A Strategy for Preparative Separation of 10 Lignans from Justicia procumbens L. by High-Speed Counter-Current Chromatography

Abstract
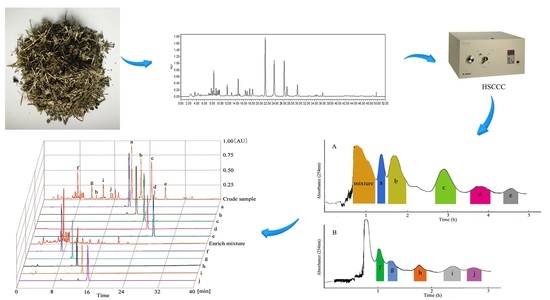
:
1. Introduction
2. Results and Discussion
2.1. Selection of the Two-Phase Solvent System
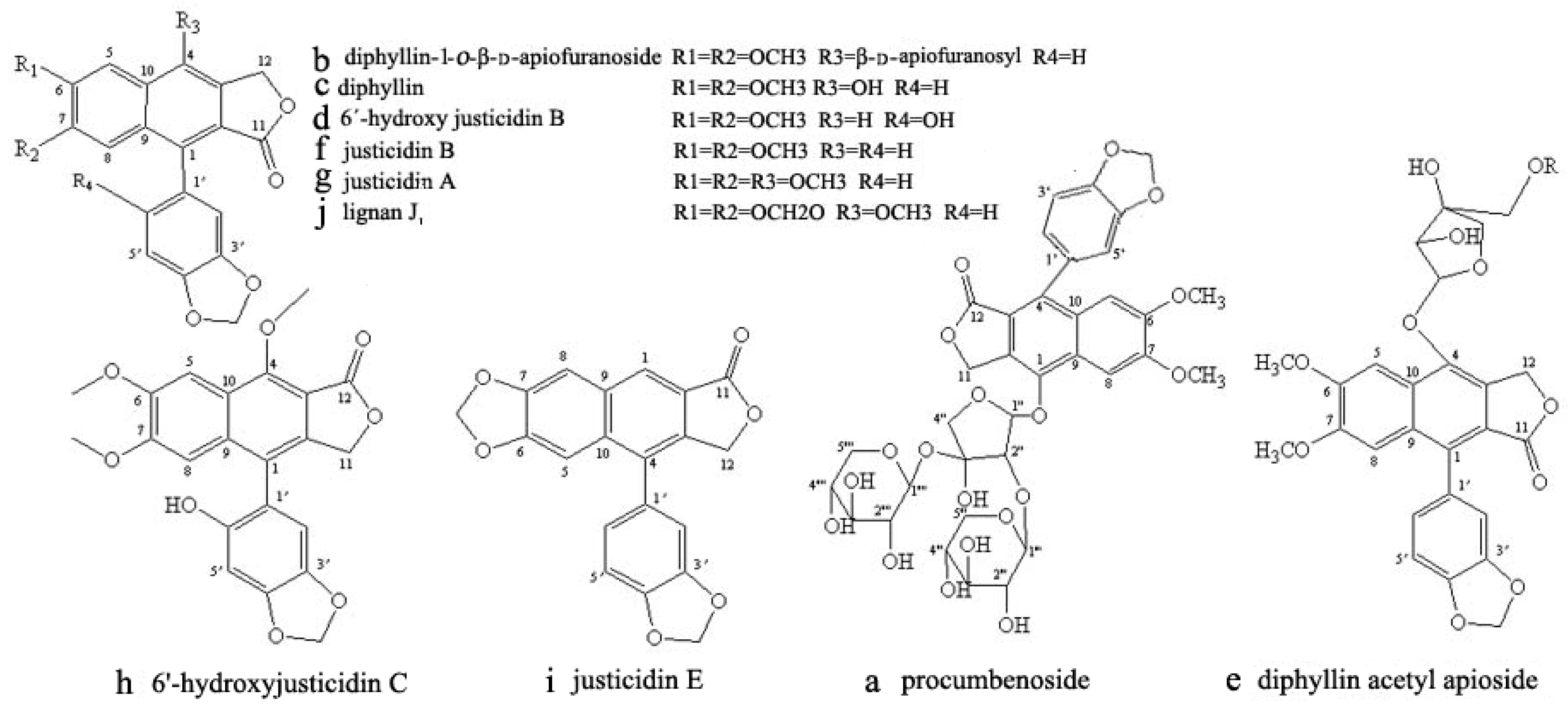
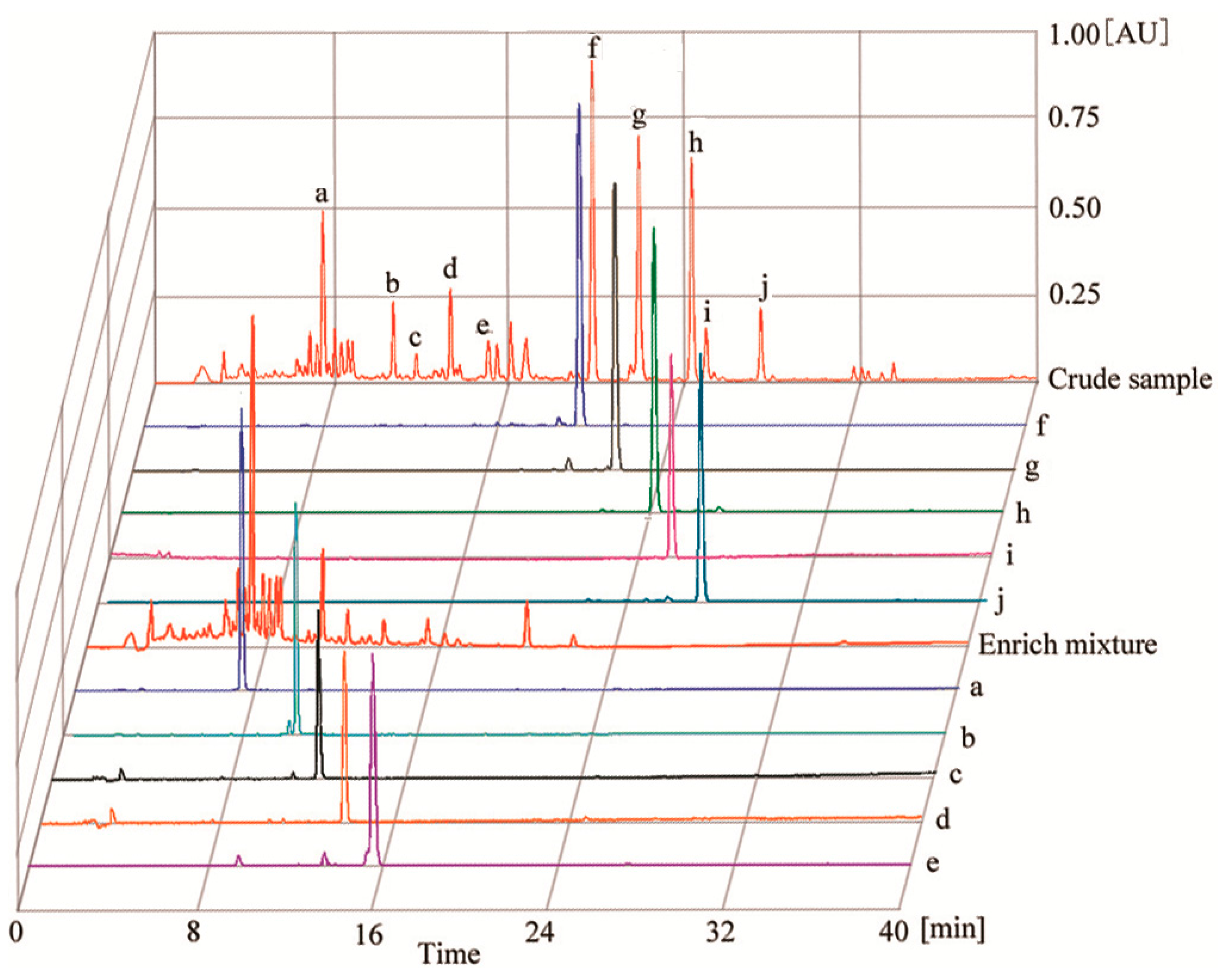
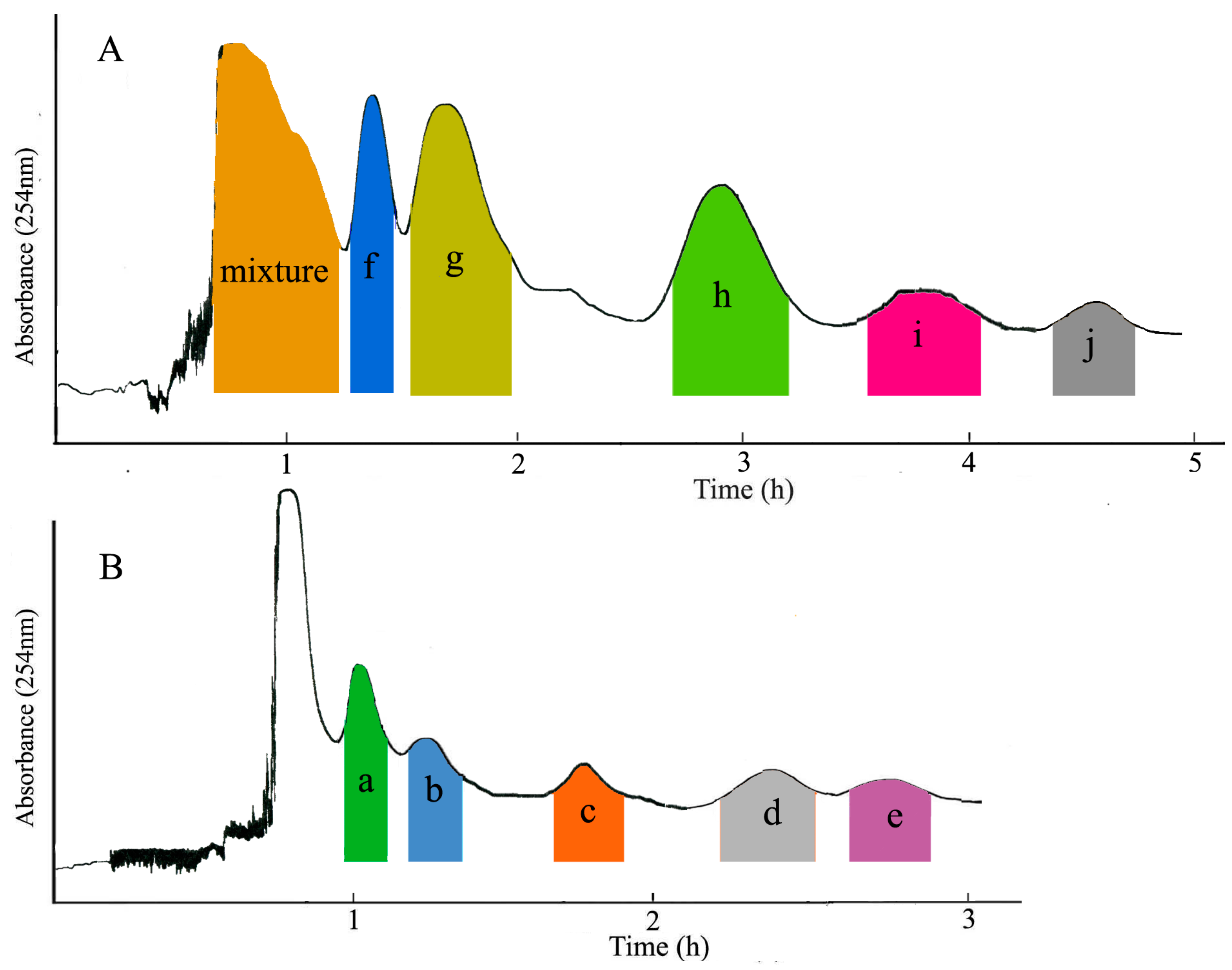
2.2. HPLC Analysis of HSCCC Peak Fractions
3. Experimental
3.1. Material and Reagents
3.2. Apparatus
3.3. Pre-Processing of Crude Sample
3.4. Measurement of the Partition Coefficients (KD)
3.5. Preparation of Solvent Systems and Sample Solutions
3.6. Separation Procedure
3.7. HPLC Analysis of CCC Separation Products
3.8. Identification of CCC Fractions
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, C.C.; Hsin, W.C.; Ko, F.N.; Huang, Y.L.; Ou, J.C.; Teng, C.M. Antiplatelet arylnaphthalide lignans from Justicia procumbens. J. Nat. Prod. 1996, 59, 1149–1150. [Google Scholar] [CrossRef] [PubMed]
- Su, C.L.; Huang, L.L.; Huang, L.M.; Lee, J.C.; Lin, C.N.; Won, S.J. Caspase-8 acts as a key upstream executor of mitochondria during justicidin A-induced apoptosis in human hepatoma cells. FEBS Lett. 2006, 580, 3185–3191. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pan, J.; Yang, M.; Wu, J.; Yang, J. Chromatographic fingerprint analysis and simultaneous determination of eight lignans in Justicia procumbens and its compound preparation by HPLC-DAD. J. Sep. Sci. 2011, 34, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Cheng, Y.X. Antibacterial Lignans and Triterpenoids from Rostellularia procumbens. Planta Med. 2007, 73, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Day, S.H.; Chiu, N.Y.; Lin, C.N. Cytotoxic Lignans of Justicia ciliate. J. Nat. Prod. 1999, 62, 1056–1058. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Kong, W.; Qiu, F.; Yang, M.; Li, Q.; Wei, R.; Yang, X.; Qin, J. Simultaneous determination of seven lignans in Justicia procumbens by high performance liquid chromatography-photodiode array detection using relative response factors. J. Sep. Sci. 2013, 36, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Willfor, S.M.; Smeds, A.I.; Holmbom, B.R. Chromatographic analysis of lignans. J. Chromatogr. A 2006, 1112, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Madl, R.L.; Takemoto, D.J.; Baybutt, R.C.; Wang, W. Lignans are involved in the antitumor activity of wheat bran in colon cancer SW480 cells. J. Nutr. 2005, 135, 598–602. [Google Scholar] [PubMed]
- Wu, M.D.; Huang, R.L.; Kuo, L.M.; Hung, C.C.; Ong, C.W.; Kuo, Y.H. The anti-HBsAg (human type B hepatitis, surface antigen) and anti-HBeAg (human type B hepatitis, e antigen) C18 dibenzocyclooctadiene lignans from Kadsura matsudai and Schizandra arisanensis. Chem. Pharm. Bull. 2003, 51, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, P.A.; Garcia, J.M.; Del Corral, M.A.; Castro, M.A. Gómez-Zurita, Podophyllotoxin: Distribution, sources, applications and new cytotoxic derivatives. Toxicon 2004, 44, 441–459. [Google Scholar] [CrossRef] [PubMed]
- Day, S.H.; Lin, Y.C.; Tsai, M.L.; Tsao, L.T.; Ko, H.H.; Chung, M.I.; Lee, J.C.; Wang, J.P.; Won, S.J.; Lin, C.N. Potent cytotoxic lignans from Justicia procumbens and their effects on nitric oxide and tumor necrosis factor-α production in mouse macrophages. J. Nat. Prod. 2002, 65, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.L.; Liu, F.; Sun, J.; Li, J.; Wang, X. Optimisation and establishment of separation conditions of organic acids from Usnea longissima Ach. by pH-zone-refining counter-current chromatography: Discussion of the eluotropic sequence. J. Chromatogr. A 2016, 1427, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Fukamiya, N.; Lee, K.H. Antitumor agents, 81. Justicidin-A and diphyllin, two cytotoxic principles from Justicia procumbens. J. Nat. Prod. 1986, 49, 348–350. [Google Scholar] [CrossRef] [PubMed]
- Skalicka-Wozniak, K.; Garrard, I. A comprehensive classification of solvent systems used for natural product purifications in countercurrent and centrifugal partition chromatography. Nat. Prod. Rep. 2015, 32, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Shehzad, O.; Khan, S.; Ha, I.J.; Park, Y.; Tosun, A.; Kim, Y.S. Dynamic pH junction high-speed counter-current chromatography coupled with microwave-assisted extraction for online separation and purification of alkaloids from Stephania cepharantha. J. Chromatogr. A 2013, 1310, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.A.; Fisher, D. Role of counter-current chromatography in the modernisation of Chinese herbal medicines. J. Chromatogr. A 2009, 1216, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, Y.; Zhang, Y.; Liu, L.; Xu, Y.X.; Xu, Y.H.; Liu, T.H. Preparative isolation and purification of five flavonoid glycosides and one benzophenone galloyl glycoside from Psidium guajava by high-speed counter-current chromatography (HSCCC). Molecules 2013, 18, 15648–15661. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Dong, H.; Liu, Y.; Yang, B.; Wang, X.; Huang, L. Application of high-speed counter-current chromatography for preparative separation of cyclic peptides from Vaccaria segetalis. J. Chromatogr. B 2011, 879, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.L.; Li, J.; Wang, D.J.; Yu, J.Q.; Wang, X.; Huang, L.Q. Preparative separation of alkaloids from Litsea cubeba using combined applications of pH-zone-refining and high-speed counter-current chromatography. RSC Adv. 2015, 92, 75831–75837. [Google Scholar] [CrossRef]
- Kotland, A.; Chollet, S.; Autret, J.M.; Diard, C.; Marchal, L.; Renault, J.H. Modeling pH-zone refining countercurrent chromatography: A dynamic approach. J. Chromatogr. A 2015, 1391, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.H.; Shu, X.K.; Jing, F.; Wang, X.; Lin, C.H.; Luo, A.Q. Preparative Separation of Alkaloids from Picrasma quassioides (D. Don) Benn. by Conventional and pH-Zone-Refining Countercurrent Chromatography. Molecules 2014, 19, 8752–8761. [Google Scholar] [CrossRef] [PubMed]
- Bakri, M.; Chen, Q.; Ma, Q.L.; Yang, Y.; Abdukadir, A.; Aisa, H.A. Separation and purification of two new and two known alkaloids from leaves of Nitraria sibirica by pH-zone-refining counter-current chromatography. J. Chromatogr. B 2015, 1006, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Z.; Zhang, Y.Q.; Liu, Q.; Sun, C.L.; Li, J.; Yang, P.; Wang, X. Preparative Separation of Phenolic Compounds from Chimonanthus praecox Flowers by High-Speed Counter-Current Chromatography Using a Stepwise Elution Mode. Molecules 2016, 21, 8752–8761. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.J.; Luo, Q.J.; Ding, L.J.; Fang, F.; Yuan, Y.; Chen, J.J.; Zhang, J.R.; Jin, H.X.; He, S. Preparative isolation and purification of lignans from Justicia procumbens using high-speed counter-current chromatography in stepwise elution mode. Molecules 2015, 20, 7048–7058. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Lu, Y.; Jiang, L.; Wu, B.; Zhang, F.; Pan, Y. Preparative isolation and purification of antioxidative stilbene oligomers from Vitis chunganeniss using high-speed counter-current chromatography in stepwise elution mode. J. Sep. Sci. 2009, 32, 2339–2345. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.T.; Li, L.X.; Cui, Y.; Luo, L.X.; Li, Y.Y.; Zhou, P.Y.; Sun, B.S. Preparative high-speed counter-current chromatography separation of grape seed proanthocyanidins according to degree of polymerization. Food Chem. 2017, 219, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.X.; Cui, Y.; Zhang, S.T.; Li, L.X.; Li, Y.Y.; Zhou, P.Y.; Sun, B.S. Preparative separation of grape skin polyphenols by high-speed counter-current chromatography. Food Chem. 2016, 212, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.L.; Oleschuk, C.J.; Si, J.; Chee, G.L.; Yang, M. Hindered rotation in arylnaphthalene lignans. J. Org. Chem. 1996, 61, 3452–3457. [Google Scholar] [CrossRef]
- Liu, G.R.; Wu, J.; Si, J.Y.; Wang, J.M.; Yang, M.H. Complete assignments of 1H and 13C-NMR data for three new arylnaphthalene lignan from Justicia procumbens. Magn. Reson. Chem. 2008, 46, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Q.; Huang, J.; Nagatsu, A.; Ogihara, Y. Two newpodophyllotoxin glucosides from Sinopodophyllum emodi (Wall.) Ying. Chem. Pharm. Bull. 2001, 49, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wu, J.; Cheng, F.; Zhou, Y. Complete assignments of 1H- and 13C-NMR data for seven arylnaphthalide lignans from Justicia procumbens. Magn. Reson. Chem. 2006, 44, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Hayat, F.; Kang, L.; Lee, C.Y.; Shin, D. Synthesis of arylnaphthalene lignan lactone using benzoin condensation, intramolecular thermal cyclization and Suzuki coupling. Tetrahedron 2015, 71, 2945. [Google Scholar] [CrossRef]
- Gopalaiah, K.; Kavitha, J.; Kanumuri, R.V.; Rajasekhar, D.; Subbaraju, G.V. Justicia lignans: Part 9—Two new lignans from Justicia neesii Ramamoorthy (white flower variety). Indian J. Chem. 2001, 40, 596–600. [Google Scholar] [CrossRef]
Sample Availability: Samples of the lignans and lignan glycosides compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Solvent System (v/v): (Pet–EtOAc–MeOH–H2O) | Partition Coefficient (KD) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a | b | c | d | e | f | g | h | i | j | ||
| Crude sample | 3:3:3:3 | 0.12 | 0.25 | 0.34 | 0.81 | 1.28 | 4.06 | 6.18 | 7.12 | 7.89 | 12.10 |
| 3:2.4:3:2.4 | 0.09 | 0.19 | 0.29 | 0.68 | 1.06 | 2.52 | 4.33 | 5.41 | 5.97 | 6.43 | |
| 3:2.1:3:2.1 | 0.07 | 0.11 | 0.18 | 0.23 | 0.42 | 0.74 | 1.35 | 1.84 | 2.18 | 2.44 | |
| 3:1.8:3:1.8 | 0.05 | 0.08 | 0.10 | 0.16 | 0.31 | 0.37 | 0.67 | 1.26 | 1.54 | 2.07 | |
| Enriched mixture | 3:3.2:3:3.2 | 0.17 | 0.30 | 0.44 | 0.93 | 1.44 | |||||
| 3:3.8:3:3.8 | 0.22 | 0.68 | 0.93 | 1.75 | 2.08 | ||||||
| 3:4.2:3:4.2 | 0.35 | 1.34 | 1.86 | 2.52 | 4.03 | ||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, J.; Dong, H.; Wang, T.; Zhao, R.; Mu, Y.; Geng, Y.; Zheng, Z.; Wang, X. A Strategy for Preparative Separation of 10 Lignans from Justicia procumbens L. by High-Speed Counter-Current Chromatography. Molecules 2017, 22, 2024. https://doi.org/10.3390/molecules22122024
Jiang J, Dong H, Wang T, Zhao R, Mu Y, Geng Y, Zheng Z, Wang X. A Strategy for Preparative Separation of 10 Lignans from Justicia procumbens L. by High-Speed Counter-Current Chromatography. Molecules. 2017; 22(12):2024. https://doi.org/10.3390/molecules22122024
Chicago/Turabian StyleJiang, Jiaojiao, Hongjing Dong, Tao Wang, Ruixuan Zhao, Yan Mu, Yanling Geng, Zhenjia Zheng, and Xiao Wang. 2017. "A Strategy for Preparative Separation of 10 Lignans from Justicia procumbens L. by High-Speed Counter-Current Chromatography" Molecules 22, no. 12: 2024. https://doi.org/10.3390/molecules22122024
APA StyleJiang, J., Dong, H., Wang, T., Zhao, R., Mu, Y., Geng, Y., Zheng, Z., & Wang, X. (2017). A Strategy for Preparative Separation of 10 Lignans from Justicia procumbens L. by High-Speed Counter-Current Chromatography. Molecules, 22(12), 2024. https://doi.org/10.3390/molecules22122024