Novel Chiral Bis-Phosphoramides as Organocatalysts for Tetrachlorosilane-Mediated Reactions



Abstract
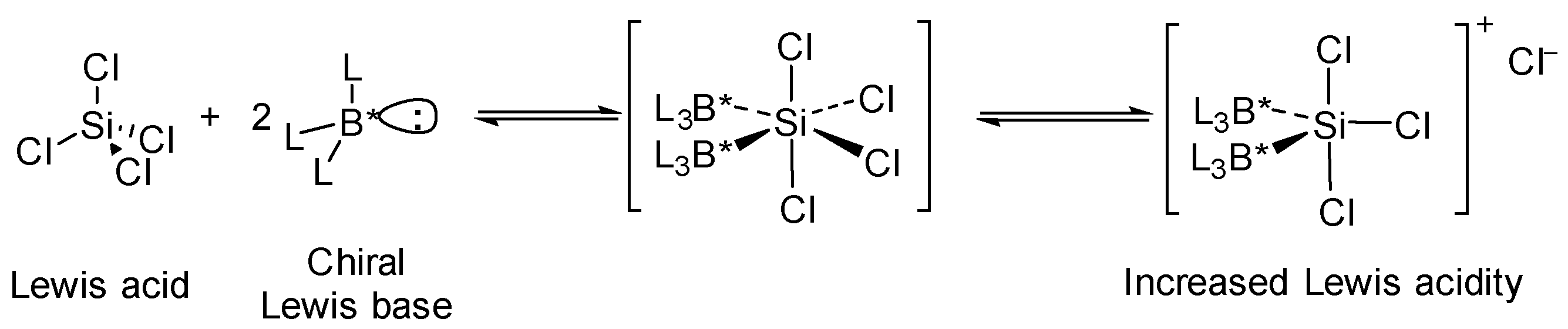
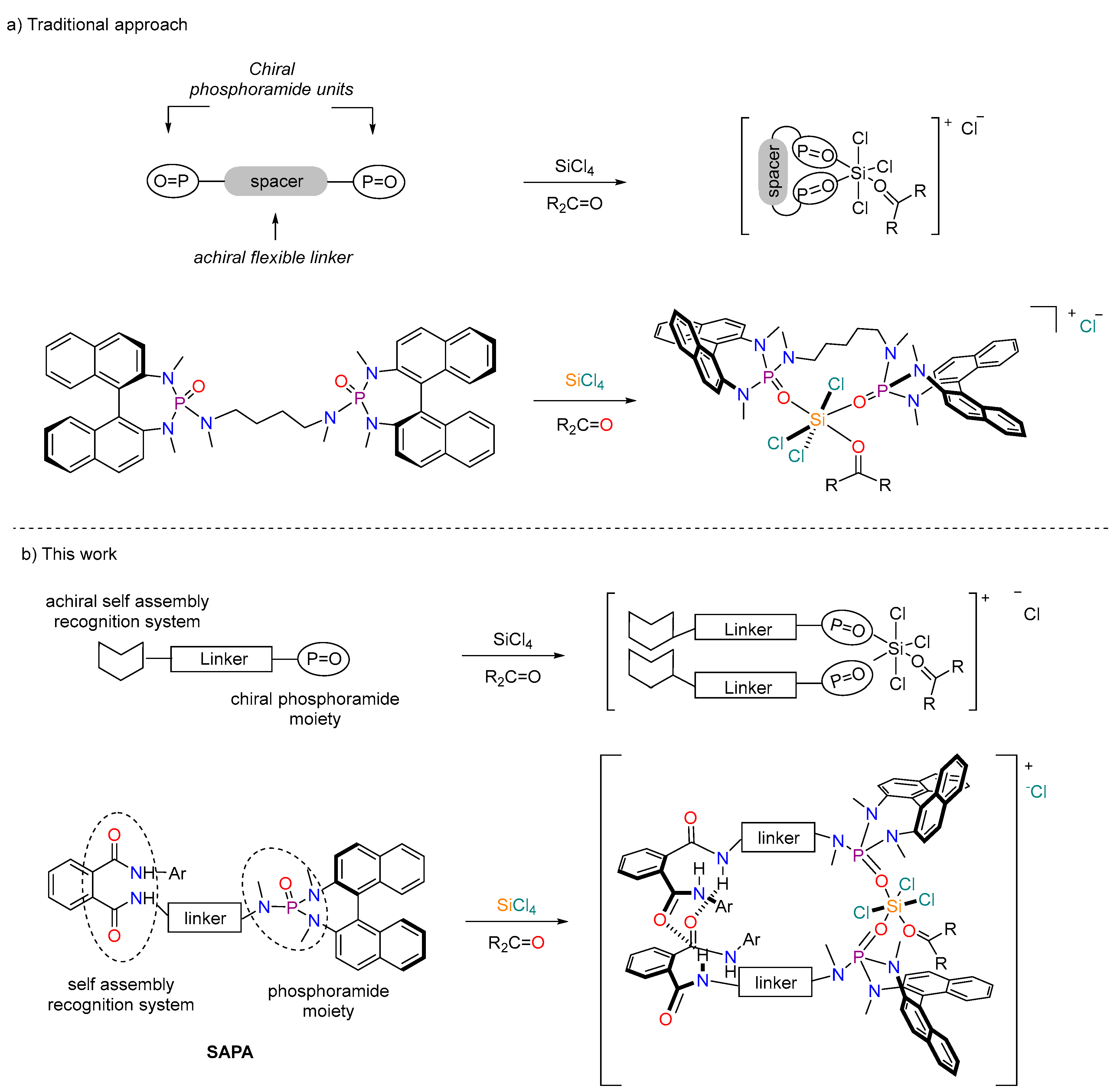
:1. Introduction
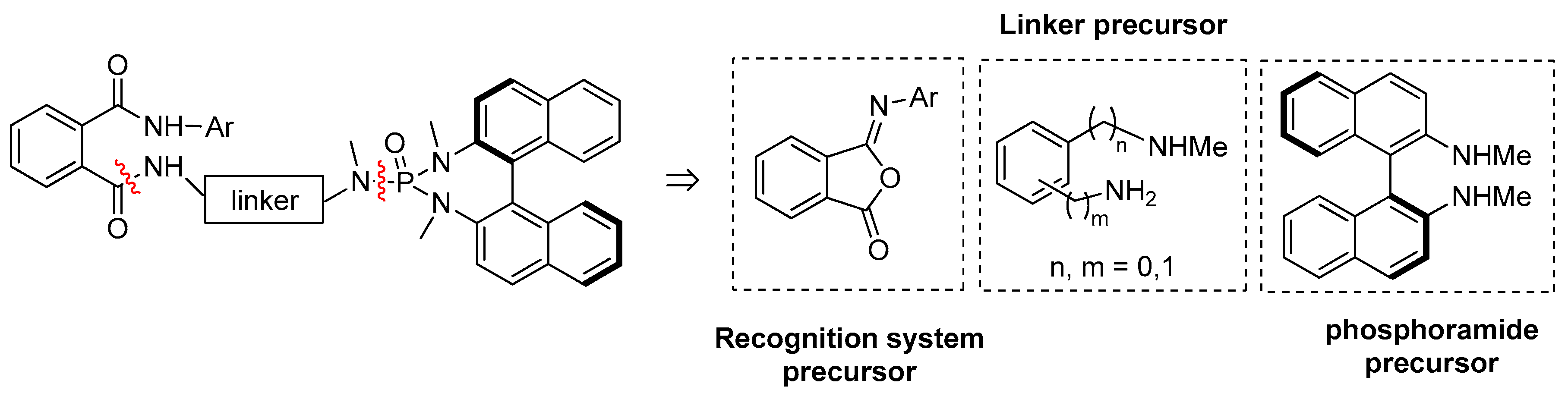
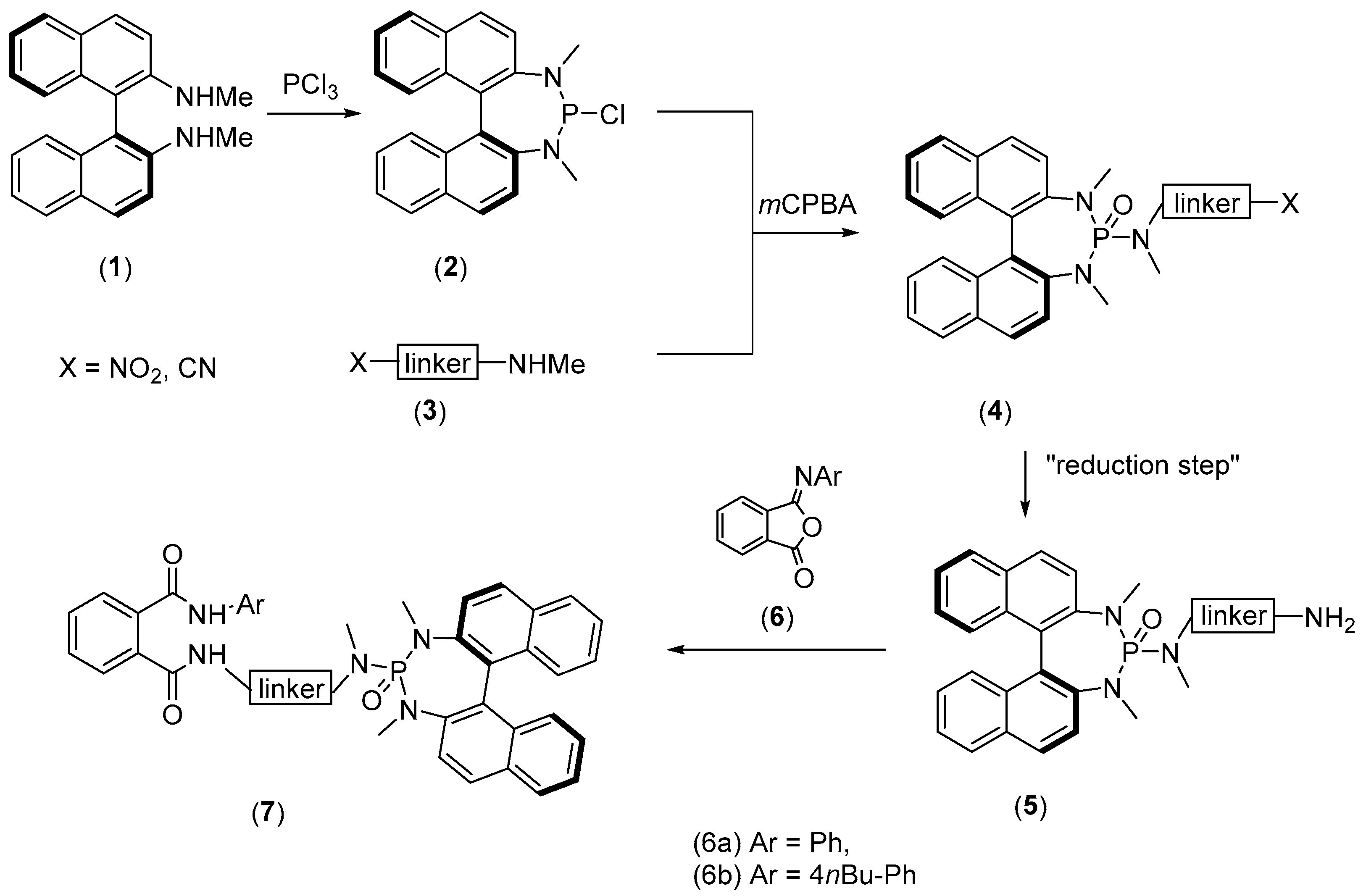
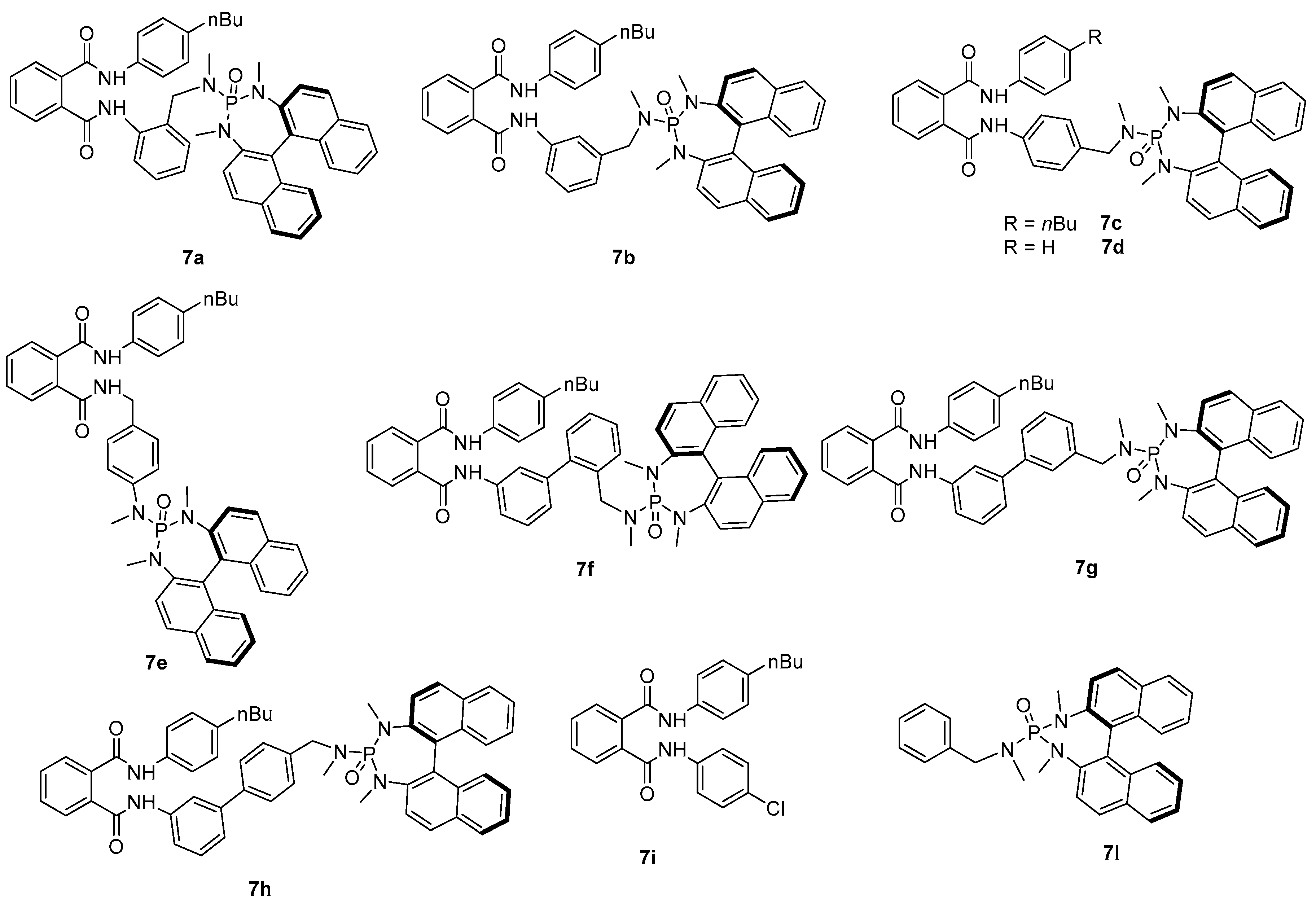
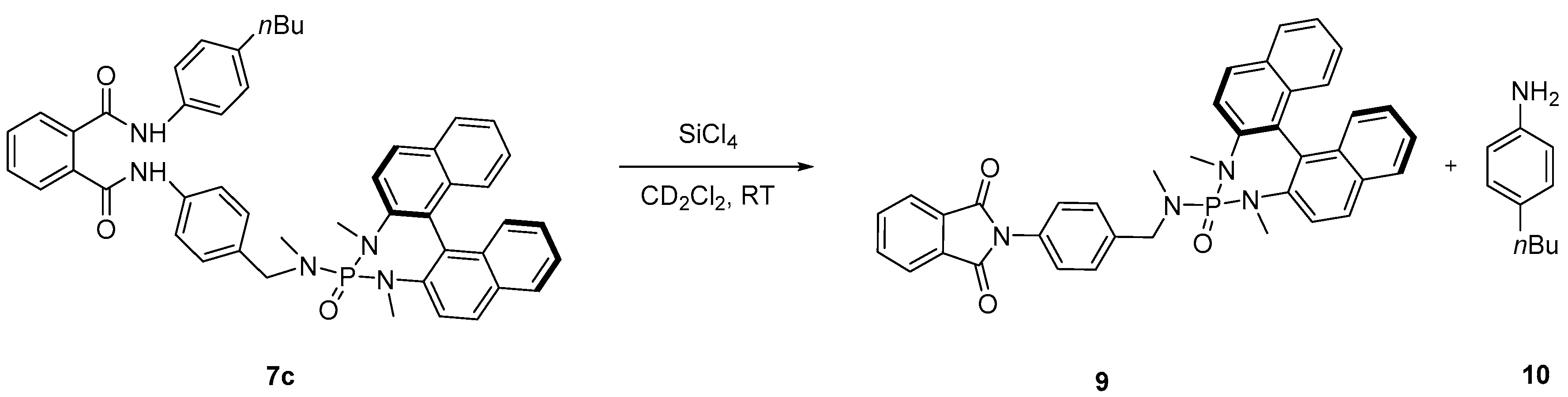
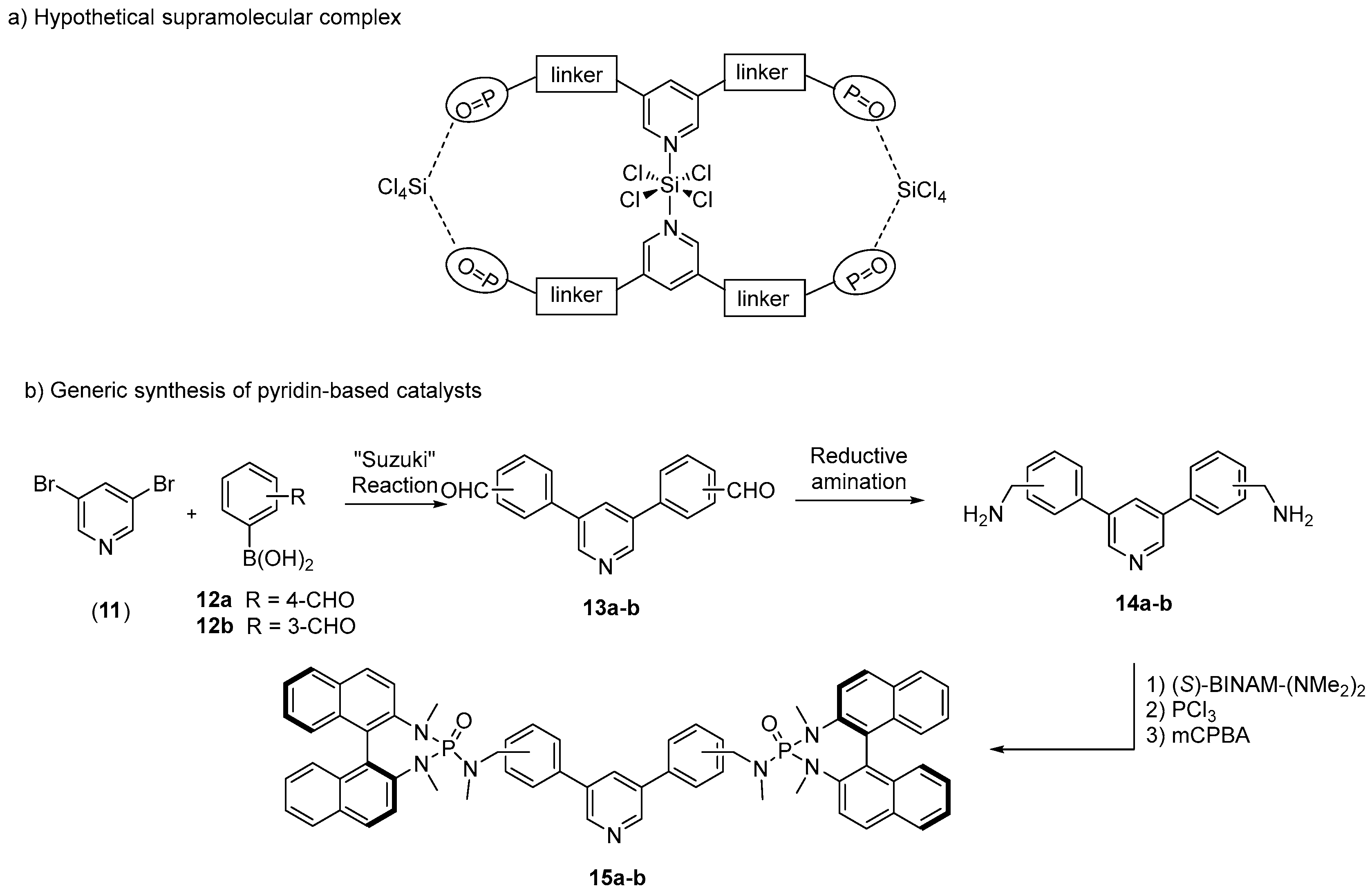
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure for the Synthesis of Nitro-Phosphoroamides (4)
3.2. General Procedure for the Synthesis of SAPAs Catalyst (7)
3.3. General Procedure for Allylation of Benzaldehyde
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Denmark, S.E.; Beutner, G.L. Lewis base catalysis in organic synthesis. Angew. Chem. Int. Ed. 2008, 47, 1560–1638. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Denmark, S.E. Chapter 21: Lewis base-catalyzed, lewis acid-mediated reactions (n →σ*). In Lewis Base Catalysis in Organic Synthesis; Vedejs, E., Denmark, S.E., Eds.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2016; Volume 2, pp. 1039–1076. [Google Scholar]
- Benaglia, M.; Rossi, S. Chiral phosphine oxides in present-day organocatalysis. Org. Biomol. Chem. 2010, 8, 3824–3830. [Google Scholar] [CrossRef] [PubMed]
- Benaglia, M.; Guizzetti, S.; Pignataro, L. Stereoselective reactions involving hypervalent silicate complexes. Coord. Chem. Rev. 2008, 252, 492–512. [Google Scholar] [CrossRef]
- Benaglia, M.; Guizzetti, S.; Rossi, S. Silicate-mediated stereoselective reactions catalyzed by chiral lewis bases. In Catalytic Methods in Asymmetric Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 579–624. [Google Scholar]
- Rossi, S.; Benaglia, M.; Genoni, A. Organic reactions mediated by tetrachlorosilane. Tetrahedron 2014, 70, 2065–2080. [Google Scholar] [CrossRef]
- Denmark, S.E.; Fu, J.; Lawler, M.J. Chiral phosphoramide-catalyzed enantioselective addition of allylic trichlorosilanes to aldehydes. Preparative studies with bidentate phosphorus-based amides. J. Org. Chem. 2006, 71, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Breit, B. Supramolecular approaches to generate libraries of chelating bidentate ligands for homogeneous catalysis. Angew. Chem. Int. Ed. 2005, 44, 6816–6825. [Google Scholar] [CrossRef] [PubMed]
- Breit, B.; Seiche, W. Hydrogen bonding as a construction element for bidentate donor ligands in homogeneous catalysis: Regioselective hydroformylation of terminal alkenes. J. Am. Chem. Soc. 2003, 125, 6608–6609. [Google Scholar] [CrossRef] [PubMed]
- Seiche, W.; Schuschkowski, A.; Breit, B. Bidentate Ligands by Self-Assembly through Hydrogen Bonding: A General Room Temperature/Ambient Pressure Regioselective Hydroformylation of Terminal Alkenes. Adv. Synth. Catal. 2005, 347, 1488–1494. [Google Scholar] [CrossRef]
- Patureau, F.W.; Kuil, M.; Sandee, A.J.; Reek, J.N. METAMORPhos: Adaptive supramolecular ligands and their mechanistic consequences for asymmetric hydrogenation. Angew. Chem. Int. Ed. 2008, 47, 3180–3183. [Google Scholar] [CrossRef] [PubMed]
- Duckmanton, P.A.; Blake, A.J.; Love, J.B. Palladium and rhodium ureaphosphine complexes: Exploring structural and catalytic consequences of anion binding. Inorg. Chem. 2005, 44, 7708–7710. [Google Scholar] [CrossRef] [PubMed]
- Knight, L.K.; Freixa, Z.; van Leeuwen, P.W.N.M.; Reek, J.N.H. Supramoleculartrans-Coordinating Phosphine Ligands. Organometallics 2006, 25, 954–960. [Google Scholar] [CrossRef]
- Sandee, A.J.; van der Burg, A.M.; Reek, J.N. UREAphos: Supramolecular bidentate ligands for asymmetric hydrogenation. Chem. Commun. 2007, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sandoval, C.A.; Yamaguchi, Y.; Zhang, X.; Wang, Z.; Kato, K.; Ding, K. Hydrogen bonding makes a difference in the rhodium-catalyzed enantioselective hydrogenation using monodentate phosphoramidites. J. Am. Chem. Soc. 2006, 128, 14212–14213. [Google Scholar] [CrossRef] [PubMed]
- Laungani, A.C.; Breit, B. Supramolecular PhanePhos-analogous ligands through hydrogen-bonding for asymmetric hydrogenation. Chem. Commun. 2008, 844–846. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, L.; Carboni, S.; Civera, M.; Colombo, R.; Piarulli, U.; Gennari, C. PhthalaPhos: Chiral supramolecular ligands for enantioselective rhodium-catalyzed hydrogenation reactions. Angew. Chem. Int. Ed. 2010, 49, 6633–6637. [Google Scholar] [CrossRef] [PubMed]
- Pignataro, L.; Boghi, M.; Civera, M.; Carboni, S.; Piarulli, U.; Gennari, C. Rhodium-catalyzed asymmetric hydrogenation of olefins with PhthalaPhos, a new class of chiral supramolecular ligands. Chemistry 2012, 18, 1383–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignataro, L.; Bovio, C.; Civera, M.; Piarulli, U.; Gennari, C. A library approach to the development of BenzaPhos: Highly efficient chiral supramolecular ligands for asymmetric hydrogenation. Chemistry 2012, 18, 10368–10381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, M.J.; van Leeuwen, P.W.; Reek, J.N. New directions in supramolecular transition metal catalysis. Org. Biomol. Chem. 2005, 3, 2371–2383. [Google Scholar] [CrossRef] [PubMed]
- Sandee, A.J.; Reek, J.N. Bidentate ligands by supramolecular chemistry—The future for catalysis? Dalton Trans. 2006, 28, 28–3385. [Google Scholar] [CrossRef] [PubMed]
- Goudriaan, P.E.; van Leeuwen, P.W.N.M.; Birkholz, M.N.; Reek, J.N.H. Libraries of Bidentate Phosphorus Ligands; Synthesis Strategies and Application in Catalysis. Eur. J. Inorg. Chem. 2008. [Google Scholar] [CrossRef]
- Reetz, M.T. Combinatorial transition-metal catalysis: Mixing monodentate ligands to control enantio-, diastereo-, and regioselectivity. Angew. Chem. Int. Ed. 2008, 47, 2556–2588. [Google Scholar] [CrossRef] [PubMed]
- Carboni, S.; Gennari, C.; Pignataro, L.; Piarulli, U. Supramolecular ligand-ligand and ligand-substrate interactions for highly selective transition metal catalysis. Dalton Trans. 2011, 40, 4355–4373. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P.W. Supramolecular catalysis. Part 1: Non-covalent interactions as a tool for building and modifying homogeneous catalysts. Chem. Soc. Rev. 2014, 43, 1660–1733. [Google Scholar] [CrossRef] [PubMed]
- Ohmatsu, K.; Ooi, T. Design of supramolecular chiral ligands for asymmetric metal catalysis. Tetrahedron Lett. 2015, 56, 2043–2048. [Google Scholar] [CrossRef]
- Vaquero, M.; Rovira, L.; Vidal-Ferran, A. Supramolecularly fine-regulated enantioselective catalysts. Chem. Commun. 2016, 52, 11038–11051. [Google Scholar] [CrossRef] [PubMed]
- Anebouselvy, K.; Shruthi, K.S.; Ramachary, D.B. Asymmetric Supramolecular Organocatalysis: A Complementary Upgrade to Organocatalysis. Eur. J. Org. Chem. 2017. [Google Scholar] [CrossRef]
- Denmark, S.E.; Wilson, T. Construction of Quaternary Stereogenic Carbon Centers by the Lewis Base Catalyzed Conjugate Addition of Silyl Ketene Imines to α,β-Unsaturated Aldehydes and Ketones. Synlett 2010, 11, 1723–1728. [Google Scholar] [CrossRef]
- Denmark, S.E.; Barsanti, P.A.; Beutner, G.L.; Wilson, T.W. Enantioselective Ring Opening of Epoxides with Silicon Tetrachloride in the Presence of a Chiral Lewis Base: Mechanism Studies. Adv. Synth. Catal. 2007, 349, 567–582. [Google Scholar] [CrossRef]
- Denmark, S.E.; Rossi, S.; Webster, M.P.; Wang, H. Catalytic, enantioselective sulfenylation of ketone-derived enoxysilanes. J. Am. Chem. Soc. 2014, 136, 13016–13028. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Wynn, T. Lewis Base Activation of Lewis Acids: Catalytic Enantioselective Allylation and Propargylation of Aldehydes. J. Am. Chem. Soc. 2001, 123, 6199–6200. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Eklov, B.M. Neutral and cationic phosphoramide adducts of silicon tetrachloride: Synthesis and characterization of their solution and solid-state structures. Chemistry 2008, 14, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Beutner, G.L.; Wynn, T.; Eastgate, M.D. Lewis base activation of Lewis acids: Catalytic, enantioselective addition of silyl ketene acetals to aldehydes. J. Am. Chem. Soc. 2005, 127, 3774–3789. [Google Scholar] [CrossRef] [PubMed]
- Fester, G.W.; Eckstein, J.; Gerlach, D.; Wagler, J.; Brendler, E.; Kroke, E. Reactions of hydridochlorosilanes with 2,2′-bipyridine and 1,10-phenanthroline: Complexation versus dismutation and metal-catalyst-free 1,4-hydrosilylation. Inorg. Chem. 2010, 49, 2667–2673. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, H.; Hensen, K.; Stumpf, T. The First SiH22+ Complex, Dihydridotetrakis(3-picoline)silicon Dichloride–Tetrakis(chloroform), [H2Si(3pic)4]Cl2·4 CHCl3: Formation, Chemical Equilibria, and Structural Investigation by NMR Spectroscopy and Single-Crystal X-ray Diffraction. Eur. J. Inorg. Chem. 1996, 129, 765–771. [Google Scholar] [CrossRef]
- Bain, V.A.; Killean, R.C.G.; Webster, M. The crystal and molecular structure of tetrafluorobispyridinesilicon(IV). Acta Crystallogr. B 1969, 25, 156–159. [Google Scholar] [CrossRef]
- Reynolds, J.E. LXV.—Silicon researches. Part XII. The action of silicochloroform on potassium pyrrole. J. Chem. Soc. Trans. 1909, 95, 508–512. [Google Scholar] [CrossRef]
- Harden, A. IV.—On the action of silicon tetrachloride on the aromatic amido-compounds. J. Chem. Soc. Trans. 1887, 51, 40–47. [Google Scholar] [CrossRef]
- Beattie, I.R.; Leigh, G.J. The interaction of certain chloro compounds of the elements of group IV with tertiary amines. J. Inorg. Nucl. Chem. 1961, 23, 55–62. [Google Scholar] [CrossRef]
- Piper, T.S.; Rochow, E.G. Addition Compounds of Silicon Tetrahalides. J. Am. Chem. Soc. 1954, 76, 4318–4320. [Google Scholar] [CrossRef]
- Feshin, V.P.; Feshina, E.V.; Zhizhina, L.I. ab initio calculations of complexes of group IVA tetrachlorides: I. Dynamics of complex formation of SiCl4 with pyridine. Russ. J. Gen. Chem. 2006, 76, 1571–1575. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Conc [M] | Yield (%) | ee (%) 1 |
|---|---|---|---|---|
| 1 | 7i | 0.5 | / | / |
| 2 | 7a | 0.5 | 83 | 13 (S) |
| 3 | 7b | 0.5 | 58 | 60 (S) |
| 4 | 7c | 0.5 | 73 | 70 (S) |
| 5 | 7c | 0.05 | 28 | 73 (S) |
| 6 | 7d | 0.5 | 78 | 68 (S) |
| 7 | 7e | 0.5 | 90 | Rac |
| 8 | 7f | 0.5 | 45 | 44 (S) |
| 9 | 7g | 0.5 | 80 | 34 (S) |
| 10 | 7h | 0.5 | 79 | 60 (S) |
| 11 | 7l | 0.5 | 98 | 65 (S) |

| Entry | Catalyst | R | Product | Yield (%) | ee (%) 1 |
|---|---|---|---|---|---|
| 1 | 15a | H | 8a | 71 | 61 (S) |
| 2 | 15b | H | 8a | 30 | 29 (S) |
| 3 | 15a | Cl | 8b | 83 | 62 (S) |
| 4 | 15a | OMe | 8c | 31 | rac |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, S.; Ziliani, M.; Annunziata, R.; Benaglia, M. Novel Chiral Bis-Phosphoramides as Organocatalysts for Tetrachlorosilane-Mediated Reactions. Molecules 2017, 22, 2181. https://doi.org/10.3390/molecules22122181
Rossi S, Ziliani M, Annunziata R, Benaglia M. Novel Chiral Bis-Phosphoramides as Organocatalysts for Tetrachlorosilane-Mediated Reactions. Molecules. 2017; 22(12):2181. https://doi.org/10.3390/molecules22122181
Chicago/Turabian StyleRossi, Sergio, Marco Ziliani, Rita Annunziata, and Maurizio Benaglia. 2017. "Novel Chiral Bis-Phosphoramides as Organocatalysts for Tetrachlorosilane-Mediated Reactions" Molecules 22, no. 12: 2181. https://doi.org/10.3390/molecules22122181
APA StyleRossi, S., Ziliani, M., Annunziata, R., & Benaglia, M. (2017). Novel Chiral Bis-Phosphoramides as Organocatalysts for Tetrachlorosilane-Mediated Reactions. Molecules, 22(12), 2181. https://doi.org/10.3390/molecules22122181