A New Series of Pyrrole-Based Chalcones: Synthesis and Evaluation of Antimicrobial Activity, Cytotoxicity, and Genotoxicity

 ,
,  ,
,  ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
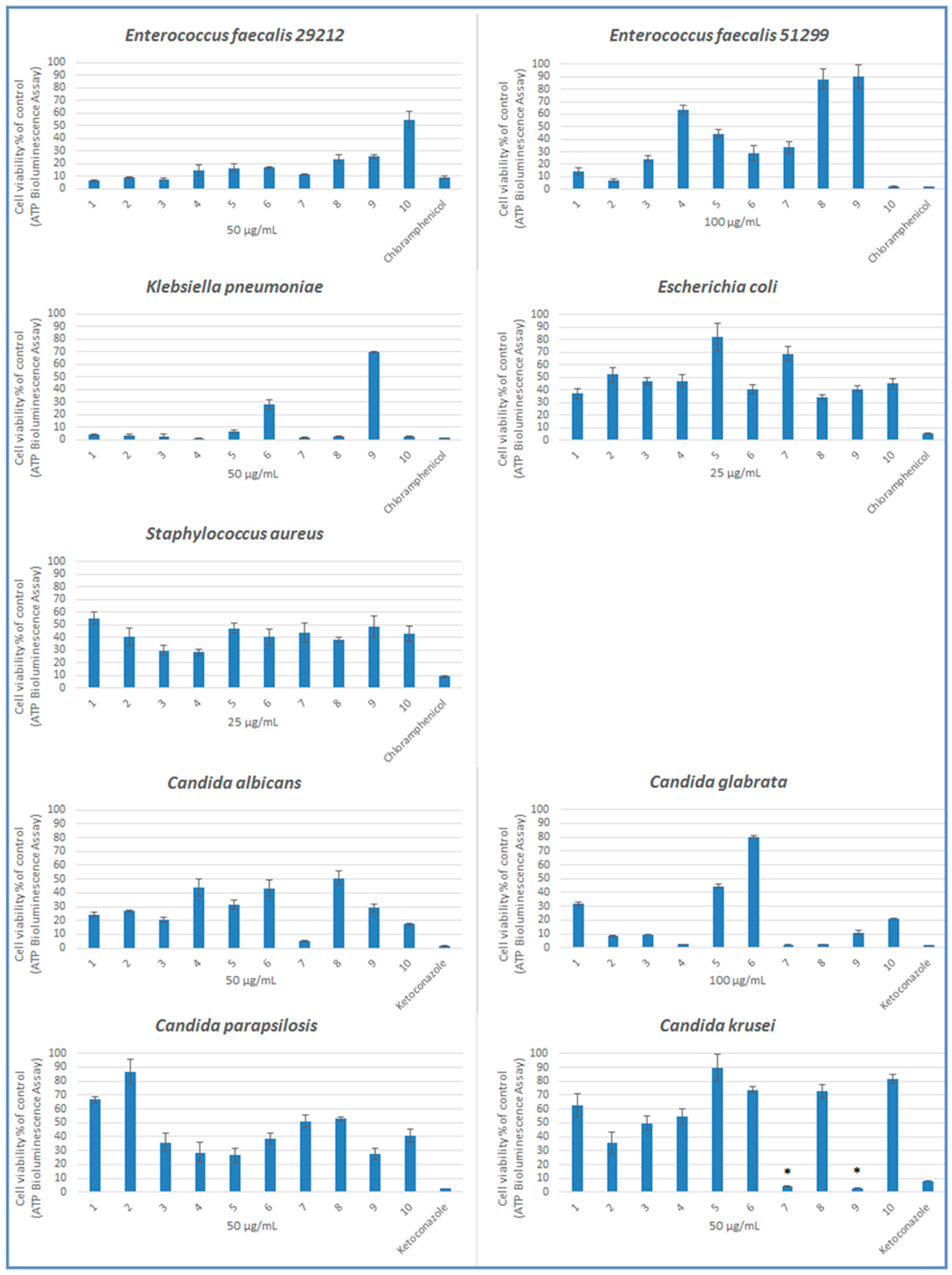
2.2. Microbiology
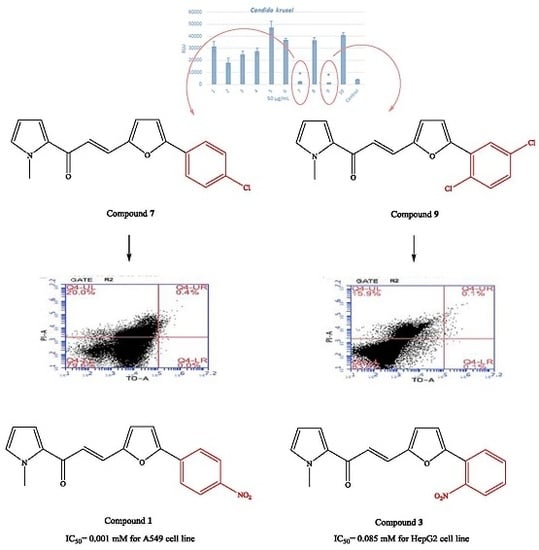
2.3. Cytotoxicity
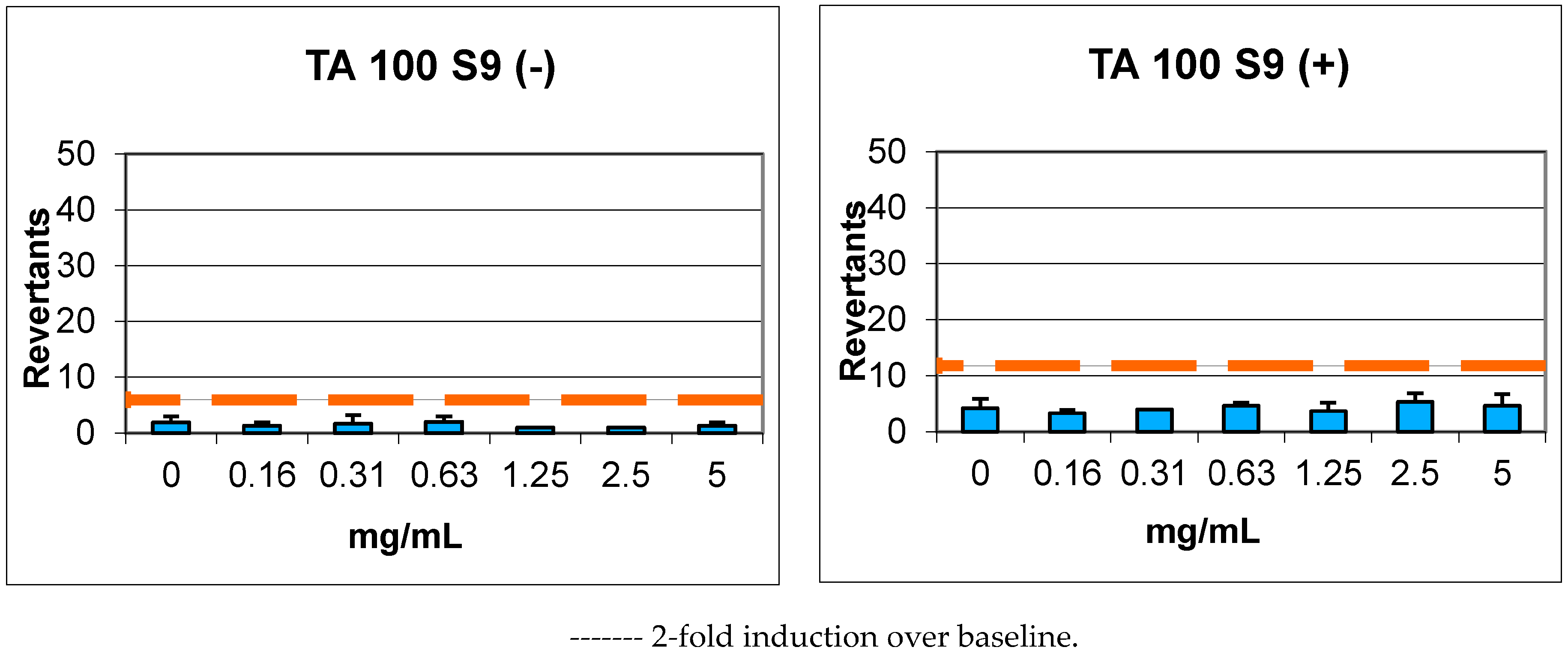
2.4. Genotoxicity
3. Materials and Methods
3.1. Chemistry
3.2. Microbiology
3.2.1. Strains and Growth Conditions
3.2.2. Microdilution Assay to Determine MICs
3.2.3. ATP Luminescence Assay
3.2.4. Flow Cytometry
3.2.5. Statistical Analysis of the Quantitative Data
3.3. Cytotoxicity
3.4. Genotoxicity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Woodford, N.; Livermore, D.M. Infections caused by gram-positive bacteria: A review of the global challenge. J. Infect. 2009, 59, S4–S16. [Google Scholar] [CrossRef]
- Tenover, F.C. Mechanisms of antimicrobial resistance in bacteria. Am. J. Med. 2006, 119, S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hou, Y.; Yue, L.; Liu, S.; Du, J.; Sun, S. Potential targets for antifungal drug discovery based on growth and virulence in Candida albicans. Antimicrob. Agents Chemother. 2015, 59, 5885–5891. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.L.; Polvi, E.J.; Shekhar-Guturja, T.; Cowen, L.E. Elucidating drug resistance in human fungal pathogens. Future Microbiol. 2014, 9, 523–542. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.S.; Robbins, N.; Cowen, L.E. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol. Mol. Biol. Rev. 2011, 75, 213–267. [Google Scholar] [CrossRef] [PubMed]
- Pierce, C.G.; Lopez-Ribot, J.L. Candidiasis drug discovery and development: New approaches targeting virulence for discovering and identifying new drugs. Expert Opin. Drug Discov. 2013, 8, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Popat, K.; McQueen, K.; Feeley, T.W. The global burden of cancer. Best Pract. Res. Clin. Anaesthesiol. 2013, 27, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged structure in medicinal chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Jandial, D.D.; Blair, C.A.; Zhang, S.; Krill, L.S.; Zhang, Y.B.; Zi, X. Molecular targeted approaches to cancer therapy and prevention using chalcones. Curr. Cancer Drug Targets 2014, 14, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kumar, R.; Kodwani, R.; Kapoor, S.; Khare, A.; Bansal, R.; Khurana, S.; Singh, S.; Thomas, J.; Roy, B.; et al. A review on mechanisms of anti tumor activity of chalcones. Anti-Cancer Agents Med. Chem. 2016, 16, 200–211. [Google Scholar] [CrossRef]
- Sahu, N.K.; Balbhadra, S.S.; Choudhary, J.; Kohli, D.V. Exploring pharmacological significance of chalcone scaffold: A review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, C.; Moorthy, N.S.; Ramasamy, S.; Vanam, U.; Manivannan, E.; Karunagaran, D.; Trivedi, P. Advances in chalcones with anticancer activities. Recent Pat. Anti-Cancer Drug Discov. 2015, 10, 97–115. [Google Scholar] [CrossRef]
- Ritter, M.; Martins, R.M.; Dias, D.; Pereira, C.M.P. Recent Advances on the synthesis of chalcones with antimicrobial activities: A brief review. Lett. Org. Chem. 2014, 11, 498–508. [Google Scholar] [CrossRef]
- Budhiraja, A.; Kadian, K.; Kaur, M.; Aggarwal, V.; Garg, A.; Sapra, S.; Nepali, K.; Suri, O.P.; Dhar, K.L. Synthesis and biological evaluation of naphthalene, furan and pyrrole based chalcones as cytotoxic and antimicrobial agents. Med. Chem. Res. 2012, 21, 2133–2140. [Google Scholar] [CrossRef]
- Nowakowska, Z. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem. 2007, 42, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Dong, H.-H.; Zhao, F.; Wang, J.; Yan, F.; Jiang, Y.-Y.; Jin, Y.-S. The synthesis and synergistic antifungal effects of chalcones against drug resistant Candida albicans. Bioorg. Med. Chem. Lett. 2016, 26, 3098–3102. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Rao, K.B.; Kushwaha, P.; Modukuri, R.K.; Singh, P.; Soni, I.; Shukla, P.K.; Chopra, S.; Pasupuleti, M. Novel chalcone-Thiazole hybrids as potent inhibitors of drug resistant Staphylococcus aureus. ACS Med. Chem. Lett. 2015, 6, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, A.; Altıntop, M.D.; Cantürk, Z.; Kaplancıklı, Z.A. Synthesis and in vitro evaluation of furan-based chalcone derivatives as antimicrobial agents. Lett. Drug Des. Discov. 2015, 12, 607–611. [Google Scholar] [CrossRef]
- López, S.N.; Castelli, M.V.; Zacchino, S.A.; Domínguez, J.N.; Lobo, G.; Charris-Charris, J.; Cortés, J.C.; Ribas, J.C.; Devia, C.; Rodríguez, A.M.; et al. In vitro antifungal evaluation and structure-activity relationships of a new series of chalcone derivatives and synthetic analogues, with inhibitory properties against polymers of the fungal cell wall. Bioorg. Med. Chem. 2001, 9, 1999–2013. [Google Scholar] [CrossRef]
- Crouch, S.P.M.; Kozlowski, R.; Slater, K.J.; Fletcher, J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. [Google Scholar] [CrossRef]
- Swenson, J.M.; Clark, N.C.; Daniel, F.S.; Ferraro, M.J.; Doern, G.; Hindler, J.; Jorgensen, J.H.; Pfaller, M.A.; Reller, L.B.; Weinstein, M.P.; et al. Molecular characterization and multilaboratory evaluation of Enterococcus faecalis ATCC 51299 for quality control of screening tests for vancomycin and high-level aminoglycoside resistance in Enterococci. J. Clin. Microbiol. 1995, 33, 3019–3021. [Google Scholar] [PubMed]
- Leitão, J.M.; Esteves da Silva, J. Firefly luciferase inhibition. J. Photochem. Photobiol. B. 2010, 101, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.W.; Chen, H.; Yu, L.F.; Lv, F.; Xing, Y.J.; Liu, T.; Xie, J.; Tang, J.; Yi, Z.; Yang, F. Novel 3,4-seco bile acid diamides as selective anticancer proliferation and migration agents. Eur. J. Med. Chem. 2016, 122, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.Y.; Dong, F.Q.; Du, X.L.; Zhou, Z.K.; Huo, H.R.; Wang, W.H.; Zhan, H.D.; Dai, Y.F.; Meng, J.; Sui, Y.P.; et al. Antitumor activities of biscoumarin and dihydropyran derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 3876–3880. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, A.; Altintop, M.D.; Kaplancıklı, Z.A.; Turan-Zitouni, G.; Karaca, H.; Tunalı, Y. Synthesis and biological evaluation of pyrazoline derivatives bearing an indole moiety as new antimicrobial agents. Arch. Pharm. Chem. Life Sci. 2013, 346, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, Approved Standard-Ninth Edition; CLSI document M07-A9; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012; pp. 16–20. ISBN 1-56238-784-7. [Google Scholar]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts, Approved Standard-Second Edition; CLSI document M27-A2; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2002; pp. 4–9. ISBN 1-56238-469-4. [Google Scholar]
- Kapoor, R.; Yadav, J. Development of a rapid ATP luminescence assay for biocidal susceptibility testing of rapidly growing Mycobecteria. J. Clin. Microbiol. 2010, 48, 3725–3728. [Google Scholar] [CrossRef] [PubMed]
- Kilic, A.; Dogan, E.; Kaya, S.; Oren, S.; Tok, D.; Ardic, N.; Baysallar, M. Rapid identification of Klebsiella pneumoniae by matrix-assisted laser desorption/Ionization—Time of flight mass spectrometry and detection of monopenem resistance by flow cytometry. J. Clin. Lab. Anal. 2016, 30, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Saint-Ruf, C.; Crussard, S.; Franceschi, C.; Orenga, S.; Quattara, J.; Ramjeet, M.; Surre, J.; Matic, I. Antibiotic susceptibility testing of the gram negative bacteria based on flow cytometry. Front. Microbiol. 2016, 7, 1121. [Google Scholar] [CrossRef] [PubMed]
- Altıntop, M.D.; Atlı, Ö.; Ilgın, S.; Demirel, R.; Özdemir, A.; Kaplancıklı, Z.A. Synthesis and biological evaluation of new naphthalene substituted thiosemicarbazone derivatives as potent antifungal and anticancer agents. Eur. J. Med. Chem. 2016, 108, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Altıntop, M.D.; Özdemir, A.; Atlı, Ö.; Cantürk, Z.; Baysal, M.; Kaplancıklı, Z.A. Synthesis and evaluation of new thiazole derivatives as potential antimicrobial agents. Lett. Drug Des. Discov. 2016, 13, 903–911. [Google Scholar] [CrossRef]
- Chandrasekaran, C.V.; Sundarajan, K.; Gupta, A.; Srikanth, H.S.; Edwin, J.; Agarwal, A. Evaluation of the genotoxic potential of standardized extract of Glycyrrhiza glabra (GutGard™). Regul. Toxicol. Pharmacol. 2011, 61, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Flückiger-Isler, S.; Kamber, M. Direct comparison of the Ames microplate format (MPF) test in liquid medium with the standard Ames pre-incubation assay on agar plates by use of equivocal to weakly positive test compounds. Mutat. Res. 2012, 747, 36–45. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1–10 are available from the authors. |








{kind=link}
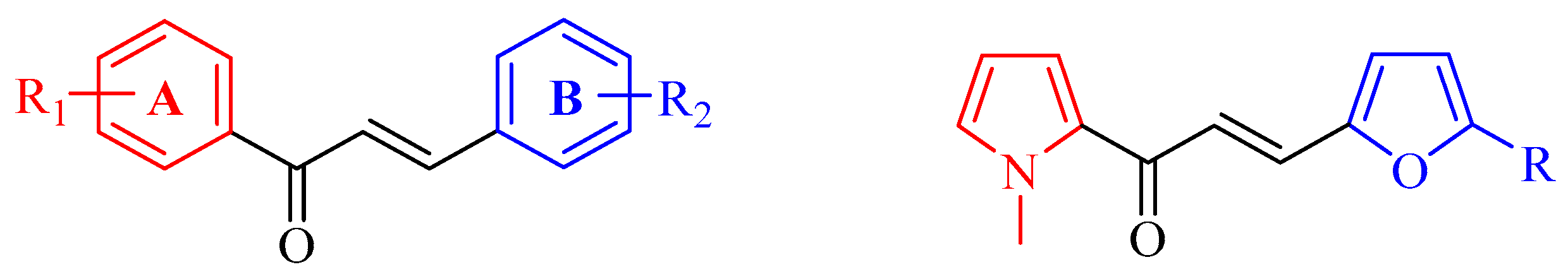{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MIC Values in μg/mL | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bacteria * | Yeasts ** | ||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| 1 | 200 | 200 | 100 | 100 | 200 | 100 | 200 | 100 | 200 |
| 2 | 200 | 100 | 100 | 100 | 200 | 100 | 100 | 100 | 100 |
| 3 | 100 | 200 | 100 | 100 | 200 | 50 | 100 | 50 | 100 |
| 4 | 100 | 200 | 200 | 100 | 100 | 200 | 100 | 50 | 200 |
| 5 | 400 | 200 | 200 | 400 | 200 | 200 | 200 | 100 | 200 |
| 6 | 200 | 200 | 200 | 100 | 200 | 200 | 200 | 100 | 200 |
| 7 | 400 | 200 | 100 | 400 | 100 | 50 | 100 | 100 | 25 |
| 8 | 400 | 400 | 200 | 100 | 200 | 200 | 100 | 100 | 50 |
| 9 | 200 | 400 | 200 | 100 | 200 | 200 | 100 | 50 | 25 |
| 10 | 200 | 100 | 400 | 100 | 200 | 100 | 200 | 100 | 200 |
| Control | 25 | 100 | 50 | 25 | 50 | 50 | 100 | 50 | 50 |
| Live Cell % | Dead Cell % | |
|---|---|---|
| Antibiotic-free control | 98.9 | 0.1 |
| Ketoconazole-treated cell | 81.2 | 18.4 |
| Compound 7-treated cell | 79.6 | 20.0 |
| Compound 9-treated cell | 83.9 | 15.9 |
| Compound | IC50 (μg/mL) | |||
|---|---|---|---|---|
| HepG2 Cell Line | C6 Cell Line | A549 Cell Line | NIH/3T3 Cell Line | |
| 1 | 322 | 84 | 0.3 | 109 |
| 2 | 277 | 258 | 322 | >322 |
| 3 | 27 | >322 | >322 | >322 |
| 4 | 100 | >357 | >357 | 36 |
| 5 | 31 | >311 | 311 | 45 |
| 6 | 98 | 103 | 98 | 38 |
| 7 | 23 | 311 | 98 | 40 |
| 8 | 106 | 90 | 225 | 277 |
| 9 | 101 | 109 | >346 | 109 |
| 10 | 100 | 100 | 346 | 148 |
| Cisplatin | 38 | 46 | 19 | >300 |
| Compound | SI Values | ||
|---|---|---|---|
| HepG2 Cell Line | C6 Cell Line | A549 Cell Line | |
| 1 | 0.339 | 1.298 | 363.33 |
| 2 | 1.162 | 1.248 | 1 |
| 3 | 11.926 | 1 | 1 |
| 4 | 0.360 | 0.100 | 0.100 |
| 5 | 1.450 | 0.145 | 0.145 |
| 6 | 0.388 | 0.369 | 0.388 |
| 7 | 1.739 | 0.129 | 0.408 |
| 8 | 2.613 | 3.077 | 1.231 |
| 9 | 1.079 | 1 | 0.315 |
| 10 | 1.480 | 1.480 | 0.428 |
| Cisplatin | 7.895 | 6.520 | 15.789 |
| Compound | Concentrations (mg/mL) | REVERTANTS Fold Increase (Over Baseline) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | TA 98 | Baseline | TA 100 | ||||||
| S9+ | S9− | S9+ | S9− | S9+ | S9− | S9+ | S9− | ||
| 7 | 0.16 | 3.95 | 3.04 | 0.68 | 0.66 | 5.91 | 3.00 | 0.56 | 0.44 |
| 0.31 | 0.76 | 0.33 | 0.68 | 0.56 | |||||
| 0.63 | 1.01 | 0.44 | 0.79 | 0.67 | |||||
| 1.25 | 0.68 | 0.66 | 0.62 | 0.33 | |||||
| 2.5 | 0.51 | 0.33 | 0.90 | 0.33 | |||||
| 5 | 0.25 | 0.00 * | 0.79 | 0.44 | |||||
| 9 | 0.16 | 5.92 | 5.60 | 0.51 | 0.89 | 5.92 | 5.60 | 0.51 | 0.48 |
| 0.31 | 0.79 | 0.83 | 0.73 | 0.54 | |||||
| 0.63 | 0.34 * | 0.89 | 0.68 | 0.54 | |||||
| 1.25 | 0.39 | 1.07 | 0.68 | 0.54 | |||||
| 2.5 | 0.79 | 0.24 * | 0.79 | 0.30 * | |||||
| 5 | 0.51 | 0.54 | 0.90 | 0.12 * | |||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Özdemir, A.; Altıntop, M.D.; Sever, B.; Gençer, H.K.; Kapkaç, H.A.; Atlı, Ö.; Baysal, M. A New Series of Pyrrole-Based Chalcones: Synthesis and Evaluation of Antimicrobial Activity, Cytotoxicity, and Genotoxicity. Molecules 2017, 22, 2112. https://doi.org/10.3390/molecules22122112
Özdemir A, Altıntop MD, Sever B, Gençer HK, Kapkaç HA, Atlı Ö, Baysal M. A New Series of Pyrrole-Based Chalcones: Synthesis and Evaluation of Antimicrobial Activity, Cytotoxicity, and Genotoxicity. Molecules. 2017; 22(12):2112. https://doi.org/10.3390/molecules22122112
Chicago/Turabian StyleÖzdemir, Ahmet, Mehlika Dilek Altıntop, Belgin Sever, Hülya Karaca Gençer, Handan Açelya Kapkaç, Özlem Atlı, and Merve Baysal. 2017. "A New Series of Pyrrole-Based Chalcones: Synthesis and Evaluation of Antimicrobial Activity, Cytotoxicity, and Genotoxicity" Molecules 22, no. 12: 2112. https://doi.org/10.3390/molecules22122112
APA StyleÖzdemir, A., Altıntop, M. D., Sever, B., Gençer, H. K., Kapkaç, H. A., Atlı, Ö., & Baysal, M. (2017). A New Series of Pyrrole-Based Chalcones: Synthesis and Evaluation of Antimicrobial Activity, Cytotoxicity, and Genotoxicity. Molecules, 22(12), 2112. https://doi.org/10.3390/molecules22122112