
Construction and Biological Evaluation of a Novel Integrin ανβ3-Specific Carrier for Targeted siRNA Delivery In Vitro


Abstract
:1. Introduction
2. Results
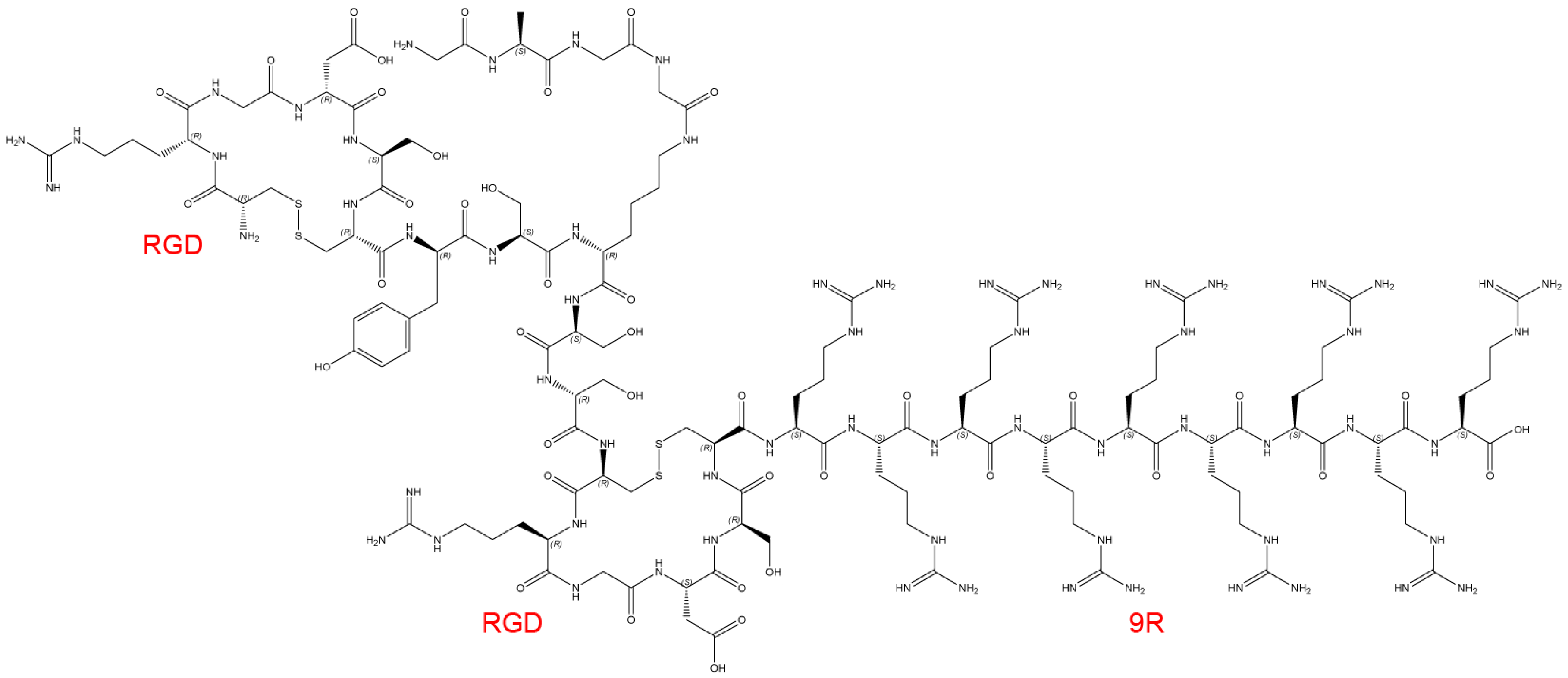
2.1. Structure of the Delivery Carrier and siRNA
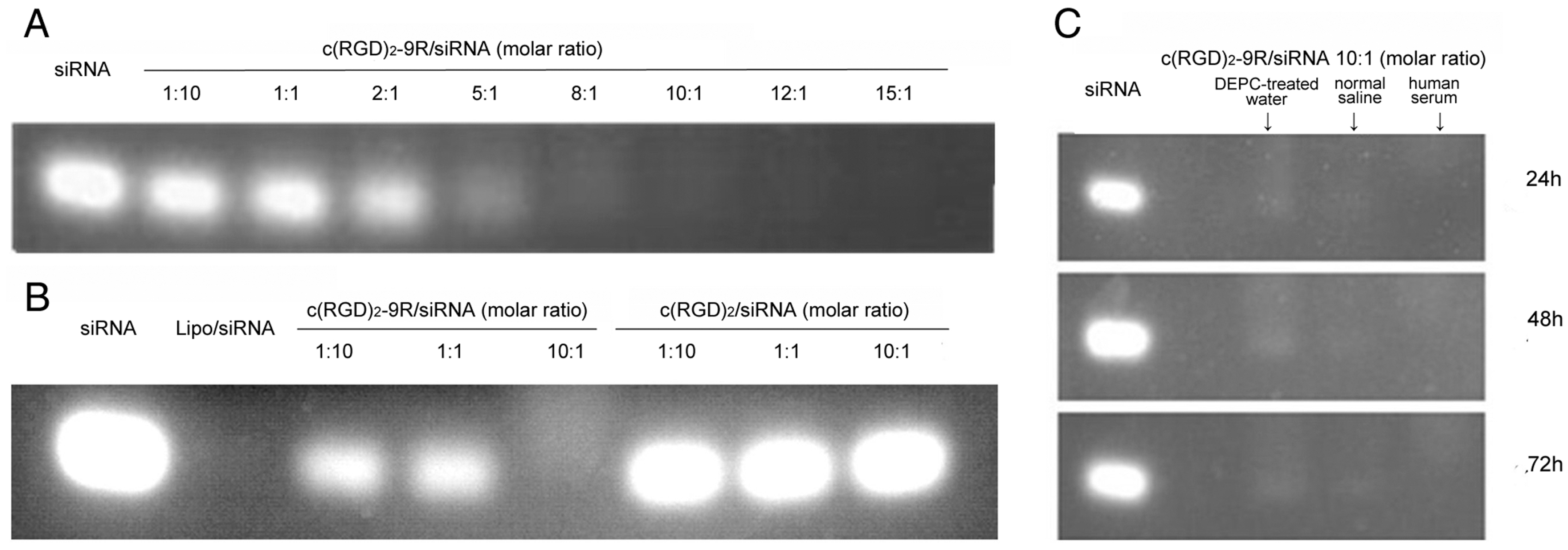
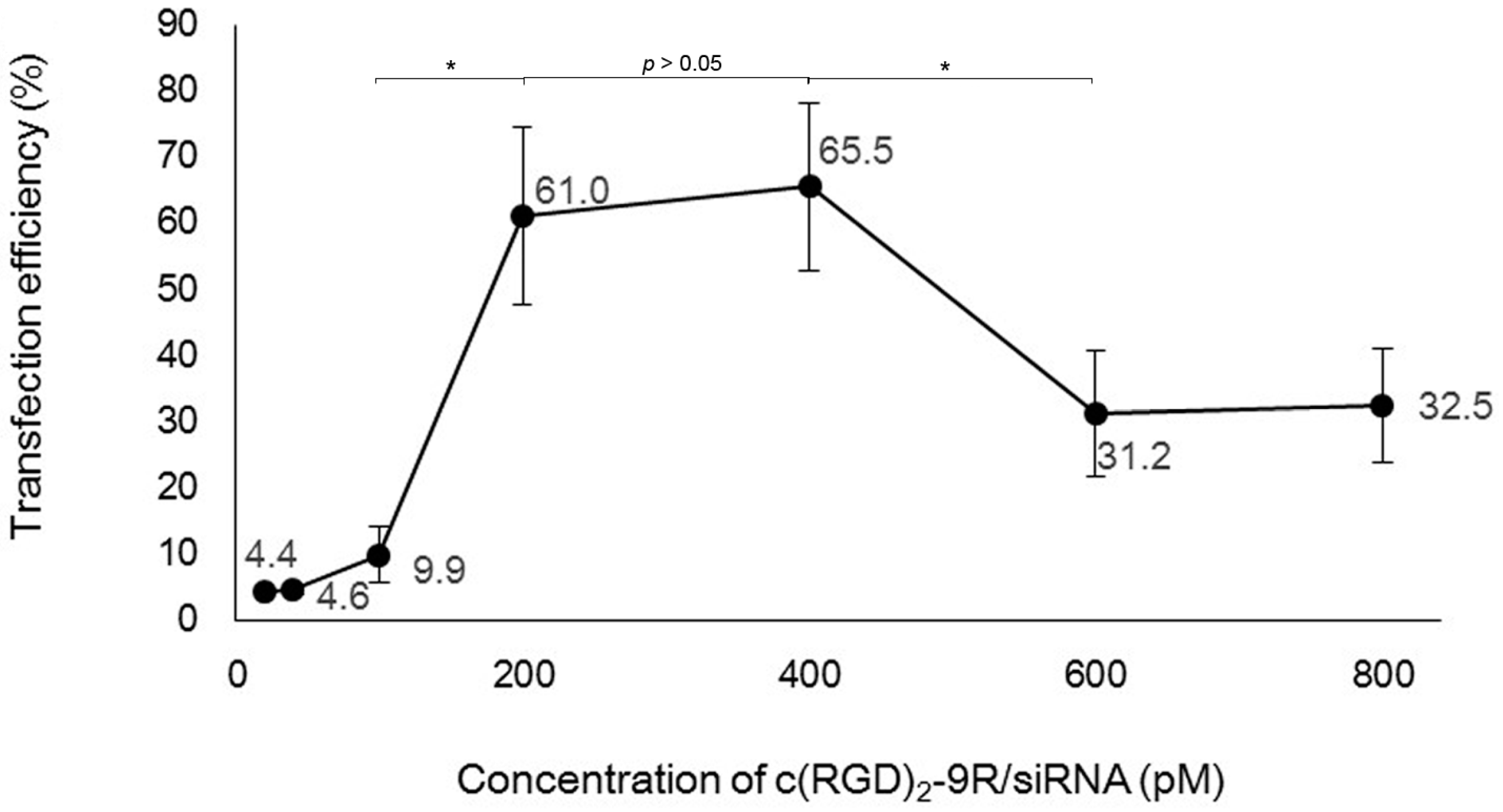
2.2. Preparation of Delivery Carrier with siRNA
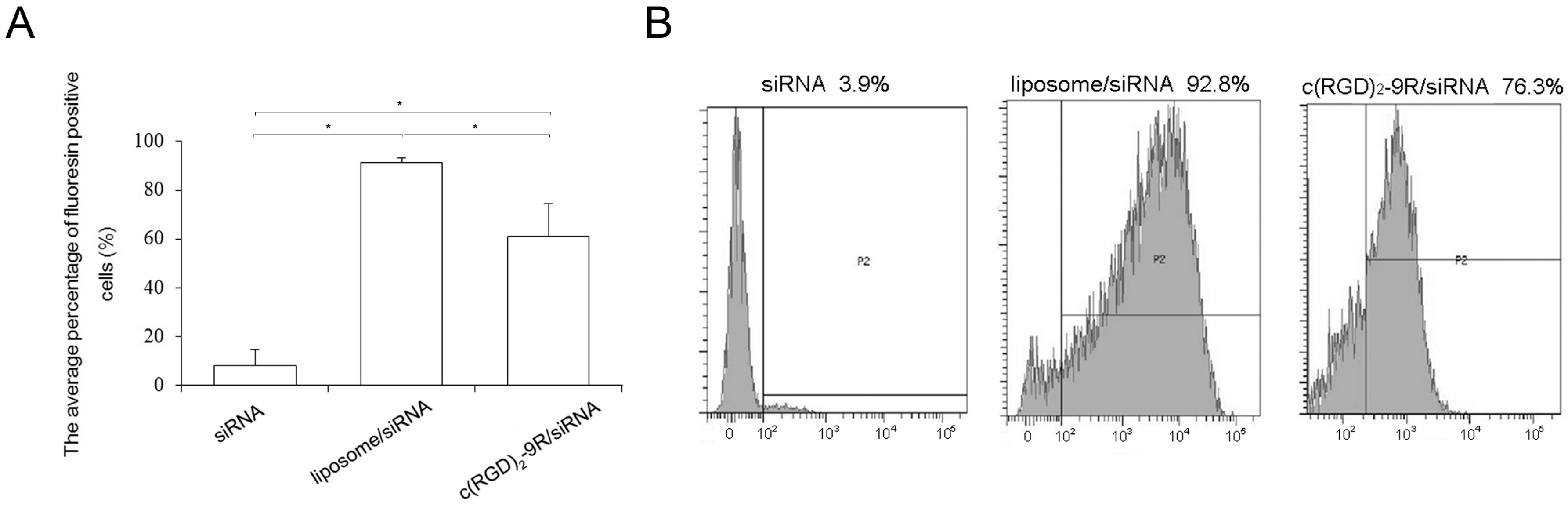
2.3. Flow Cytometry
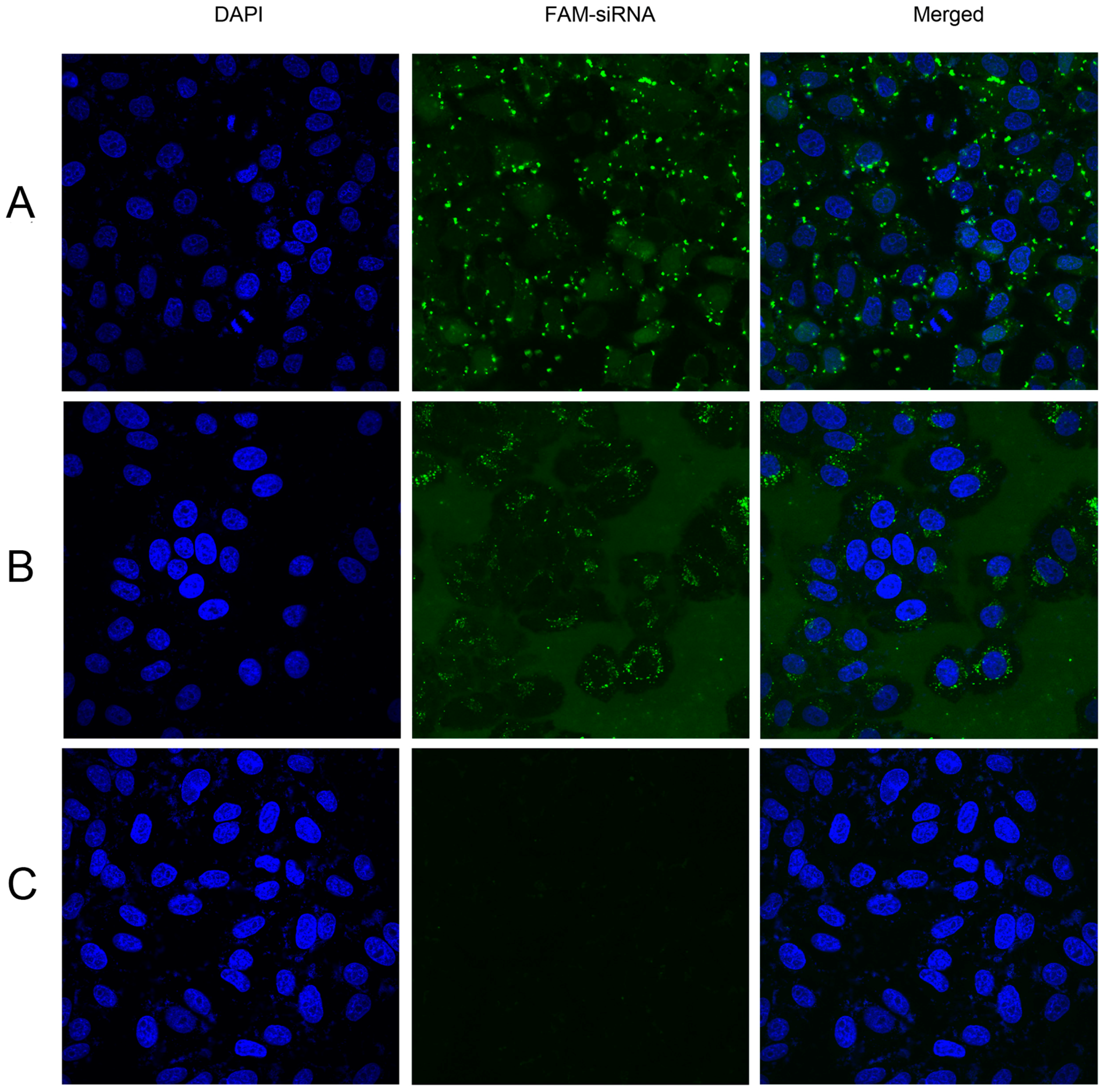
2.4. Confocal Microscopy
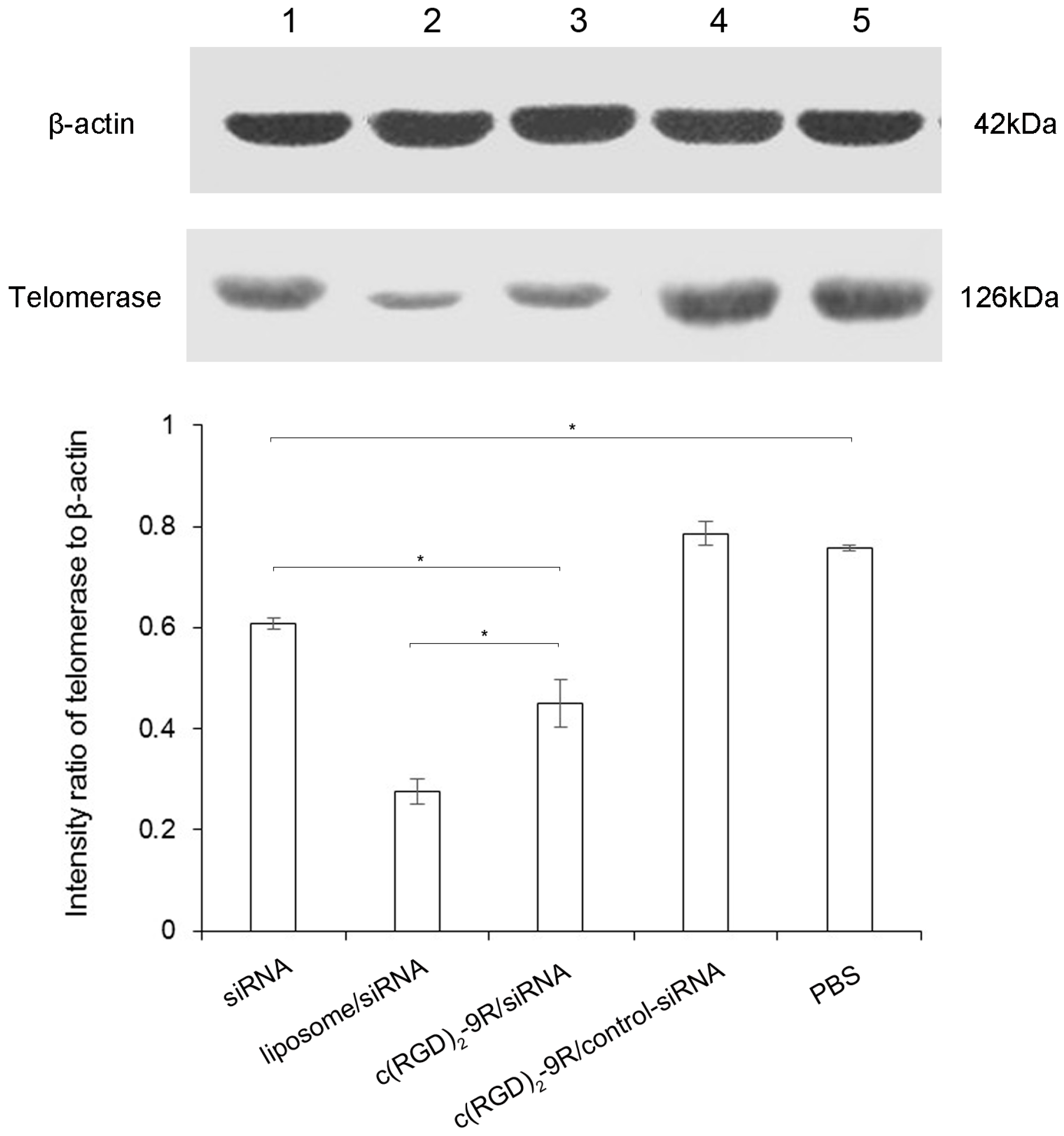
2.5. Western Blot
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of Delivery Carrier with siRNA
4.3. Cell Culture
4.4. Cell Transfection
4.5. Flow Cytometry
4.6. Confocal Microscopy
4.7. Western Blot
4.8. Statistical Analyses
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jones, S.W.; Souza, P.M.; Lindsay, M.A. siRNA for gene silencing: A route to drug target discovery. Curr. Opin. Pharmacol. 2004, 4, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.; Robbins, P.D. Cell-type specific penetrating peptides: Therapeutic promises and challenges. Molecules 2015, 20, 13055–13070. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Tanaka, G.; Futaki, S. Cell-penetrating peptides (CPPs) as a vector for the delivery of siRNAs into cells. Mol. Biosyst. 2013, 9, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Endoh, T.; Ohtsuki, T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv. Drug. Deliv. Rev. 2009, 61, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Zeller, S.; Choi, C.S.; Uchil, P.D.; Ban, H.S.; Siefert, A.; Fahmy, T.M.; Mothes, W.; Lee, S.K.; Kumar, P. Attachment of cell-binding ligands to arginine-rich cell-penetrating peptides enables cytosolic translocation of complexed siRNA. Chem. Biol. 2015, 22, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Ban, H.S.; Kim, S.S.; Wu, H.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.; Haridas, V.; Habiro, K.; et al. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell 2008, 134, 577–586. [Google Scholar] [CrossRef]
- Lee, Y.K.; Kim, K.S.; Kim, J.S.; Baek, J.E.; Park, S.I.; Jeong, H.Y.; Yoon, S.S.; Jung, K.C.; Song, H.G.; Park, Y.S. Leukemia-specific siRNA delivery by immunonanoplexes consisting of anti-JL1 minibody conjugated to oligo-9 Arg-peptides. Mol. Cells 2010, 29, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Kowolik, C.M.; Swiderski, P.M.; Kortylewski, M.; Yu, H.; Horne, D.A.; Jove, R.; Caballero, O.L.; Simpson, A.J.; Lee, F.T.; et al. Humanized Lewis-Y specific antibody based delivery of STAT3 siRNA. ACS Chem. Biol. 2011, 6, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, F.C.; Kessler, H.; Wester, H.J.; Schwaiger, M.; Beer, A.J. Radiolabelled RGD peptides for imaging and therapy. Eur. J. Nucl. Med. Mol. Imaging 2012, 39 (Suppl. 1), S126–S138. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome. Med. 2016, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Hao, P.; Zhang, L.; Ma, C.; Yan, P.; Wang, R.F.; Zhang, C.L. A new cyclic RGD peptide dimer for integrin αvβ3 imaging. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 613–619. [Google Scholar] [PubMed]
- Kang, L.; Wang, R.F.; Yan, P.; Liu, M.; Zhang, C.L.; Yu, M.M.; Cui, Y.G.; Xu, X.J. Noninvasive visualization of RNA delivery with 99mTc-radiolabeled small-interference RNA in tumor xenografts. J. Nucl. Med. 2010, 51, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, K.; Yu, E.Y.; Khurts, S.; Ben-Porath, I.; Currier, J.L.; Metz, G.B.; Brooks, M.W.; Kaneko, S.; Murakami, S.; de Caprio, J.A.; et al. Telomerase maintains telomere structure in normal human cells. Cell 2003, 114, 241–253. [Google Scholar] [CrossRef]
- Ku, S.H.; Jo, S.D.; Lee, Y.K.; Kim, K.; Kim, S.H. Chemical and structural modifications of RNAi therapeutics. Adv. Drug. Deliv. Rev. 2016, 104, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Chernolovskaya, E.L.; Zenkova, M.A. Chemical modification of siRNA. Curr. Opin. Mol. Ther. 2010, 12, 158–167. [Google Scholar] [PubMed]
- Kraynack, B.A.; Baker, B.F. Small interfering RNAs containing full 2′-O-methylribonucleotide-modified sense strands display Argonaute2/eIF2C2-dependent activity. RNA 2006, 12, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Rana, T.M. siRNA function in RNAi: a chemical modification analysis. RNA 2003, 9, 1034–1048. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Bourre, L.; Soden, D.M.; O’Sullivan, G.C.; O’Driscoll, C. Can non-viral technologies knockdown the barriers to siRNA delivery and achieve the next generation of cancer therapeutics? Biotechnol. Adv. 2011, 29, 402–417. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Burgess, D.J.; Patil, S.D. Physicochemical characterization techniques for lipid based delivery systems for siRNA. Int. J. Pharm. 2012, 427, 35–57. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug. Deliv. Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Adamo, G.; Grimaldi, N.; Campora, S.; Bulone, D.; Bondi, M.L.; Al-Sheikhly, M.; Sabatino, M.A.; Dispenza, C.; Ghersi, G. Multi-Functional Nanogels for Tumor Targeting and Redox-Sensitive Drug and siRNA Delivery. Molecules 2016, 21, 1594. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Killinger, B.; Moszczynska, A.; Merkel, O.M. Targeted Delivery of siRNA to Transferrin Receptor Overexpressing Tumor Cells via Peptide Modified Polyethylenimine. Molecules 2016, 21, 1334. [Google Scholar] [CrossRef] [PubMed]
- Ibaraki, H.; Kanazawa, T.; Takashima, Y.; Okada, H.; Seta, Y. Development of an Innovative Intradermal siRNA Delivery System Using a Combination of a Functional Stearylated Cytoplasm-Responsive Peptide and a Tight Junction-Opening Peptide. Molecules 2016, 21, 1279. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, A.N.; Paquin, K.L.; Howlett, N.G.; Mandal, D.; Parang, K. Cyclic peptide-capped gold nanoparticles for enhanced siRNA delivery. Molecules 2014, 19, 13319–13331. [Google Scholar] [CrossRef] [PubMed]
- Merkel, O.M.; Zheng, M.; Mintzer, M.A.; Pavan, G.M.; Librizzi, D.; Maly, M.; Hoffken, H.; Danani, A.; Simanek, E.E.; Kissel, T. Molecular modeling and in vivo imaging can identify successful flexible triazine dendrimer-based siRNA delivery systems. J. Control. Release 2011, 153, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Bruno, K. Using drug-excipient interactions for siRNA delivery. Adv. Drug. Deliv. Rev. 2011, 63, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | |
|---|---|---|
| FAM-labeled siRNA | Sense | 5’-FAM UUU CAU CAG CAA GUU UGG AdTdT-3’ |
| (FAM-siRNA) | Antisense | 5’-UCC AAA CUU GCU GAU GAA AdTdT-3’ |
| Negative siRNA | Sense | 5’-UUC UCC GAA CGU GUC ACG UdTdT-3’ |
| Antisense | 5’-ACG UGA CAC GUU CGG AGA AdTdT-3’ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Liu, M.; Wang, R.; Yan, P.; Zhang, C.; Ma, C.; Yin, L. Construction and Biological Evaluation of a Novel Integrin ανβ3-Specific Carrier for Targeted siRNA Delivery In Vitro. Molecules 2017, 22, 231. https://doi.org/10.3390/molecules22020231
Chen X, Liu M, Wang R, Yan P, Zhang C, Ma C, Yin L. Construction and Biological Evaluation of a Novel Integrin ανβ3-Specific Carrier for Targeted siRNA Delivery In Vitro. Molecules. 2017; 22(2):231. https://doi.org/10.3390/molecules22020231
Chicago/Turabian StyleChen, Xueqi, Meng Liu, Rongfu Wang, Ping Yan, Chunli Zhang, Chao Ma, and Lei Yin. 2017. "Construction and Biological Evaluation of a Novel Integrin ανβ3-Specific Carrier for Targeted siRNA Delivery In Vitro" Molecules 22, no. 2: 231. https://doi.org/10.3390/molecules22020231
APA StyleChen, X., Liu, M., Wang, R., Yan, P., Zhang, C., Ma, C., & Yin, L. (2017). Construction and Biological Evaluation of a Novel Integrin ανβ3-Specific Carrier for Targeted siRNA Delivery In Vitro. Molecules, 22(2), 231. https://doi.org/10.3390/molecules22020231