Incorporating Methyl and Phenyl Substituted Stannylene Units into Oligosilanes. The Influence on Optical Absorption Properties



Abstract
:1. Introduction
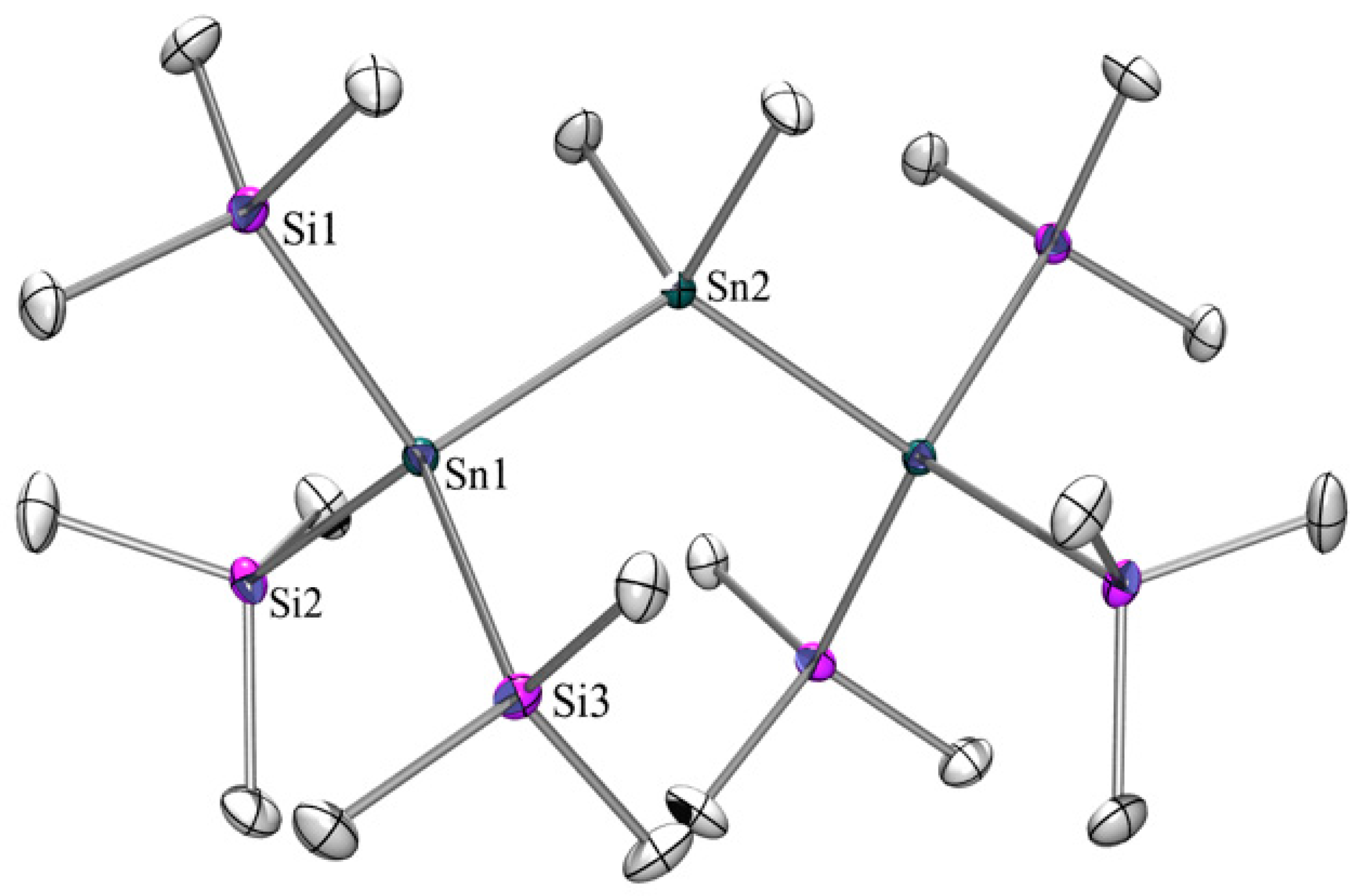
2. Results and Discussion
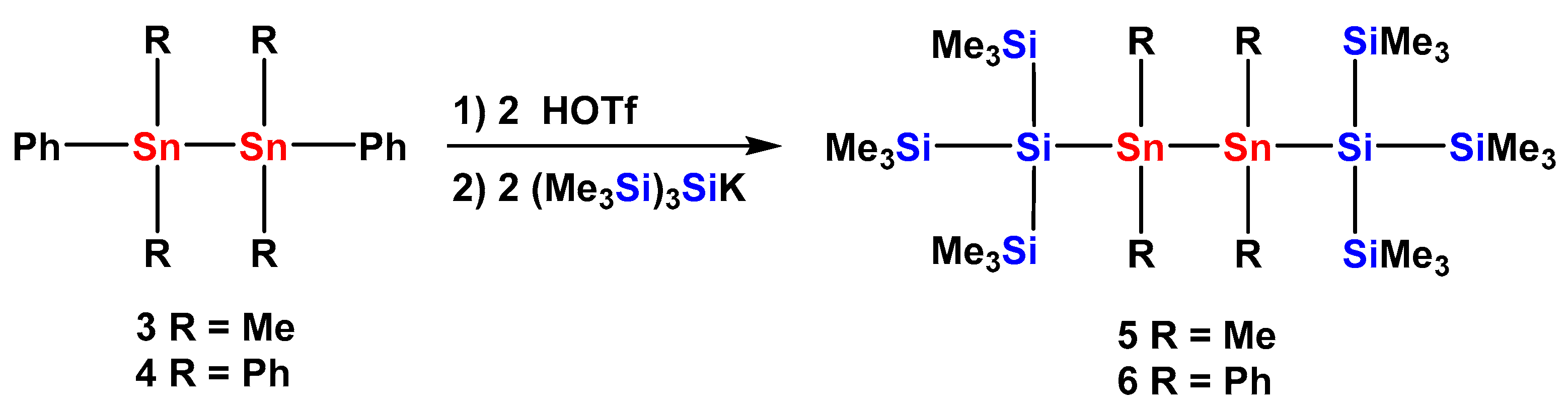
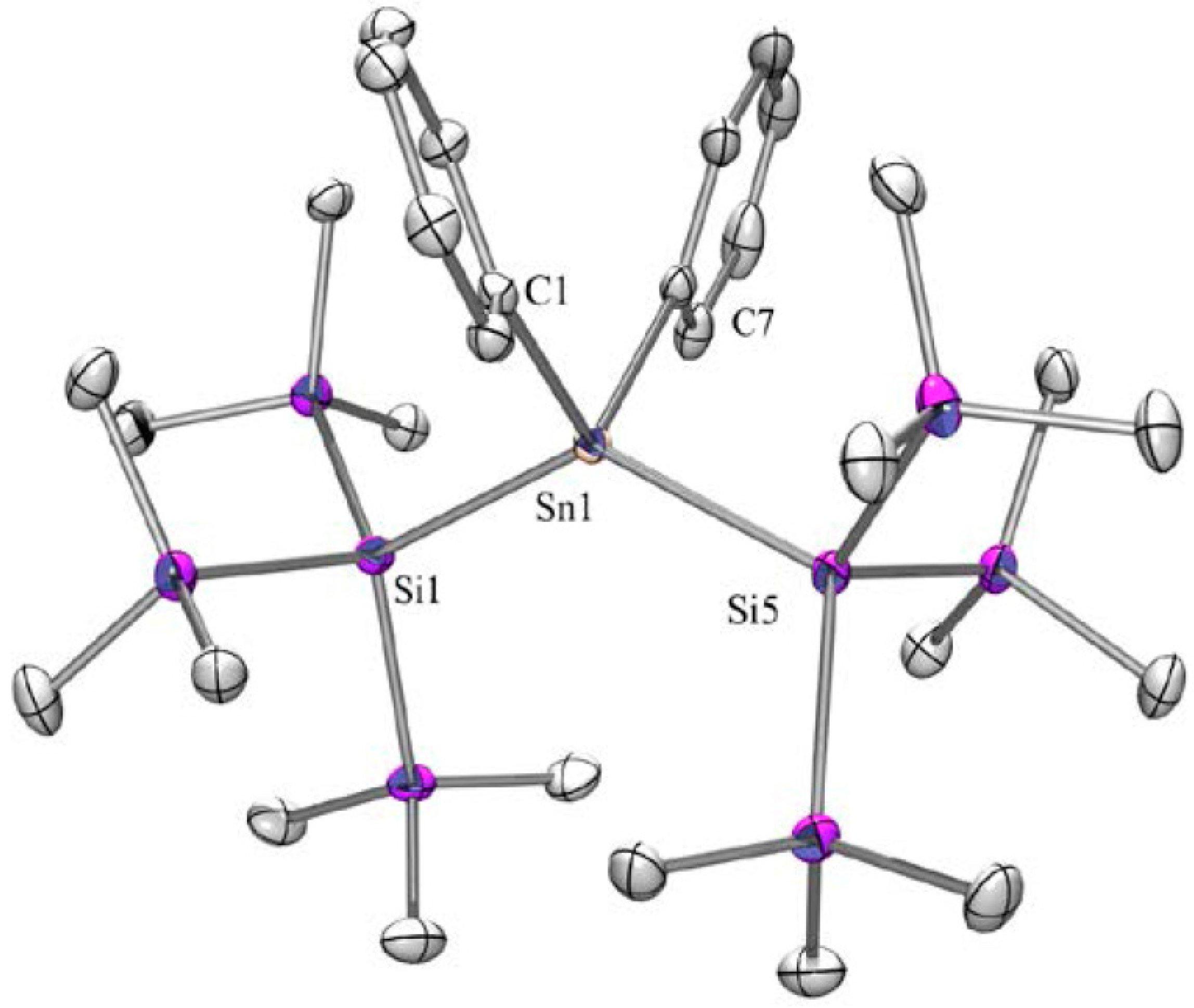
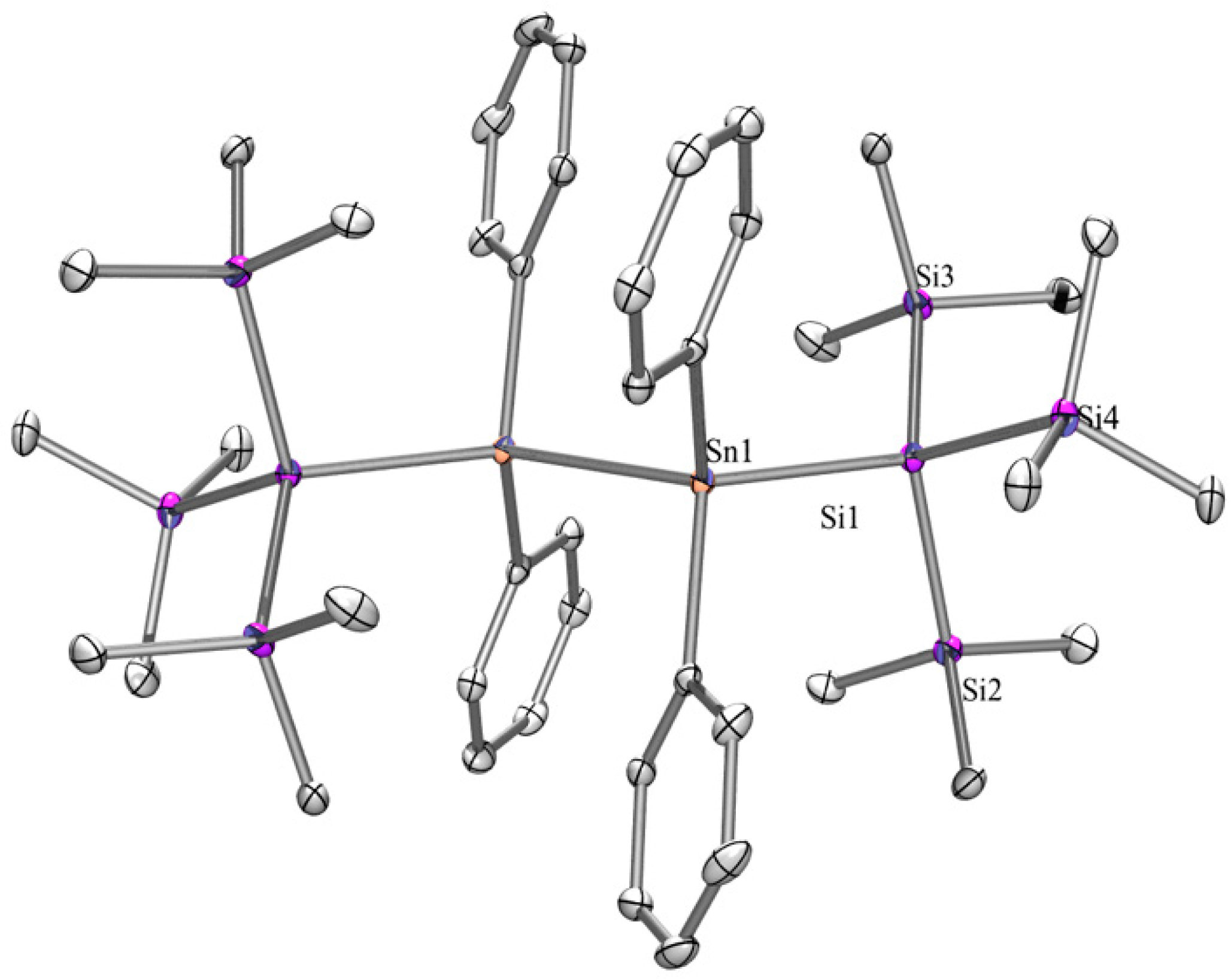
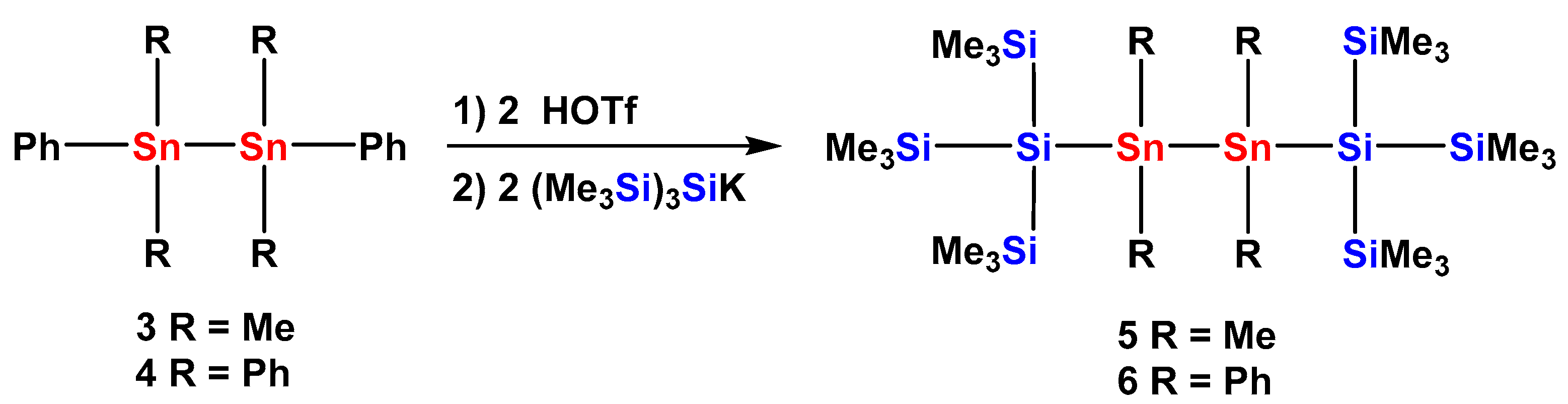
2.1. Synthesis
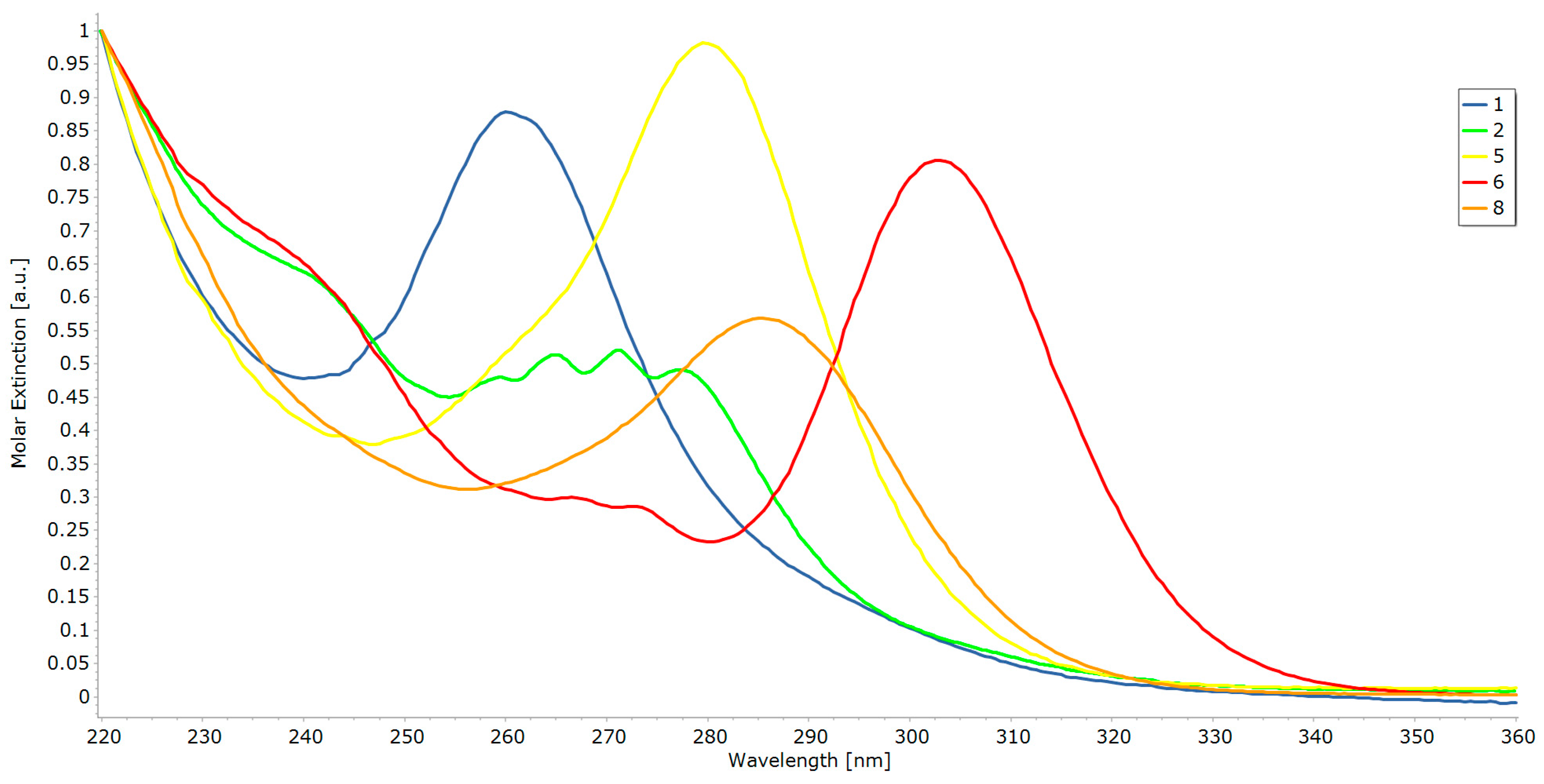
2.2. UV-Vis Spectroscopy
3. Materials and Methods
Experimental Section
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fogarty, H.A.; Casher, D.L.; Imhof, R.; Schepers, T.; Rooklin, D.W.; Michl, J. σ Bonds: Electronic Structure, Photophysics, and Photochemistry of Oligosilanes. Pure Appl. Chem. 2009, 75, 999–1020. [Google Scholar] [CrossRef]
- Miller, R.D.; Michl, J. Polysilane High Polymers. Chem. Rev. 1989, 89, 1359–1410. [Google Scholar] [CrossRef]
- Gilman, H.; Atwell, W.H.; Schwebke, G.L. Ultraviolet Properties of Compounds Containing the Silicon-Silicon Bond. J. Organomet. Chem. 1964, 2, 369–371. [Google Scholar] [CrossRef]
- Sakurai, H.; Kumada, M. The Ultraviolet Spectra of Some Polysilanes. Bull. Chem. Soc. Jpn. 1964, 37, 1894–1895. [Google Scholar] [CrossRef]
- Sakurai, H.; Yamamori, H.; Kumada, M. The Substituent Effect on the Ultraviolet Spectrum of 1,2-Diphenyltetramethyldisilane. Bull. Chem. Soc. Jpn. 1965, 38, 2024. [Google Scholar] [CrossRef]
- Michl, J.; West, R. Conformations of Linear Chains. Systematics and Suggestions for Nomenclature. Acc. Chem. Res. 2000, 33, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Michl, J.; Tamao, K. Recent Experimental and Theoretical Aspects of the Conformational Dependence of UV Absorption of Short Chain Peralkylated Oligosilanes. J. Organomet. Chem. 2003, 685, 9–14. [Google Scholar] [CrossRef]
- Jovanovic, M.; Antic, D.; Rooklin, D.; Bande, A.; Michl, J. Intuitive Understanding of σ Delocalization in Loose and σ Localization in Tight Helical Conformations of an Oligosilane Chain. Chem. Asian J. 2017, 12, 1250–1263. [Google Scholar] [CrossRef] [PubMed]
- Tamao, K.; Tsuji, H.; Terada, M.; Asahara, M.; Yamaguchi, S.; Toshimitsu, A. Conformation Control of Oligosilanes Based on Configurationally Constrained Bicyclic Disilane Units. Angew. Chem. Int. Ed. 2000, 39, 3287–3290. [Google Scholar] [CrossRef]
- Tsuji, H.; Terada, M.; Toshimitsu, A.; Tamao, K. σσ* Transition in anti,cisoid Alternating Oligosilanes: Clear-Cut Evidence for Suppression of Conjugation Effect by a cisoid Turn. J. Am. Chem. Soc. 2003, 125, 7486–7487. [Google Scholar] [CrossRef] [PubMed]
- Mallesha, H.; Tsuji, H.; Tamao, K. UV Absorption and Mass Spectra of n-Alkylsilyl End-Capped Anti, Cisoid-Alternating Oligosilanes up to Docosasilane (Si22). Organometallics 2004, 23, 1639–1642. [Google Scholar] [CrossRef]
- Tsuji, H.; Fukazawa, A.; Yamaguchi, S.; Toshimitsu, A.; Tamao, K. All-anti-Pentasilane: Conformation Control of Oligosilanes Based on the Bis(tetramethylene)-Tethered Trisilane Unit. Organometallics 2004, 23, 3375–3377. [Google Scholar] [CrossRef]
- Fukazawa, A.; Tsuji, H.; Tamao, K. All-anti-Octasilane: Conformation Control of Silicon Chains Using the Bicyclic Trisilane as a Building Block. J. Am. Chem. Soc. 2006, 128, 6800–6801. [Google Scholar] [CrossRef] [PubMed]
- Marschner, C.; Baumgartner, J.; Wallner, A. Structurally and Conformationally Defined Small Methyl Polysilanes. Dalton Trans. 2006, 5667–5674. [Google Scholar] [CrossRef] [PubMed]
- Wallner, A.; Wagner, H.; Baumgartner, J.; Marschner, C.; Rohm, H.W.; Kockerling, M.; Krempner, C. Structure, Conformation, and UV Absorption Behavior of Partially Trimethylsilylated Oligosilane Chains. Organometallics 2008, 27, 5221–5229. [Google Scholar] [CrossRef]
- Wallner, A.; Hlina, J.; Konopa, T.; Wagner, H.; Baumgartner, J.; Marschner, C.; Flörke, U. Cyclic and Bicyclic Methylpolysilanes and Some Oligosilanylene-Bridged Derivatives. Organometallics 2010, 29, 2660–2675. [Google Scholar] [CrossRef]
- Wallner, A.; Hlina, J.; Wagner, H.; Baumgartner, J.; Marschner, C. Conformational Control of Polysilanes: The Use of CH2-Spacers in the Silicon Backbone. Organometallics 2011, 30, 3930–3938. [Google Scholar] [CrossRef] [PubMed]
- Emanuelsson, R.; Löfås, H.; Wallner, A.; Nauroozi, D.; Baumgartner, J.; Marschner, C.; Ahuja, R.; Ott, S.; Grigoriev, A.; Ottosson, H. Configuration- and Conformation-Dependent Electronic-Structure Variations in 1,4-Disubstituted Cyclohexanes Enabled by a Carbon-to-Silicon Exchange. Chem. Eur. J. 2014, 20, 9304–9311. [Google Scholar] [CrossRef] [PubMed]
- Hlina, J.; Stella, F.; Aghazadeh Meshgi, M.; Marschner, C.; Baumgartner, J. σ-Bond Electron Delocalization in Oligosilanes as Function of Substitution Pattern, Chain Length, and Spatial Orientation. Molecules 2016, 21, 1079. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh Meshgi, M.; Baumgartner, J.; Marschner, C. Oligosilanylsilatranes. Organometallics 2015, 34, 3721–3731. [Google Scholar] [CrossRef] [PubMed]
- Hlina, J.; Zitz, R.; Wagner, H.; Stella, F.; Baumgartner, J.; Marschner, C. σ-Bond Electron Delocalization of Branched Oligogermanes and Germanium Containing Oligosilanes. Inorg. Chim. Acta 2014, 422, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Amadoruge, M.L.; Gardinier, J.R.; Weinert, C.S. Substituent Effects in Linear Organogermanium Catenates. Organometallics 2008, 27, 3753–3760. [Google Scholar] [CrossRef]
- Weinert, C.S. Syntheses, Structures and Properties of Linear and Branched Oligogermanes. Dalton Trans. 2009, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Amadoruge, M.L.; Short, E.K.; Moore, C.; Rheingold, A.L.; Weinert, C.S. Structural, Spectral, and Electrochemical Investigations of para-Tolyl-Substituted Oligogermanes. J. Organomet. Chem. 2010, 695, 1813–1823. [Google Scholar] [CrossRef]
- Samanamu, C.R.; Amadoruge, M.L.; Yoder, C.H.; Golen, J.A.; Moore, C.E.; Rheingold, A.L.; Materer, N.F.; Weinert, C.S. Syntheses, Structures, and Electronic Properties of the Branched Oligogermanes (Ph3Ge)3GeH and (Ph3Ge)3GeX (X = Cl, Br, I). Organometallics 2011, 30, 1046–1058. [Google Scholar] [CrossRef]
- Weinert, C.S. Synthetic, Structural, and Physical Aspects of Organo-Oligogermanes. Comments Inorg. Chem. 2011, 32, 55–87. [Google Scholar] [CrossRef]
- Samanamu, C.R.; Amadoruge, M.L.; Schrick, A.C.; Chen, C.; Golen, J.A.; Rheingold, A.L.; Materer, N.F.; Weinert, C.S. Synthetic, Structural, and Physical Investigations of the Large Linear and Branched Oligogermanes Ph3GeGePh2GePh2GePh2H, Ge5Ph12, and (Ph3Ge)4Ge. Organometallics 2012, 31, 4374–4385. [Google Scholar] [CrossRef]
- Roewe, K.D.; Rheingold, A.L.; Weinert, C.S. A Luminescent and Dichroic Hexagermane. Chem. Commun. 2013, 49, 8380–8382. [Google Scholar] [CrossRef] [PubMed]
- Roewe, K.D.; Golen, J.A.; Rheingold, A.L.; Weinert, C.S. Synthesis, Structure, and Properties of the Hexagermane Pri3Ge(GePh2)4GePri3. Can. J. Chem. 2014, 533–541. [Google Scholar] [CrossRef]
- Komanduri, S.P.; Shumaker, F.A.; Roewe, K.D.; Wolf, M.; Uhlig, F.; Moore, C.E.; Rheingold, A.L.; Weinert, C.S. A Series of Isopropyl-Substituted Oligogermanes Pri3Ge(GePh2)nGePri3 (n = 0–3) Featuring a Luminescent and Dichroic Pentagermane Pri3Ge(GePh2)3GePri3. Organometallics 2016, 35, 3240–3247. [Google Scholar] [CrossRef]
- Komanduri, S.P.; Shumaker, F.A.; Hallenbeck, S.A.; Knight, C.J.; Yoder, C.H.; Buckwalter, B.A.; Dufresne, C.P.; Fernandez, E.J.; Kaffel, C.A.; Nazareno, R.E.; et al. Structure/Property Relationships in Branched Oligogermanes. Preparation of (Me3Ge)3GePh, (Me2ButGe)3GePh, and (Me2PhGe)3GePh and Investigation of Their Properties by Spectroscopic, Spectrometric and Electrochemical Methods. J. Organomet. Chem. 2017, 848, 104–113. [Google Scholar] [CrossRef]
- Marschner, C. Preparation and Reactions of Polysilanyl Anions and Dianions. Organometallics 2006, 25, 2110–2125. [Google Scholar] [CrossRef]
- Fischer, J.; Baumgartner, J.; Marschner, C. Silylgermylpotassium Compounds. Organometallics 2005, 24, 1263–1268. [Google Scholar] [CrossRef]
- Fischer, R.; Baumgartner, J.; Marschner, C.; Uhlig, F. Tris(trimethylsilyl)stannyl Alkali Derivatives: Syntheses and NMR Spectroscopic Properties. Inorg. Chim. Acta 2005, 358, 3174–3182. [Google Scholar] [CrossRef]
- Marschner, C. A New and Easy Route to Polysilanylpotassium Compounds. Eur. J. Inorg. Chem. 1998, 221–226. [Google Scholar] [CrossRef]
- Kayser, C.; Fischer, R.; Baumgartner, J.; Marschner, C. Tailor-made Oligosilyl Potassium Compounds. Organometallics 2002, 21, 1023–1030. [Google Scholar] [CrossRef]
- Mallela, S.P.; Geanangel, R.A. Preparation and Characterization of Tris(trimethylsilyl)silyl Derivatives of Tin. X-Ray Crystal Structure of Cl2Sn[Si(Si(CH3)3)3]2. Inorg. Chem. 1990, 29, 3525–3528. [Google Scholar] [CrossRef]
- Krempner, C. Polysilane Dendrimers. Polymers 2012, 4, 408–447. [Google Scholar] [CrossRef]
- Marsmann, H.C.; Uhlig, F. Further advances in germanium, tin and lead NMR. In The Chemistry of Organic Germanium, Tin and Lead Compounds; Rappoport, Z., Ed.; Wiley: Chichester, UK; New York, NY, USA, 2002; Volume 2, pp. 399–460. ISBN 0-471-49738-X. [Google Scholar]
- Arif, A.M.; Cowley, A.H.; Elkins, T.M. A Bulky Silyl Derivative of Tin(II). J. Organomet. Chem. 1987, 325, C11–C13. [Google Scholar] [CrossRef]
- Klinkhammer, K.W.; Schwarz, W. Bis(hypersilyl)tin and Bis(hypersilyl)lead, Two Electron-Rich Carbene Homologs. Angew. Chem. Int. Ed. Engl. 1995, 34, 1334–1336. [Google Scholar] [CrossRef]
- Katir, N.; Matioszek, D.; Ladeira, S.; Escudié, J.; Castel, A. Stable N-Heterocyclic Carbene Complexes of Hypermetallyl Germanium(II) and Tin(II) Compounds. Angew. Chem. Int. Ed. 2011, 50, 5352–5355. [Google Scholar] [CrossRef] [PubMed]
- Arp, H.; Marschner, C.; Baumgartner, J.; Zark, P.; Müller, T. Coordination Chemistry of Disilylated Stannylenes with Group 10 d10 Transition Metals: Silastannene vs Stannylene Complexation. J. Am. Chem. Soc. 2013, 135, 7949–7959. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Frank, D.; Gaderbauer, W.; Kayser, C.; Mechtler, C.; Baumgartner, J.; Marschner, C. α,ω-Oligosilyl Dianions and Their Application in the Synthesis of Homo- and Heterocyclosilanes. Organometallics 2003, 22, 3723–3731. [Google Scholar] [CrossRef]
- Baumgartner, J.; Fischer, R.; Fischer, J.; Wallner, A.; Marschner, C.; Flörke, U. Structural Aspects of Trimethylsilylated Branched Group 14 Compounds. Organometallics 2005, 24, 6450–6457. [Google Scholar] [CrossRef]
- Schmidt, R.K.; Oestreich, M. Transition-Metal-Free Conjugate Stannyl Transfer to α,β-Unsaturated Carbonyl and Carboxyl Compounds in Basic Aqueous Media. Synlett 2008, 1690–1692. [Google Scholar] [CrossRef]
- Ascher, K.R.S.; Eliyahu, M.; Bultent, E.J.; Meinema, H.A. Antifeedant Effects of Mixed Methylphenyl Hexaorganoditin Compounds of the Type PhnMe6-nSnSn on Larvae of Spodoptera littoralis and Epilachna varivestis. Appl. Organomet. Chem. 1987, 1, 303–309. [Google Scholar] [CrossRef]
- Whittaker, S.M.; Brun, M.-C.; Cervantes-Lee, F.; Pannell, K.H. Synthesis, Structure, and Reactivity of the Permethylated Decasilane (Me3Si)3SiSiMe2SiMe2SiMe3)3. J. Organomet. Chem. 1995, 499, 247–252. [Google Scholar] [CrossRef]
- Lambert, J.B.; Pflug, J.L.; Allgeier, A.M.; Campbell, D.J.; Higgins, T.B.; Singewald, E.T.; Stern, C.L. A Branched Polysilane. Acta Cryst. C 1995, 51, 713–715. [Google Scholar] [CrossRef]
- Baumgartner, J.; Frank, D.; Kayser, C.; Marschner, C. Comparative Study of Structural Aspects of Branched Oligosilanes. Organometallics 2005, 24, 750–761. [Google Scholar] [CrossRef]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Becker, G.; Gekeler, M.; Hartmann, H.-M.; Mundt, O.; Westerhausen, M. Tetrakis(trimethylsilyl)silane, -germane, and -stannane and Some Complexes of Lithium Tris(trimethylsilyl)silanide, -germanide and -stannanide—M[Si(CH3)3]4 (M = Si, Ge, Sn), LxLi–M[Si(CH3)3]3. In Synthetic Methods of Organometallic and Inorganic Chemistry. Groups 1, 2, 13, and 14; Herrmann, W., Auner, N., Klingebiel, U., Eds.; Thieme: Stuttgart, Germany, 1996; Volume 2, pp. 186–192. [Google Scholar]
- Morris, G.A.; Freeman, R. Enhancement of Nuclear Magnetic Resonance Signals by Polarization Transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Helmer, B.J.; West, R. Enhancement of 29Si NMR Signals by Proton Polarization Transfer. Organometallics 1982, 1, 877–879. [Google Scholar] [CrossRef]
- Menges, F. Spectragryph—Optical Spectroscopy Software; Spectroscopy Ninja: Oberstdorf, Germany, 2017. [Google Scholar]
- SAINTPLUS: Software Reference Manual, version 6.45; Bruker-AXS: Madison, WI, USA, 1997–2003.
- Sheldrick, G.M. SADABS, version 2.10; Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Cryst. A 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POVRAY, version 3.6; Persistence of Vision Pty. Ltd.: Williamstown, Victoria, Australia, 2004; Available online: http://www.povray.org/download/ (accessed on 9 July 2008).
- Apodaca, P.; Cervantes-Lee, F.; Pannell, K.H. Mixed Aryl-Alkyl Organotin Compounds. Main Group Met. Chem. 2011, 24, 597–602. [Google Scholar] [CrossRef]
- Zarl, E.; Baumgartner, J.; Decker, K.; Fischer, R.; Seibt, B.; Uhlig, F. Solvent Influence in Reactions of Fluoroalkyl Sulfonic Acids with Phenyldistannanes. Phosphorus Sulfur Silicon Relat. Elem. 2008, 183, 1923–1934. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
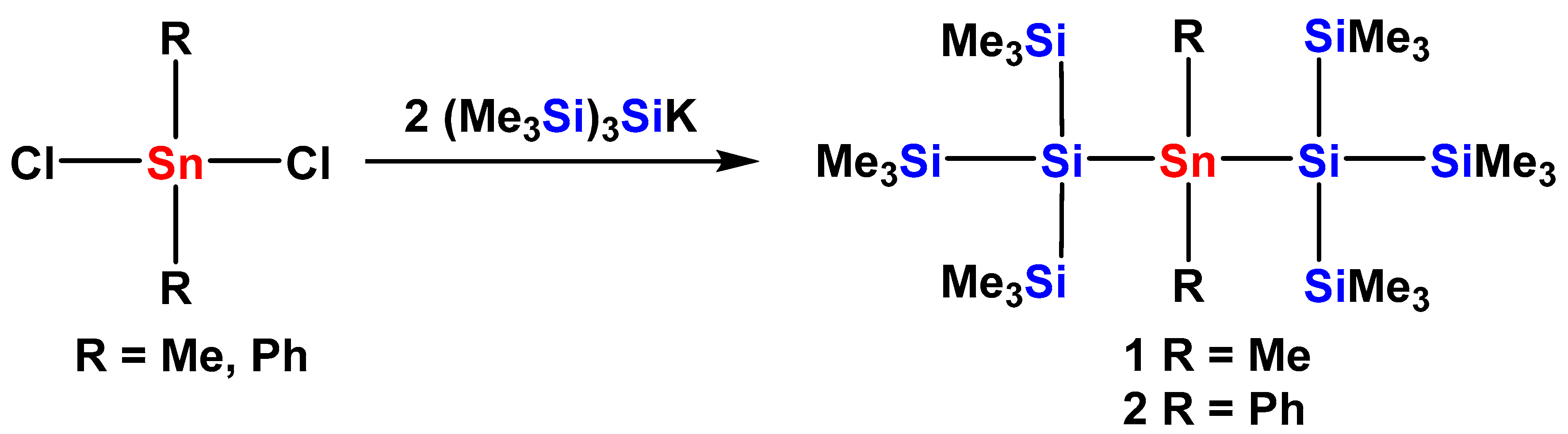{kind=link}
{kind=link}
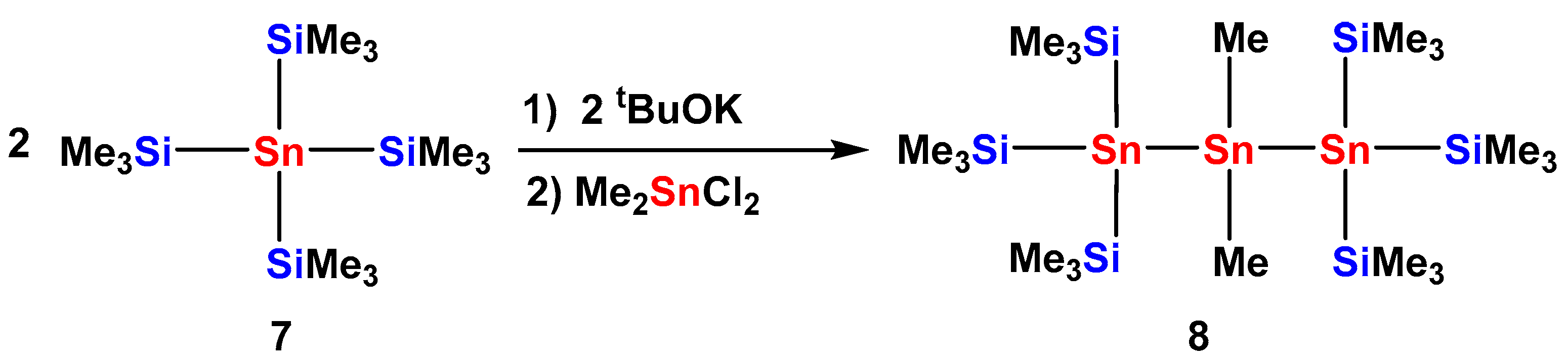{kind=link}
| Compound | 29Si (SiMe3) | 29Si Si(EMe3) | 119Sn |
|---|---|---|---|
| 1 | −7.7 | −123.7 | −176 |
| 2 | −9.9 | −133.9 | −174 |
| 5 | −7.8 | −128.7 | −68 |
| 6 | −8.4 | −131.8 | −114 |
| 8 | −7.1 | - | −277 (SnMe), −464 (SnSi) |
| Dihedral Angles ω [°] Determined by X-ray Crystallography | |||||
|---|---|---|---|---|---|
| Compound | ω1 | ω2 | ω3 | ω4 | Conformation 1 |
| 2 | 153 | 143 | DD | ||
| 5 | 159 | 180 | 159 | TAT | |
| 6 | 160 | 180 | 160 | TAT | |
| 6a | 162 | 162 | 161 | 148 | TTTD |
| 8 | 170 | 170 | TT | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stella, F.; Marschner, C.; Baumgartner, J. Incorporating Methyl and Phenyl Substituted Stannylene Units into Oligosilanes. The Influence on Optical Absorption Properties. Molecules 2017, 22, 2212. https://doi.org/10.3390/molecules22122212
Stella F, Marschner C, Baumgartner J. Incorporating Methyl and Phenyl Substituted Stannylene Units into Oligosilanes. The Influence on Optical Absorption Properties. Molecules. 2017; 22(12):2212. https://doi.org/10.3390/molecules22122212
Chicago/Turabian StyleStella, Filippo, Christoph Marschner, and Judith Baumgartner. 2017. "Incorporating Methyl and Phenyl Substituted Stannylene Units into Oligosilanes. The Influence on Optical Absorption Properties" Molecules 22, no. 12: 2212. https://doi.org/10.3390/molecules22122212
APA StyleStella, F., Marschner, C., & Baumgartner, J. (2017). Incorporating Methyl and Phenyl Substituted Stannylene Units into Oligosilanes. The Influence on Optical Absorption Properties. Molecules, 22(12), 2212. https://doi.org/10.3390/molecules22122212