
Impact of Age and Insulin-Like Growth Factor-1 on DNA Damage Responses in UV-Irradiated Human Skin



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. UV-Induced DNA Damage Formation, Repair, and Checkpoint Signaling
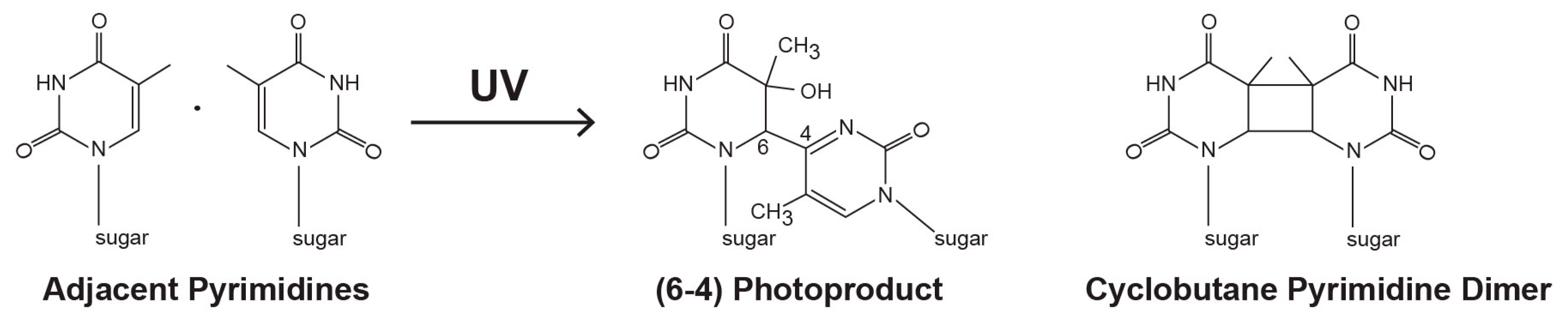
2.1. UV-Induced DNA Photoproduct Formation
2.2. Removal of UV Photoproducts by Nucleotide Excision Repair (NER)
2.3. Suppression of DNA Synthesis and Cell Cycle Progression by the DNA Damage Checkpoint
3. Effect of Aging on DNA Damage Responses in UV-Irradiated Human Epidermis
3.1. Effect of Age on UV Photoproduct Formation in the Epidermis
3.2. Effect of Age on UV Photoproduct Repair in the Epidermis
3.3. Effects of UVR on DNA Synthesis and DNA Damage Checkpoints in the Epidermis
4. Effect of Aging on Insulin-Like Growth Factor-1 (IGF-1) Production in the Skin
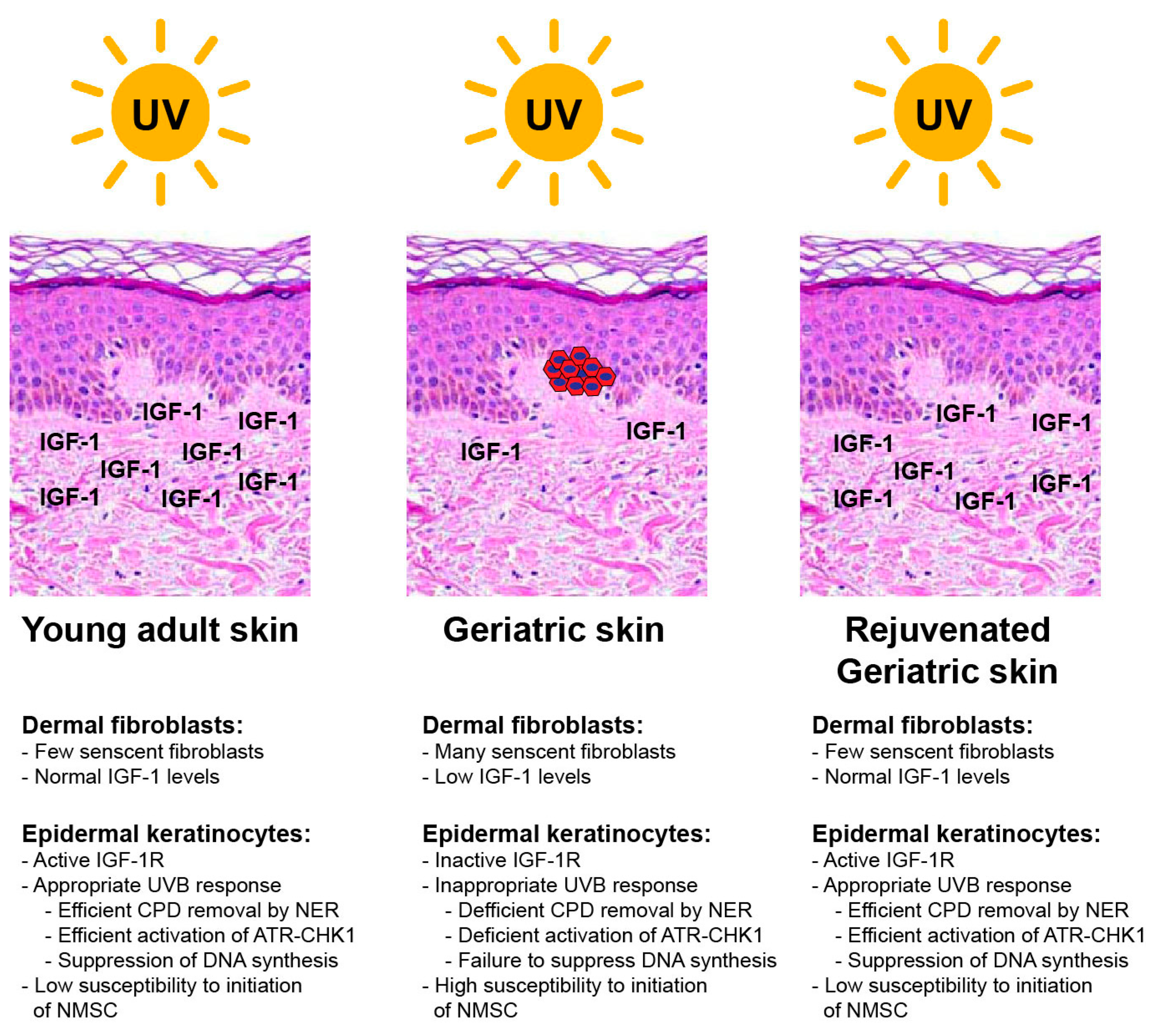
4.1. Epidermal Keratinocyte IGF-1 Receptor (IGF-1R) Activation Is Altered in Aged Skin
4.2. The IGF-1/IGF-1R System Affects Cell Fate Following Exposure to UVR
4.3. The Removal of UV-Induced CPDs Is Affected by IGF-1R Status in Human Keratinocytes
4.4. Disruption of ATR-CHK1 Kinase Signaling and the Suppression of DNA Synthesis in Keratinocytes with Inactive IGF-1Rs
5. Dermal Wounding as a Preventive Approach for NMSC
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Cancer Society. Key Statistics for Basal and Squamous Cell Skin Cancers. Available online: http://www.cancer.org/cancer/skincancer-basalandsquamouscell/detailedguide/skin-cancer-basal-and-squamous-cell-key-statistics (accessed on 28 December 2016).
- Karia, P.S.; Han, J.; Schmults, C.D. Cutaneous Squamous Cell Carcinoma: Estimated Incidence of Disease, Nodal Metastasis, and Deaths from Disease in the United States, 2012. J. Am. Acad. Dermatol. 2013, 68, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, K.H. Sunlight and Skin Cancer: Another Link Revealed. Proc. Natl. Acad. Sci. USA 1997, 94, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Naylor, M.F.; Boyd, A.; Smith, D.W.; Cameron, G.S.; Hubbard, D.; Neldner, K.H. High Sun Protection Factor Sunscreens in the Suppression of Actinic Neoplasia. Arch. Dermatol. 1995, 131, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.C.; Jolley, D.; Marks, R. Reduction of Solar Keratoses by Regular Sunscreen Use. N. Engl. J. Med. 1993, 329, 1147–1151. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, C.; Jurgensen, J.S.; Degen, A.; Hackethal, M.; Ulrich, M.; Patel, M.J.; Eberle, J.; Terhorst, D.; Sterry, W.; Stockfleth, E. Prevention of Non-Melanoma Skin Cancer in Organ Transplant Patients by Regular use of a Sunscreen: A 24 Months, Prospective, Case-Control Study. Br. J. Dermatol. 2009, 161 (Suppl. 3), 78–84. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Travers, J.B.; Spandau, D.F. A New Paradigm for the Role of Aging in the Development of Skin Cancer. J. Invest. Dermatol. 2009, 129, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Travers, J.B.; Somani, A.K.; Spandau, D.F. The IGF-1/IGF-1R Signaling Axis in the Skin: A New Role for the Dermis in Aging-Associated Skin Cancer. Oncogene 2010, 29, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.B.; Spandau, D.F.; Lewis, D.A.; Machado, C.; Kingsley, M.; Mousdicas, N.; Somani, A.K. Fibroblast Senescence and Squamous Cell Carcinoma: How Wounding Therapies could be Protective. Dermatol. Surg. 2013, 39, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Sage, E.; Douki, T. Ultraviolet Radiation-Mediated Damage to Cellular DNA. Mutat. Res. 2005, 571, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Grand, A.; Douki, T. Solar UV Radiation-Induced DNA Bipyrimidine Photoproducts: Formation and Mechanistic Insights. Top. Curr. Chem. 2015, 356, 249–275. [Google Scholar] [PubMed]
- Douki, T.; Reynaud-Angelin, A.; Cadet, J.; Sage, E. Bipyrimidine Photoproducts rather than Oxidative Lesions are the Main Type of DNA Damage Involved in the Genotoxic Effect of Solar UVA Radiation. Biochemistry 2003, 42, 9221–9226. [Google Scholar] [CrossRef] [PubMed]
- Mouret, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane Pyrimidine Dimers are Predominant DNA Lesions in Whole Human Skin Exposed to UVA Radiation. Proc. Natl. Acad. Sci. USA 2006, 103, 13765–13770. [Google Scholar] [CrossRef] [PubMed]
- Premi, S.; Wallisch, S.; Mano, C.M.; Weiner, A.B.; Bacchiocchi, A.; Wakamatsu, K.; Bechara, E.J.; Halaban, R.; Douki, T.; Brash, D.E. Photochemistry. Chemiexcitation of Melanin Derivatives Induces DNA Photoproducts Long After UV Exposure. Science 2015, 347, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Premi, S.; Brash, D.E. Chemical Excitation of Electrons: A Dark Path to Melanoma. DNA Repair (Amst.) 2016, 44, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Cannistraro, V.J.; Taylor, J.S. Acceleration of 5-Methylcytosine Deamination in Cyclobutane Dimers by G and its Implications for UV-Induced C-to-T Mutation Hotspots. J. Mol. Biol. 2009, 392, 1145–1157. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Taylor, J.S. In Vivo Evidence that UV-Induced C→T Mutations at Dipyrimidine Sites could Result from the Replicative Bypass of Cis-Syn Cyclobutane Dimers Or their Deamination Products. Biochemistry 1993, 32, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Shaw, B.R. Accelerated Deamination of Cytosine Residues in UV-Induced Cyclobutane Pyrimidine Dimers Leads to CC→TT Transitions. Biochemistry 1996, 35, 10172–10181. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Dammann, R.; Pfeifer, G.P. Sequence and Time-Dependent Deamination of Cytosine Bases in UVB-Induced Cyclobutane Pyrimidine Dimers in Vivo. J. Mol. Biol. 1998, 284, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. UV Signature Mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.D.; Bose, K.K.; Rabkin, S.D.; Strauss, B.S. Sites of Termination of in Vitro DNA Synthesis on Ultraviolet- and N-Acetylaminofluorene-Treated Phi X174 Templates by Prokaryotic and Eukaryotic DNA Polymerases. Proc. Natl. Acad. Sci. USA 1981, 78, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M.; Lane, D.P. Transcription-Guarding the Genome by Sensing DNA Damage. Nat. Rev. Cancer. 2004, 4, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M. The Transcription Stress Response. Cell. Cycle 2007, 6, 2252–2257. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.F.; Woodgate, R. Translesion DNA Polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef] [PubMed]
- Klarer, A.C.; McGregor, W. Replication of Damaged Genomes. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E. Translesion DNA Synthesis and Mutagenesis in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.L.; Orr-Weaver, T.L. Replication Fork Instability and the Consequences of Fork Collisions from Rereplication. Genes Dev. 2016, 30, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Labib, K.; Hodgson, B. Replication Fork Barriers: Pausing for a Break or Stalling for Time? EMBO Rep. 2007, 8, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.K. The Human Intra-S Checkpoint Response to UVC-Induced DNA Damage. Carcinogenesis 2010, 31, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Batista, L.F.; Kaina, B.; Meneghini, R.; Menck, C.F. How DNA Lesions are turned into Powerful Killing Structures: Insights from UV-Induced Apoptosis. Mutat. Res. 2009, 681, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A. Mechanisms of DNA Repair by Photolyase and Excision Nuclease (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2016, 55, 8502–8527. [Google Scholar] [CrossRef] [PubMed]
- Reardon, J.T.; Sancar, A. Nucleotide Excision Repair. Prog. Nucleic Acid Res. Mol. Biol. 2005, 79, 183–235. [Google Scholar] [PubMed]
- Scharer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Hu, J. PostExcision Events in Human Nucleotide Excision Repair. Photochem. Photobiol. 2017, 93, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E.; Lam, E.T.; Revet, I. Disorders of Nucleotide Excision Repair: The Genetic and Molecular Basis of Heterogeneity. Nat. Rev. Genet. 2009, 10, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding Nucleotide Excision Repair and its Roles in Cancer and Ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making it Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an Essential Kinase that is Regulated by Atr and Required for the G(2)/M DNA Damage Checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [PubMed]
- Guo, Z.; Kumagai, A.; Wang, S.X.; Dunphy, W.G. Requirement for Atr in Phosphorylation of Chk1 and Cell Cycle Regulation in Response to DNA Replication Blocks and UV-Damaged DNA in Xenopus Egg Extracts. Genes Dev. 2000, 14, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Piwnica-Worms, H. ATR-Mediated Checkpoint Pathways Regulate Phosphorylation and Activation of Human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, T.P.; Simpson, D.A.; Frank, A.R.; Heinloth, A.N.; Paules, R.S.; Cordeiro-Stone, M.; Kaufmann, W.K. An ATR- and Chk1-Dependent S Checkpoint Inhibits Replicon Initiation Following UVC-Induced DNA Damage. Mol. Cell. Biol. 2002, 22, 8552–8561. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, T.P.; Unsal-Kacmaz, K.; Heinloth, A.N.; Simpson, D.A.; Paules, R.S.; Sancar, A.; Cordeiro-Stone, M.; Kaufmann, W.K. Cdc7-Dbf4 and the Human S Checkpoint Response to UVC. J. Biol. Chem. 2007, 282, 9458–9468. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.K.; Cleaver, J.E. Mechanisms of Inhibition of DNA Replication by Ultraviolet Light in Normal Human and Xeroderma Pigmentosum Fibroblasts. J. Mol. Biol. 1981, 149, 171–187. [Google Scholar] [CrossRef]
- Kaufmann, W.K.; Cleaver, J.E.; Painter, R.B. Ultraviolet Radiation Inhibits Replicon Initiation in S Phase Human Cells. Biochim. Biophys. Acta 1980, 608, 191–195. [Google Scholar] [CrossRef]
- Painter, R.B. Inhibition and Recovery of DNA Synthesis in Human Cells after Exposure to Ultraviolet Light. Mutat. Res. 1985, 145, 63–69. [Google Scholar] [CrossRef]
- Miao, H.; Seiler, J.A.; Burhans, W.C. Regulation of Cellular and SV40 Virus Origins of Replication by Chk1-Dependent Intrinsic and UVC Radiation-Induced Checkpoints. J. Biol. Chem. 2003, 278, 4295–4304. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.A.; Cortez, D. ATR Signalling: More than Meeting at the Fork. Biochem. J. 2011, 436, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An Essential Regulator of Genome Integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, M.; Follonier, C.; Tourriere, H.; Puddu, F.; Lazzaro, F.; Pasero, P.; Lopes, M.; Plevani, P.; Muzi-Falconi, M. Exo1 Competes with Repair Synthesis, Converts NER Intermediates to Long ssDNA Gaps, and Promotes Checkpoint Activation. Mol. Cell 2010, 40, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Sertic, S.; Pizzi, S.; Cloney, R.; Lehmann, A.R.; Marini, F.; Plevani, P.; Muzi-Falconi, M. Human Exonuclease 1 Connects Nucleotide Excision Repair (NER) Processing with Checkpoint Activation in Response to UV Irradiation. Proc. Natl. Acad. Sci. USA 2011, 108, 13647–13652. [Google Scholar] [CrossRef] [PubMed]
- Lindsey-Boltz, L.A.; Kemp, M.G.; Reardon, J.T.; DeRocco, V.; Iyer, R.R.; Modrich, P.; Sancar, A. Coupling of Human DNA Excision Repair and the DNA Damage Checkpoint in a Defined in Vitro System. J. Biol. Chem. 2014, 289, 5074–5082. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.C.; Walter, J.C.; Cimprich, K.A. Functional Uncoupling of MCM Helicase and DNA Polymerase Activities Activates the ATR-Dependent Checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- MacDougall, C.A.; Byun, T.S.; Van, C.; Yee, M.C.; Cimprich, K.A. The Structural Determinants of Checkpoint Activation. Genes Dev. 2007, 21, 898–903. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. RPA-Coated Single-Stranded DNA as a Platform for Post-Translational Modifications in the DNA Damage Response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Oakley, G.G.; Patrick, S.M. Replication Protein A: Directing Traffic at the Intersection of Replication and Repair. Front. Biosci. 2010, 15, 883–900. [Google Scholar] [CrossRef]
- Wold, M.S. Replication Protein A: A Heterotrimeric, Single-Stranded DNA-Binding Protein Required for Eukaryotic DNA Metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Fanning, E.; Klimovich, V.; Nager, A.R. A Dynamic Model for Replication Protein A (RPA) Function in DNA Processing Pathways. Nucleic Acids Res. 2006, 34, 4126–4137. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA Damage through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Vaithiyalingam, S.; Glick, G.G.; Mordes, D.A.; Chazin, W.J.; Cortez, D. The Basic Cleft of RPA70N Binds Multiple Checkpoint Proteins, Including RAD9, to Regulate ATR Signaling. Mol. Cell. Biol. 2008, 28, 7345–7353. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, J.; Yan, S.; Michael, W.M. Direct Binding to Replication Protein A (RPA)-Coated Single-Stranded DNA Allows Recruitment of the ATR Activator TopBP1 to Sites of DNA Damage. J. Biol. Chem. 2016, 291, 13124–13131. [Google Scholar] [CrossRef] [PubMed]
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 Acts at Stalled Replication Forks to Maintain Genome Integrity. Nat. Cell Biol. 2016, 18, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Raschle, M.; Mailand, N. Activation of the ATR Kinase by the RPA-Binding Protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Zhou, Q.; Chen, J.; Yuan, J. RPA-Binding Protein ETAA1 is an ATR Activator Involved in DNA Replication Stress Response. Curr. Biol. 2016, 26, 3257–3268. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Akan, Z.; Yilmaz, S.; Grillo, M.; Smith-Roe, S.L.; Kang, T.H.; Cordeiro-Stone, M.; Kaufmann, W.K.; Abraham, R.T.; Sancar, A.; et al. Tipin-Replication Protein A Interaction Mediates Chk1 Phosphorylation by ATR in Response to Genotoxic Stress. J. Biol. Chem. 2010, 285, 16562–16571. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.A.; Oehler, H.; Voigt, A.; Dalic, D.; Freiwald, A.; Serve, H.; Beli, P. ATR Inhibition Rewires Cellular Signaling Networks Induced by Replication Stress. Proteomics 2016, 16, 402–416. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, F.M.B.; Kim, D.; Cussiol, J.R.; Das, J.; Jeong, M.C.; Doerfler, L.; Schmidt, K.H.; Yu, H.; Smolka, M.B. Phosphoproteomics Reveals Distinct Modes of Mec1/ATR Signaling during DNA Replication. Mol. Cell 2015, 57, 1124–1132. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Rush, J.; Macneill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; et al. Profiling of UV-Induced ATM/ATR Signaling Pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 19855–19860. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 Checkpoint Pathway in Mammals: Linkage of DNA Damage to Cdk Regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Kumagai, A.; Schlacher, K.; Shevchenko, A.; Shevchenko, A.; Dunphy, W.G. Interaction of Chk1 with Treslin Negatively Regulates the Initiation of Chromosomal DNA Replication. Mol. Cell 2015, 57, 492–505. [Google Scholar] [CrossRef] [PubMed]
- Boos, D.; Sanchez-Pulido, L.; Rappas, M.; Pearl, L.H.; Oliver, A.W.; Ponting, C.P.; Diffley, J.F. Regulation of DNA Replication through Sld3-Dpb11 Interaction is Conserved from Yeast to Humans. Curr. Biol. 2011, 21, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Hassan, B.H.; Lindsey-Boltz, L.A.; Kemp, M.G.; Sancar, A. Direct Role for the Replication Protein Treslin (Ticrr) in the ATR Kinase-Mediated Checkpoint Response. J. Biol. Chem. 2013, 288, 18903–18910. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Besteiro, M.A.; Gottifredi, V. The Fork and the Kinase: A DNA Replication Tale from a CHK1 Perspective. Mutat. Res. Rev. Mutat. Res. 2015, 763, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Elvers, I.; Hagenkort, A.; Johansson, F.; Djureinovic, T.; Lagerqvist, A.; Schultz, N.; Stoimenov, I.; Erixon, K.; Helleday, T. CHK1 Activity is Required for Continuous Replication Fork Elongation but Not Stabilization of Post-Replicative Gaps After UV Irradiation. Nucleic Acids Res. 2012, 40, 8440–8448. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Betous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR Phosphorylates SMARCAL1 to Prevent Replication Fork Collapse. Genes Dev. 2013, 27, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Mayca Pozo, F.; Wisotsky, J.N.; Wang, B.; Jacobberger, J.W.; Zhang, Y. Phosphorylation of Minichromosome Maintenance 3 (MCM3) by Checkpoint Kinase 1 (Chk1) Negatively Regulates DNA Replication and Checkpoint Activation. J. Biol. Chem. 2015, 290, 12370–12378. [Google Scholar] [CrossRef] [PubMed]
- Chastain, P.D., 2nd; Heffernan, T.P.; Nevis, K.R.; Lin, L.; Kaufmann, W.K.; Kaufman, D.G.; Cordeiro-Stone, M. Checkpoint Regulation of Replication Dynamics in UV-Irradiated Human Cells. Cell. Cycle 2006, 5, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Unsal-Kacmaz, K.; Chastain, P.D.; Qu, P.P.; Minoo, P.; Cordeiro-Stone, M.; Sancar, A.; Kaufmann, W.K. The Human Tim/Tipin Complex Coordinates an Intra-S Checkpoint Response to UV that Slows Replication Fork Displacement. Mol. Cell. Biol. 2007, 27, 3131–3142. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.H.; Liu, Q.; Elledge, S.J.; Rosen, J.M. Chk1 is Haploinsufficient for Multiple Functions Critical to Tumor Suppression. Cancer Cell. 2004, 6, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tsao, C.C.; Goodman, B.K.; Furumai, R.; Tirado, C.A.; Abraham, R.T.; Wang, X.F. ATR Functions as a Gene Dosage-Dependent Tumor Suppressor on a Mismatch Repair-Deficient Background. EMBO J. 2004, 23, 3164–3174. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Baltimore, D. ATR Disruption Leads to Chromosomal Fragmentation and Early Embryonic Lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar] [PubMed]
- Tho, L.M.; Libertini, S.; Rampling, R.; Sansom, O.; Gillespie, D.A. Chk1 is Essential for Chemical Carcinogen-Induced Mouse Skin Tumorigenesis. Oncogene 2012, 31, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Branchet, M.C.; Boisnic, S.; Frances, C.; Robert, A.M. Skin Thickness Changes in Normal Aging Skin. Gerontology 1990, 36, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Fenske, N.A.; Lober, C.W. Structural and Functional Changes of Normal Aging Skin. J. Am. Acad. Dermatol. 1986, 15, 571–585. [Google Scholar] [CrossRef]
- Cerimele, D.; Celleno, L.; Serri, F. Physiological Changes in Ageing Skin. Br. J. Dermatol. 1990, 122 (Suppl. 35), 13–20. [Google Scholar] [CrossRef] [PubMed]
- Marks, R. Measurement of Biological Ageing in Human Epidermis. Br. J. Dermatol. 1981, 104, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Gilchrest, B.A.; Blog, F.B.; Szabo, G. Effects of Aging and Chronic Sun Exposure on Melanocytes in Human Skin. J. Invest. Dermatol. 1979, 73, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Snellman, E.; Bykov, V.J.; Jansen, C.T.; Hemminki, K. Effect of Age on the Formation and Repair of UV Photoproducts in Human Skin in Situ. Mutat. Res. 2000, 459, 195–202. [Google Scholar] [CrossRef]
- Hu, J.; Lieb, J.D.; Sancar, A.; Adar, S. Cisplatin DNA Damage and Repair Maps of the Human Genome at Single-Nucleotide Resolution. Proc. Natl. Acad. Sci. USA 2016, 113, 11507–11512. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Adar, S. The Cartography of UV-Induced DNA Damage Formation and DNA Repair. Photochem. Photobiol. 2016, 93, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Smerdon, M.J.; Roberts, S.A.; Wyrick, J.J. Chromosomal Landscape of UV Damage Formation and Repair at Single-Nucleotide Resolution. Proc. Natl. Acad. Sci. USA 2016, 113, 9057–9062. [Google Scholar] [CrossRef] [PubMed]
- Zavala, A.G.; Morris, R.T.; Wyrick, J.J.; Smerdon, M.J. High-Resolution Characterization of CPD Hotspot Formation in Human Fibroblasts. Nucleic Acids Res. 2014, 42, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; Denissenko, M.F.; Tang, M.S. PCR-Based Approaches to Adduct Analysis. Toxicol. Lett. 1998, 102–103, 447–451. [Google Scholar] [CrossRef]
- Besaratinia, A.; Pfeifer, G.P. Measuring the Formation and Repair of UV Damage at the DNA Sequence Level by Ligation-Mediated PCR. Methods Mol. Biol. 2012, 920, 189–202. [Google Scholar] [PubMed]
- Hart, R.W.; Setlow, R.B. DNA Repair in Late-Passage Human Cells. Mech. Ageing Dev. 1976, 5, 67–77. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA Repair during Aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.E. Variations in Excision Repair of UVB-Induced Pyrimidine Dimers in DNA of Human Skin in Situ. J. Invest. Dermatol. 1988, 90, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Setlow, R.B.; Setlow, J.K.; Carrier, W.L. Endonuclease from Micrococcus Luteus which has Activity Toward Ultraviolet-Irradiated Deoxyribonucleic Acid: Its Action on Transforming Deoxyribonucleic Acid. J. Bacteriol. 1970, 102, 187–192. [Google Scholar] [PubMed]
- Yamada, M.; Udono, M.U.; Hori, M.; Hirose, R.; Sato, S.; Mori, T.; Nikaido, O. Aged Human Skin Removes UVB-Induced Pyrimidine Dimers from the Epidermis More Slowly than Younger Adult Skin In Vivo. Arch. Dermatol. Res. 2006, 297, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Choi, J.H.; Gaddameedhi, S.; Kemp, M.G.; Reardon, J.T.; Sancar, A. Nucleotide Excision Repair in Human Cells: Fate of the Excised Oligonucleotide Carrying DNA Damage in Vivo. J. Biol. Chem. 2013, 288, 20918–20926. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Gaddameedhi, S.; Kim, S.Y.; Hu, J.; Kemp, M.G.; Sancar, A. Highly Specific and Sensitive Method for Measuring Nucleotide Excision Repair Kinetics of Ultraviolet Photoproducts in Human Cells. Nucleic Acids Res. 2014, 42, e29. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Gaddameedhi, S.; Choi, J.H.; Hu, J.; Sancar, A. DNA Repair Synthesis and Ligation Affect the Processing of Excised Oligonucleotides Generated by Human Nucleotide Excision Repair. J. Biol. Chem. 2014, 289, 26574–26583. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Kim, S.Y.; Kim, S.K.; Kemp, M.G.; Sancar, A. An Integrated Approach for Analysis of the DNA Damage Response in Mammalian Cells: Nucleotide Excision Repair, DNA Damage Checkpoint, and Apoptosis. J. Biol. Chem. 2015, 290, 28812–28821. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Kemp, M.G.; Choi, J.H. Detection of the Excised, Damage-Containing Oligonucleotide Products of Nucleotide Excision Repair in Human Cells. Photochem. Photobiol. 2017, 93, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Epstein, W.L.; Fukuyama, K.; Epstein, J.H. Early Effects of Ultraviolet Light on DNA Synthesis in Human Skin in Vivo. Arch. Dermatol. 1969, 100, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Spandau, D.F.; Simman, R.; Travers, J.B. Insulin-Like Growth Factor-1 Receptor Signaling is Required for Optimal ATR-CHK1 Kinase Signaling in UVB-Irradiated Human Keratinocytes. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kawasumi, M.; Lemos, B.; Bradner, J.E.; Thibodeau, R.; Kim, Y.S.; Schmidt, M.; Higgins, E.; Koo, S.W.; Angle-Zahn, A.; Chen, A.; et al. Protection from UV-Induced Skin Carcinogenesis by Genetic Inhibition of the Ataxia Telangiectasia and Rad3-Related (ATR) Kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 13716–13721. [Google Scholar] [CrossRef] [PubMed]
- Pavey, S.; Russell, T.; Gabrielli, B. G2 Phase Cell Cycle Arrest in Human Skin Following UV Irradiation. Oncogene 2001, 20, 6103–6110. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR Kinase Activities by the Radiosensitizing Agent, Caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar] [PubMed]
- Tavakkol, A.; Elder, J.T.; Griffiths, C.E.; Cooper, K.D.; Talwar, H.; Fisher, G.J.; Keane, K.M.; Foltin, S.K.; Voorhees, J.J. Expression of Growth Hormone Receptor, Insulin-Like Growth Factor 1 (IGF-1) and IGF-1 Receptor mRNA and Proteins in Human Skin. J. Invest. Dermatol. 1992, 99, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Jensen, P.J. Epidermal Growth Factor and Insulin-Like Growth Factor I Enhance Keratinocyte Migration. J. Invest. Dermatol. 1993, 100, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Barreca, A.; De Luca, M.; Del Monte, P.; Bondanza, S.; Damonte, G.; Cariola, G.; Di Marco, E.; Giordano, G.; Cancedda, R.; Minuto, F. In Vitro Paracrine Regulation of Human Keratinocyte Growth by Fibroblast-Derived Insulin-Like Growth Factors. J. Cell. Physiol. 1992, 151, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Siddle, K. Signalling by Insulin and IGF Receptors: Supporting Acts and New Players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef] [PubMed]
- Ferber, A.; Chang, C.; Sell, C.; Ptasznik, A.; Cristofalo, V.J.; Hubbard, K.; Ozer, H.L.; Adamo, M.; Roberts, C.T., Jr.; LeRoith, D. Failure of Senescent Human Fibroblasts to Express the Insulin-Like Growth Factor-1 Gene. J. Biol. Chem. 1993, 268, 17883–17888. [Google Scholar] [PubMed]
- Lewis, D.A.; Travers, J.B.; Machado, C.; Somani, A.K.; Spandau, D.F. Reversing the Aging Stromal Phenotype Prevents Carcinoma Initiation. Aging 2011, 3, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Ben-Porath, I.; Weinberg, R.A. The Signals and Pathways Activating Cellular Senescence. Int. J. Biochem. Cell Biol. 2005, 37, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Mallette, F.A.; Gaumont-Leclerc, M.F.; Ferbeyre, G. The DNA Damage Signaling Pathway is a Critical Mediator of Oncogene-Induced Senescence. Genes Dev. 2007, 21, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Suppressing Cancer: The Importance of being Senescent. Science 2005, 309, 886–887. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P. What has Senescence Got to do with Cancer? Cancer Cell. 2005, 7, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, C.; Hurwitz, S.A.; Kumar, M.G.; Cotton, J.; Spandau, D.F. Activation of the Insulin-Like Growth Factor-1 Receptor Promotes the Survival of Human Keratinocytes Following Ultraviolet B Irradiation. Int. J. Cancer 1999, 80, 431–438. [Google Scholar] [CrossRef]
- Fernandez, T.L.; Van Lonkhuyzen, D.R.; Dawson, R.A.; Kimlin, M.G.; Upton, Z. Insulin-Like Growth Factor-I and UVB Photoprotection in Human Keratinocytes. Exp. Dermatol. 2015, 24, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Yi, Q.; Travers, J.B.; Spandau, D.F. UVB-Induced Senescence in Human Keratinocytes Requires a Functional Insulin-Like Growth Factor-1 Receptor and p53. Mol. Biol. Cell 2008, 19, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Loesch, M.M.; Collier, A.E.; Southern, D.H.; Ward, R.E.; Tholpady, S.S.; Lewis, D.A.; Travers, J.B.; Spandau, D.F. Insulin-Like Growth Factor-1 Receptor Regulates Repair of Ultraviolet B-Induced DNA Damage in Human Keratinocytes in Vivo. Mol. Oncol. 2016, 10, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Barkley, L.R.; Day, T.; Bi, X.; Slater, D.M.; Alexandrow, M.G.; Nasheuer, H.P.; Vaziri, C. The Chk1-Mediated S-Phase Checkpoint Targets Initiation Factor Cdc45 via a Cdc25A/Cdk2-Independent Mechanism. J. Biol. Chem. 2006, 281, 30631–30644. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Sancar, A. ATR Kinase Inhibition Protects Non-Cycling Cells from the Lethal Effects of DNA Damage and Transcription Stress. J. Biol. Chem. 2016, 291, 9330–9342. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.A.; Keegan, K.S.; Herendeen, D.R.; Bentley, N.J.; Carr, A.M.; Hoekstra, M.F.; Concannon, P. Protein Kinase Mutants of Human ATR Increase Sensitivity to UV and Ionizing Radiation and Abrogate Cell Cycle Checkpoint Control. Proc. Natl. Acad. Sci. USA 1998, 95, 7445–7450. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, T.P.; Kawasumi, M.; Blasina, A.; Anderes, K.; Conney, A.H.; Nghiem, P. ATR-Chk1 Pathway Inhibition Promotes Apoptosis After UV Treatment in Primary Human Keratinocytes: Potential Basis for the UV Protective Effects of Caffeine. J. Invest. Dermatol. 2009, 129, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Cliby, W.A.; Roberts, C.J.; Cimprich, K.A.; Stringer, C.M.; Lamb, J.R.; Schreiber, S.L.; Friend, S.H. Overexpression of a Kinase-Inactive ATR Protein Causes Sensitivity to DNA-Damaging Agents and Defects in Cell Cycle Checkpoints. EMBO J. 1998, 17, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Nghiem, P.; Park, P.K.; Kim, Y.; Vaziri, C.; Schreiber, S.L. ATR Inhibition Selectively Sensitizes G1 Checkpoint-Deficient Cells to Lethal Premature Chromatin Condensation. Proc. Natl. Acad. Sci. USA 2001, 98, 9092–9097. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Gutierrez-Martinez, P.; Soria, R.; Fernandez-Capetillo, O. ATR Signaling can Drive Cells into Senescence in the Absence of DNA Breaks. Genes Dev. 2008, 22, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA Damage Response Induces Inflammation and Senescence by Inhibiting Autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed]
- Auclair, Y.; Rouget, R.; Affar el, B.; Drobetsky, E.A. ATR Kinase is Required for Global Genomic Nucleotide Excision Repair Exclusively during S Phase in Human Cells. Proc. Natl. Acad. Sci. USA 2008, 105, 17896–17901. [Google Scholar] [CrossRef] [PubMed]
- Belanger, F.; Angers, J.P.; Fortier, E.; Hammond-Martel, I.; Costantino, S.; Drobetsky, E.; Wurtele, H. Mutations in Replicative Stress Response Pathways are Associated with S Phase-Specific Defects in Nucleotide Excision Repair. J. Biol. Chem. 2016, 291, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Mahrenholz, A.; Lee, S.H. RPA Stabilizes the XPA-Damaged DNA Complex through Protein-Protein Interaction. Biochemistry 2000, 39, 6433–6439. [Google Scholar] [CrossRef] [PubMed]
- Reardon, J.T.; Sancar, A. Recognition and Repair of the Cyclobutane Thymine Dimer, a Major Cause of Skin Cancers, by the Human Excision Nuclease. Genes Dev. 2003, 17, 2539–2551. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Reardon, J.T.; Lindsey-Boltz, L.A.; Sancar, A. Mechanism of Release and Fate of Excised Oligonucleotides during Nucleotide Excision Repair. J. Biol. Chem. 2012, 287, 22889–22899. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Henricksen, L.A.; Wold, M.S.; Ingles, C.J. RPA Involvement in the Damage-Recognition and Incision Steps of Nucleotide Excision Repair. Nature 1995, 374, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lu, X.; Peterson, C.A.; Legerski, R.J. An Interaction between the DNA Repair Factor XPA and Replication Protein A Appears Essential for Nucleotide Excision Repair. Mol. Cell. Biol. 1995, 15, 5396–5402. [Google Scholar] [CrossRef] [PubMed]
- Stigger, E.; Drissi, R.; Lee, S.H. Functional Analysis of Human Replication Protein A in Nucleotide Excision Repair. J. Biol. Chem. 1998, 273, 9337–9343. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, M.; Sancar, A. Assembly, Subunit Composition, and Footprint of Human DNA Repair Excision Nuclease. Proc. Natl. Acad. Sci. USA 1998, 95, 6669–6674. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, M.; Sancar, A. Order of Assembly of Human DNA Repair Excision Nuclease. J. Biol. Chem. 1999, 274, 18759–18768. [Google Scholar] [CrossRef] [PubMed]
- Patrick, S.M.; Turchi, J.J. Replication Protein A (RPA) Binding to Duplex Cisplatin-Damaged DNA is Mediated through the Generation of Single-Stranded DNA. J. Biol. Chem. 1999, 274, 14972–14978. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Park, C.H.; Bessho, T.; Mu, D.; Sancar, A. Replication Protein A Confers Structure-Specific Endonuclease Activities to the XPF-ERCC1 and XPG Subunits of Human DNA Repair Excision Nuclease. J. Biol. Chem. 1996, 271, 11047–11050. [Google Scholar] [PubMed]
- de Laat, W.L.; Appeldoorn, E.; Sugasawa, K.; Weterings, E.; Jaspers, N.G.; Hoeijmakers, J.H. DNA-Binding Polarity of Human Replication Protein A Positions Nucleases in Nucleotide Excision Repair. Genes Dev. 1998, 12, 2598–2609. [Google Scholar] [CrossRef] [PubMed]
- Shivji, M.K.; Podust, V.N.; Hubscher, U.; Wood, R.D. Nucleotide Excision Repair DNA Synthesis by DNA Polymerase Epsilon in the Presence of PCNA, RFC, and RPA. Biochemistry 1995, 34, 5011–5017. [Google Scholar] [CrossRef] [PubMed]
- Overmeer, R.M.; Moser, J.; Volker, M.; Kool, H.; Tomkinson, A.E.; van Zeeland, A.A.; Mullenders, L.H.; Fousteri, M. Replication Protein A Safeguards Genome Integrity by Controlling NER Incision Events. J. Cell Biol. 2011, 192, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Guven, M.; Brem, R.; Macpherson, P.; Peacock, M.; Karran, P. Oxidative Damage to RPA Limits the Nucleotide Excision Repair Capacity of Human Cells. J. Invest. Dermatol. 2015, 135, 2834–2841. [Google Scholar] [CrossRef] [PubMed]
- Tsaalbi-Shtylik, A.; Moser, J.; Mullenders, L.H.; Jansen, J.G.; de Wind, N. Persistently Stalled Replication Forks Inhibit Nucleotide Excision Repair in Trans by Sequestering Replication Protein A. Nucleic Acids Res. 2014, 42, 4406–4413. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Rapp, A.; Berndt, N.; Staroske, W.; Schuster, M.; Dobrick-Mattheuer, M.; Kretschmer, S.; Konig, N.; Kurth, T.; Wieczorek, D.; et al. RPA and Rad51 Constitute a Cell Intrinsic Mechanism to Protect the Cytosol from Self DNA. Nat. Commun. 2016, 7, 11752. [Google Scholar] [CrossRef] [PubMed]
- Ramos-e-Silva, M.; da Silva Carneiro, S.C. Elderly Skin and its Rejuvenation: Products and Procedures for the Aging Skin. J. Cosmet. Dermatol. 2007, 6, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.; Lippitz, J. Chemical Peels, Dermabrasion, and Laser Therapy. Dis. Mon. 2009, 55, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Loesch, M.M.; Somani, A.K.; Kingsley, M.M.; Travers, J.B.; Spandau, D.F. Skin Resurfacing Procedures: New and Emerging Options. Clin. Cosmet. Investig. Dermatol. 2014, 7, 231–241. [Google Scholar] [PubMed]
- Spandau, D.F.; Lewis, D.A.; Somani, A.K.; Travers, J.B. Fractionated Laser Resurfacing Corrects the Inappropriate UVB Response in Geriatric Skin. J. Invest. Dermatol. 2012, 132, 1591–1596. [Google Scholar] [CrossRef] [PubMed]
- Krbanjevic, A.; Travers, J.B.; Spandau, D.F. How Wounding via Lasers has Potential Photocarcinogenic Preventative Effects via Dermal Remodeling. Curr. Dermatol. Rep. 2016, 5, 222–227. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kemp, M.G.; Spandau, D.F.; Travers, J.B. Impact of Age and Insulin-Like Growth Factor-1 on DNA Damage Responses in UV-Irradiated Human Skin. Molecules 2017, 22, 356. https://doi.org/10.3390/molecules22030356
Kemp MG, Spandau DF, Travers JB. Impact of Age and Insulin-Like Growth Factor-1 on DNA Damage Responses in UV-Irradiated Human Skin. Molecules. 2017; 22(3):356. https://doi.org/10.3390/molecules22030356
Chicago/Turabian StyleKemp, Michael G., Dan F Spandau, and Jeffrey B. Travers. 2017. "Impact of Age and Insulin-Like Growth Factor-1 on DNA Damage Responses in UV-Irradiated Human Skin" Molecules 22, no. 3: 356. https://doi.org/10.3390/molecules22030356
APA StyleKemp, M. G., Spandau, D. F., & Travers, J. B. (2017). Impact of Age and Insulin-Like Growth Factor-1 on DNA Damage Responses in UV-Irradiated Human Skin. Molecules, 22(3), 356. https://doi.org/10.3390/molecules22030356