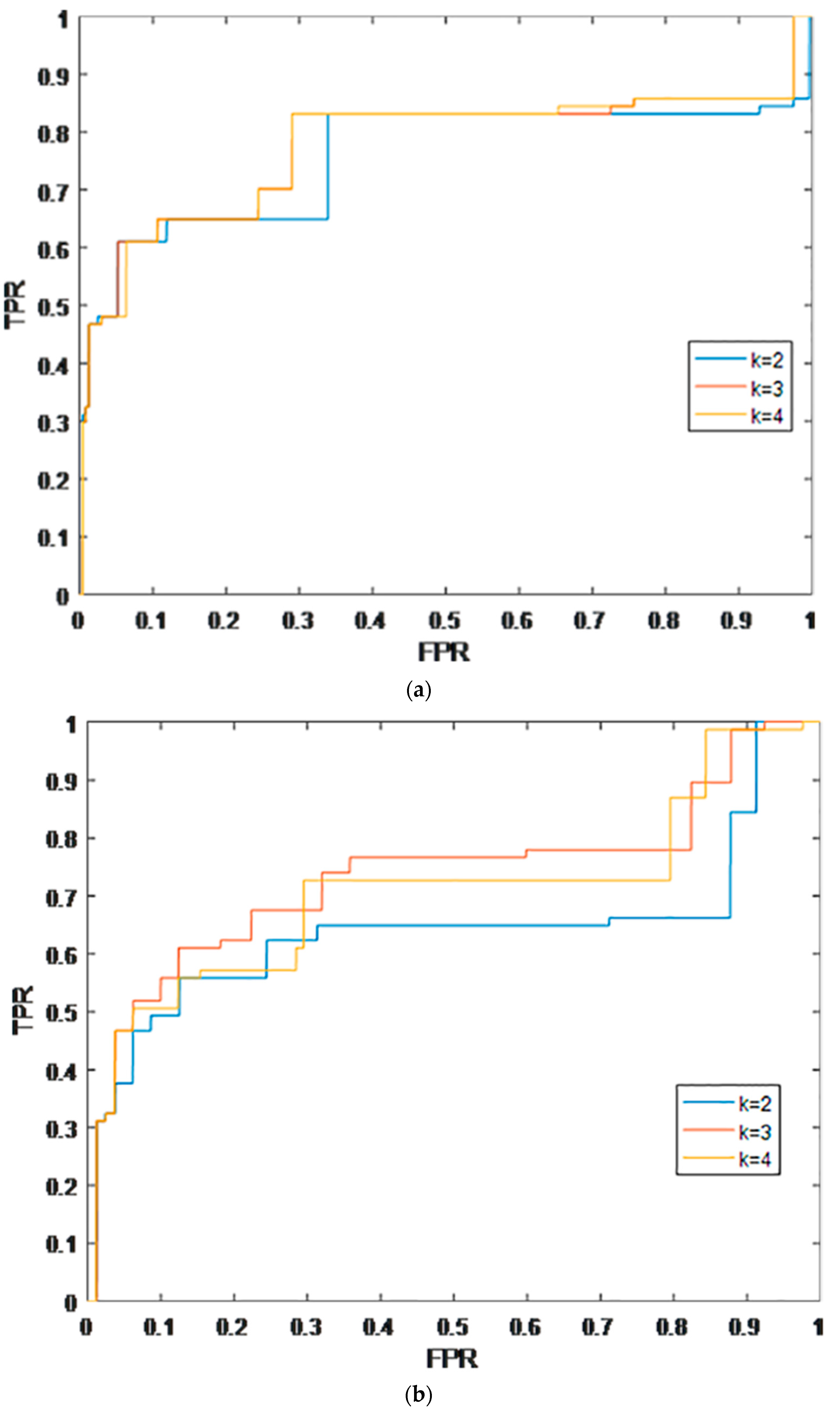
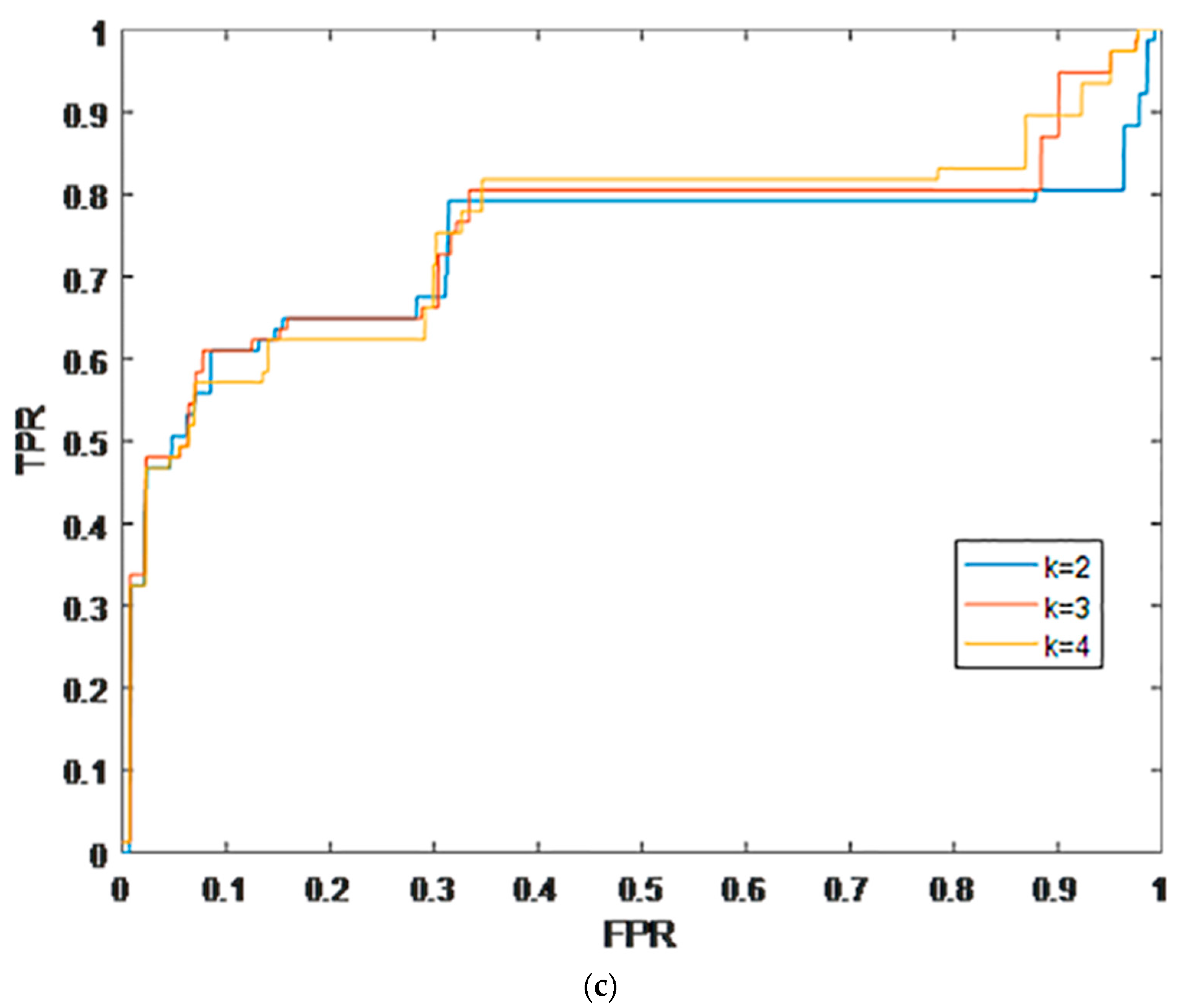
2.1. Leave-One-Out Cross Validation
2.2. K-Fold Cross Validation
3. Materials and Methods
3.1. Materials
3.2. Methods
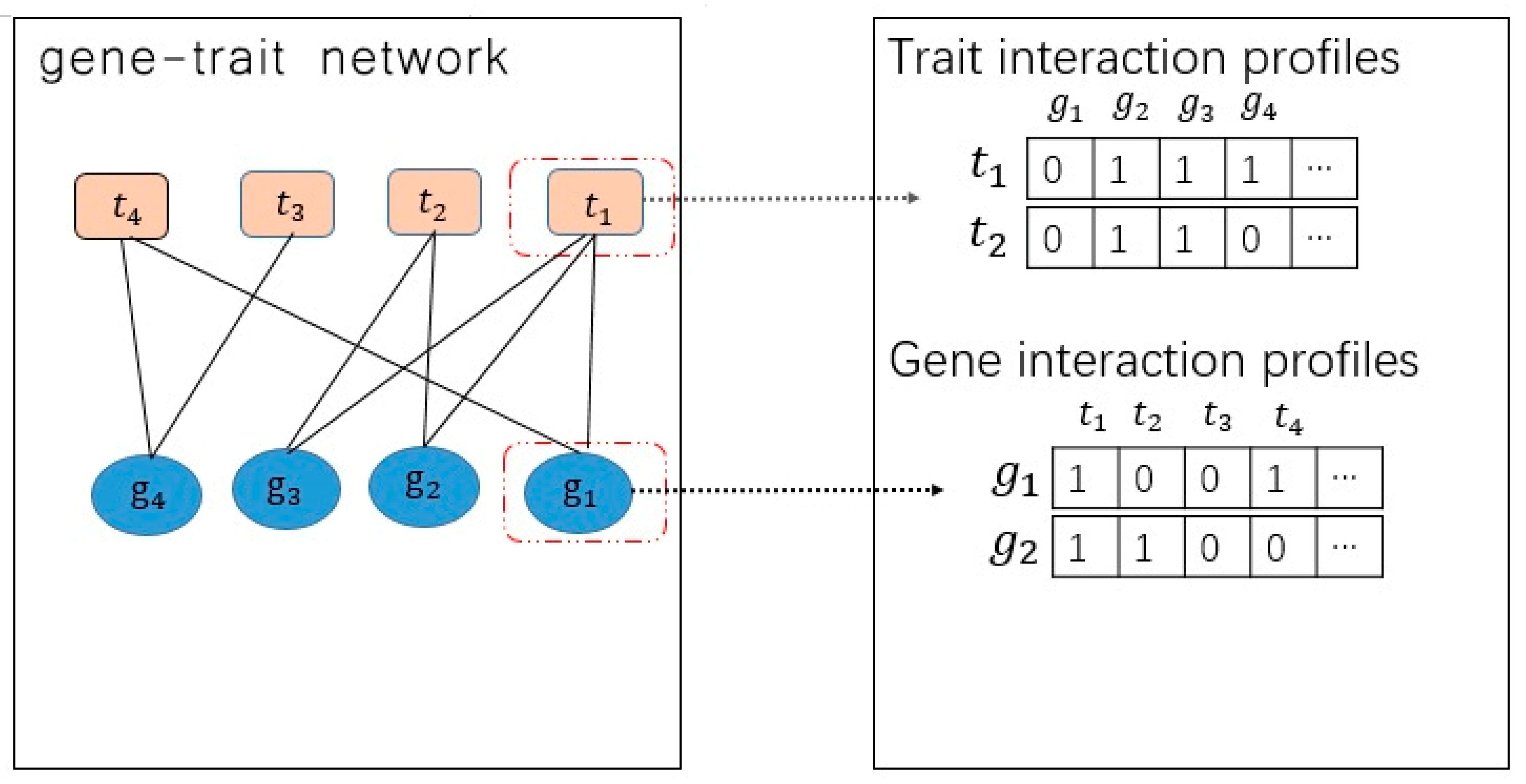
3.2.1. Problem to Formalize [8]
3.2.2. Gaussian Interaction Profile Kernel Similarity for Genes
3.2.3. Gaussian Interaction Profile Kernel Similarity for Trait
3.2.4. KATZ-YC
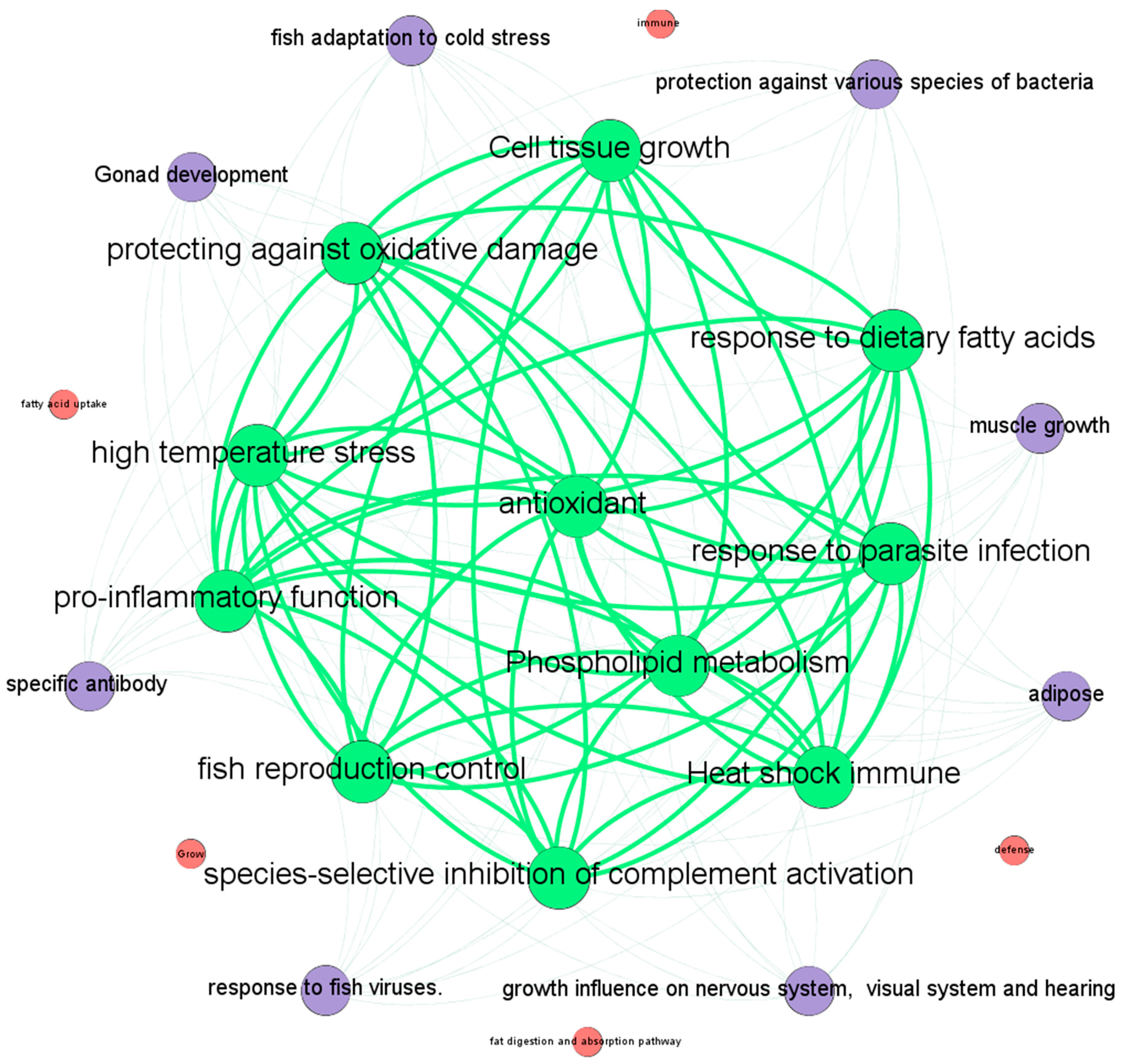
4. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brown, S.M. Molecular Biology Technology. In Essentials of Medical Genomics, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 31–51. [Google Scholar]
- Shen, B.; Hu, Y.; Zhang, S.; Zheng, J.; Zeng, L.; Zhang, J.; Zhu, A.; Wu, C. Molecular characterization and expression analyses of three RIG-I-like receptor signaling pathway genes (MDA5, LGP2 and MAVS) in Larimichthys crocea. Fish Shellfish Immunol. 2016, 55, 535. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.F.; Huang, X.N.; Yao, C.L.; Sun, Q.X.; Li, Y.; Zhu, Q.; Yu, Z.X.; Fan, Z.J. Cloning and functional characterization of IRAK4 in large yellow croaker (Larimichthys crocea) that associates with MyD88 but impairs NF-κB activation. Fish Shellfish Immunol. 2017, 63, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kang, L.; Liu, W.; Lou, B.; Wu, C.; Jiang, L. Molecular characterization and expression analysis of the large yellow croaker (Larimichthys crocea) chemokine receptors CXCR2, CXCR3, and CXCR4 after bacterial and poly I:C challenge. Fish Shellfish Immunol. 2017, 70, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Kankainen, M.; Setälä, J.; Kause, A.; Quinton, C.; Airaksinen, S.; Koskela, J. Economic values of supply chain productivity and quality traits calculated for a farmed European whitefish breeding program. Aquac. Econ. Manag. 2016, 20, 131–164. [Google Scholar] [CrossRef]
- Katz, L. A new status index derived from sociometric analysis. Psychometrika 1953, 18, 39–43. [Google Scholar] [CrossRef]
- Chen, X.; Yan, G.Y. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 2013, 29, 2617. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Q.; Rong, Z.H.; Chen, X.; Yan, G.Y.; You, Z.H. MCMDA: Matrix completion for MiRNA-disease association prediction. Oncotarget 2017, 8, 21187–21199. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Y.; Yin, H.; Le, H.; Rui, M. The Algorithm of Link Prediction on Social Network. Math. Probl. Eng. 2013, 2013, 125123. [Google Scholar] [CrossRef]
- Laarhoven, T.V.; Nabuurs, S.B.; Marchiori, E. Gaussian Interaction Profile Kernels for Predicting Drug–Target Interaction; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Shao, Z.; Meng, J.E.; Wang, N. An Efficient Leave-One-Out Cross-Validation-Based Extreme Learning Machine (ELOO-ELM) With Minimal User Intervention. IEEE Trans. Cybern. 2016, 46, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Tan, P.; Cai, Z.; Xu, H.; Li, J.; Ren, W.; Xu, H.; Zuo, R.; Zhou, J.; Mai, K.; et al. Regulation of FADS2 transcription by SREBP-1 and PPAR-α influences LC-PUFA biosynthesis in fish. Sci. Rep. 2017, 7, 40024. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; You, Z.H.; Yan, G.Y.; Gong, D.W. IRWRLDA: Improved random walk with restart for lncRNA-disease association prediction. Oncotarget 2016, 7, 57919. [Google Scholar] [CrossRef] [PubMed]
- Liao, K.; Yan, J.; Mai, K.; Ai, Q. Dietary lipid concentration affects liver mitochondrial DNA copy number, gene expression and DNA methylation in large yellow croaker (Larimichthys crocea). Comp. Biochem. Physiol. Part B 2015, 193, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Li, C.; Yu, Z.X.; Zou, P.F.; Meng, Q.X.; Yao, C.L. Molecular and immune response characterizations of IL-6 in large yellow croaker (Larimichthys crocea). Fish Shellfish Immunol. 2016, 53, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Jin, M.; Li, Y.; Lu, Y.; Hou, Y.; Zhou, Q. Dietary Lipid Sources Influence Fatty Acid Composition in Tissue of Large Yellow Croaker (Larmichthys crocea) by Regulating Triacylglycerol Synthesis and Catabolism at the Transcriptional Level. PLoS ONE 2017, 12, e0169985. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Bao, P.; Tang, B. Transcriptome analysis and discovery of genes involved in immune pathways in large yellow croaker (Larimichthys crocea) under high stocking density stress. Fish Shellfish Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, Y.; He, S.; Zhu, Z. Cloning and sequencing of the growth hormone gene of large yellow croaker and its phylogenetic significance. Biochem. Genet. 2004, 42, 365. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, Y.A.; You, Z.H.; Yan, G.Y.; Wang, X.S. A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 2016, 33, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. KATZLDA: KATZ measure for the lncRNA-disease association prediction. Sci. Rep. 2015, 5, 16840. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Wang, P.; Zhang, Y.; Fang, L.; Liu, Y.; Li, J.T.; Wang, Z.Y. Gene map of large yellow croaker (Larimichthys crocea) provides insights into teleost genome evolution and conserved regions associated with growth. Sci. Rep. 2015, 5, 18661. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |



| T | t1 | t2 | t3 | t4 | … | |
|---|---|---|---|---|---|---|
| G | ||||||
| g1 | 1 | 0 | 0 | 1 | … | |
| g2 | 1 | 1 | 0 | 0 | … | |
| g3 | 1 | 1 | 0 | 0 | … | |
| g4 | 0 | 0 | 1 | 1 | … | |
| … | … | … | … | … | … |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| K = 2 | K = 3 | K = 4 |
|---|---|---|
| 0.7564 | 0.7766 | 0.7760 |
| K = 2 | K = 3 | K = 4 |
|---|---|---|
| 0.7036 ± 0.03 | 0.7183 ± 0.03 | 0.7132 ± 0.05 |
| K = 2 | K = 3 | K = 4 |
|---|---|---|
| 0.7338 ± 0.03 | 0.7476 ± 0.03 | 0.7357 ± 0.04 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, X.; Jin, S.; Jiang, J.; Han, K.; Min, X.; Liu, X. Predict the Relationship between Gene and Large Yellow Croaker’s Economic Traits. Molecules 2017, 22, 1978. https://doi.org/10.3390/molecules22111978
Zeng X, Jin S, Jiang J, Han K, Min X, Liu X. Predict the Relationship between Gene and Large Yellow Croaker’s Economic Traits. Molecules. 2017; 22(11):1978. https://doi.org/10.3390/molecules22111978
Chicago/Turabian StyleZeng, Xiangxiang, Shuting Jin, Jing Jiang, Kunhuang Han, Xiaoping Min, and Xiangrong Liu. 2017. "Predict the Relationship between Gene and Large Yellow Croaker’s Economic Traits" Molecules 22, no. 11: 1978. https://doi.org/10.3390/molecules22111978
APA StyleZeng, X., Jin, S., Jiang, J., Han, K., Min, X., & Liu, X. (2017). Predict the Relationship between Gene and Large Yellow Croaker’s Economic Traits. Molecules, 22(11), 1978. https://doi.org/10.3390/molecules22111978