
Looking Inside the Intramolecular C−H∙∙∙O Hydrogen Bond in Lactams Derived from α-Methylbenzylamine

Abstract
:1. Introduction
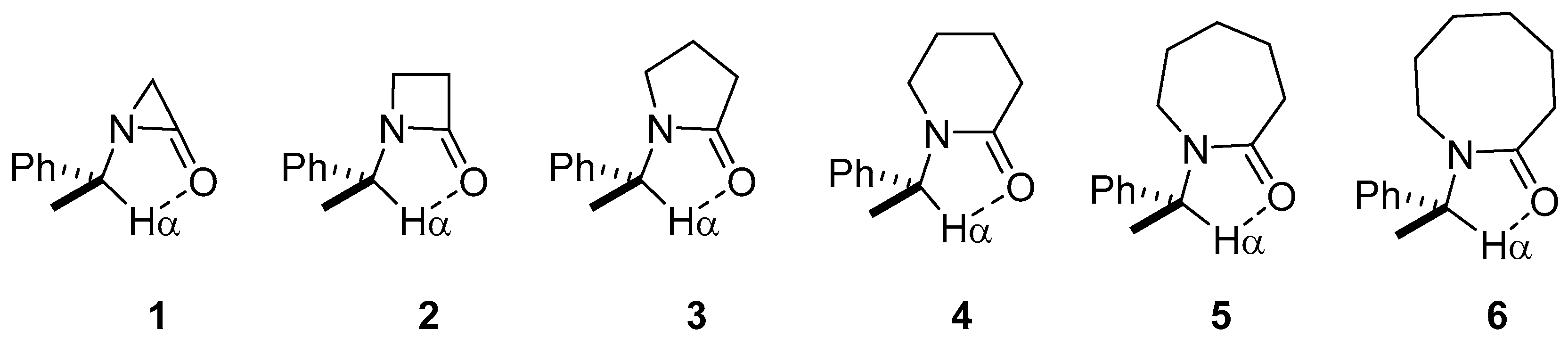
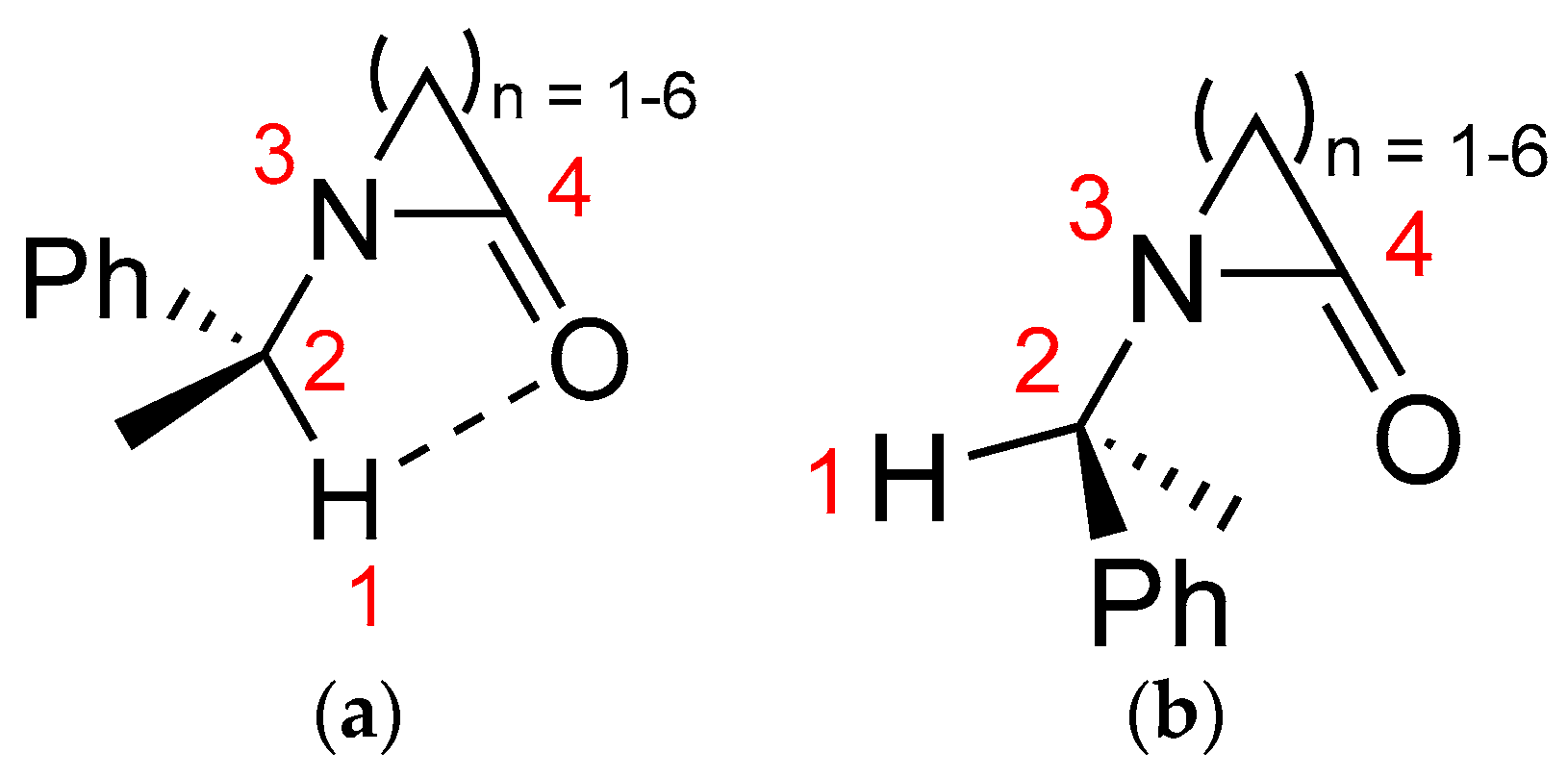
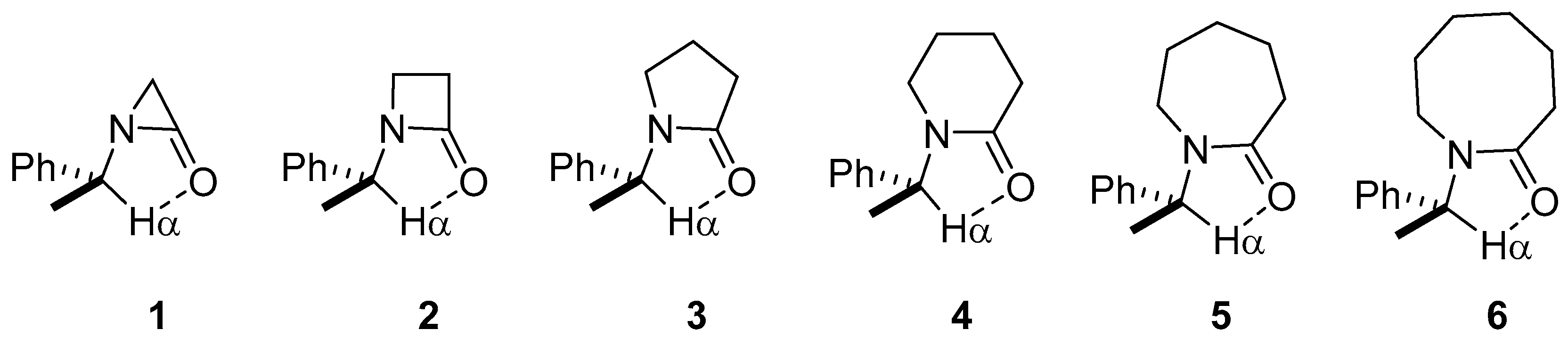
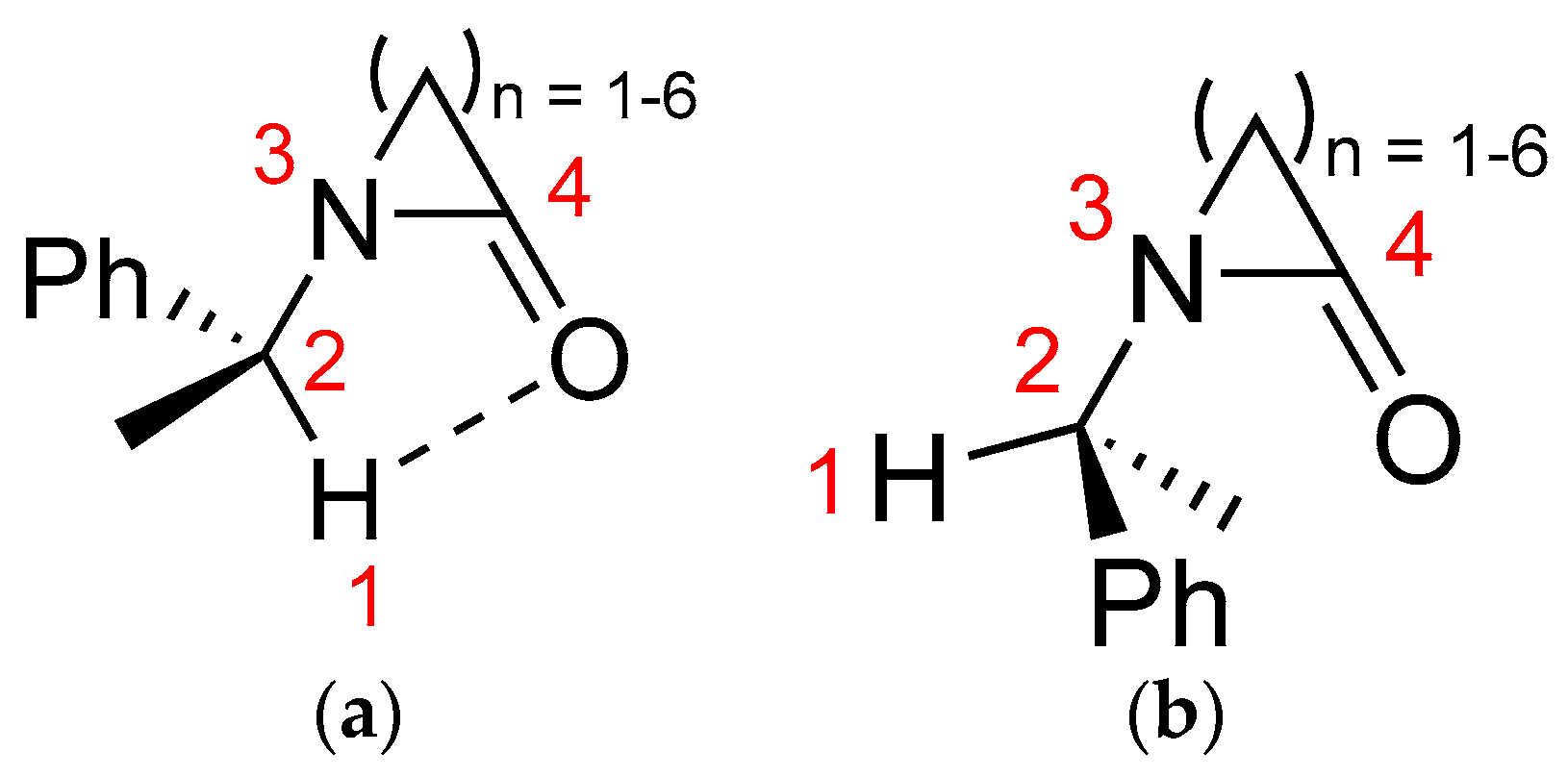
2. Results and Discussion
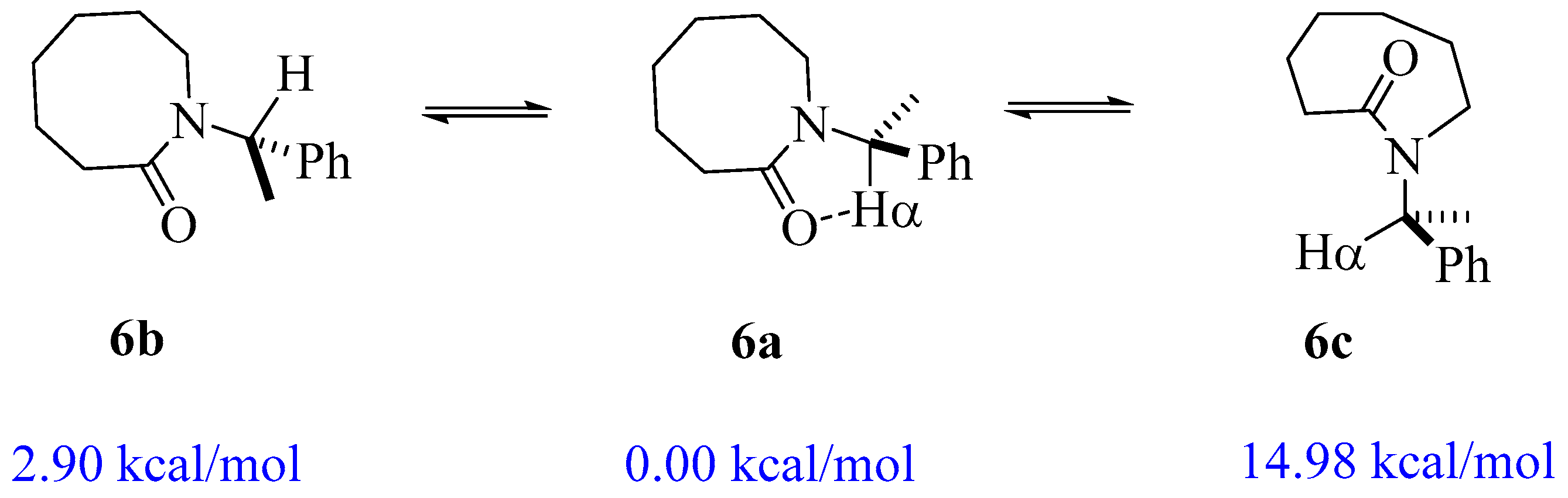
2.1. Energy and Geometrical Parameters
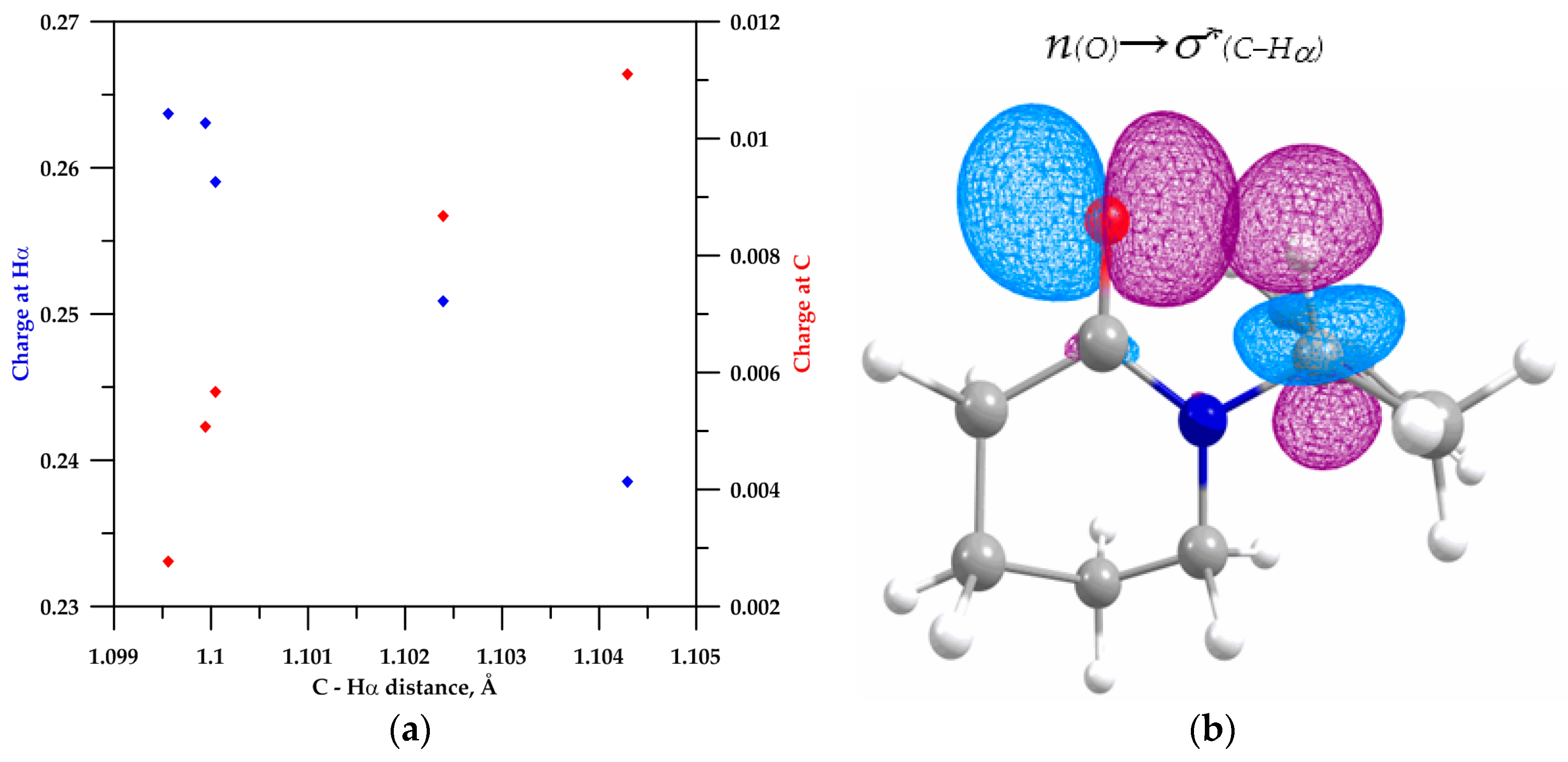
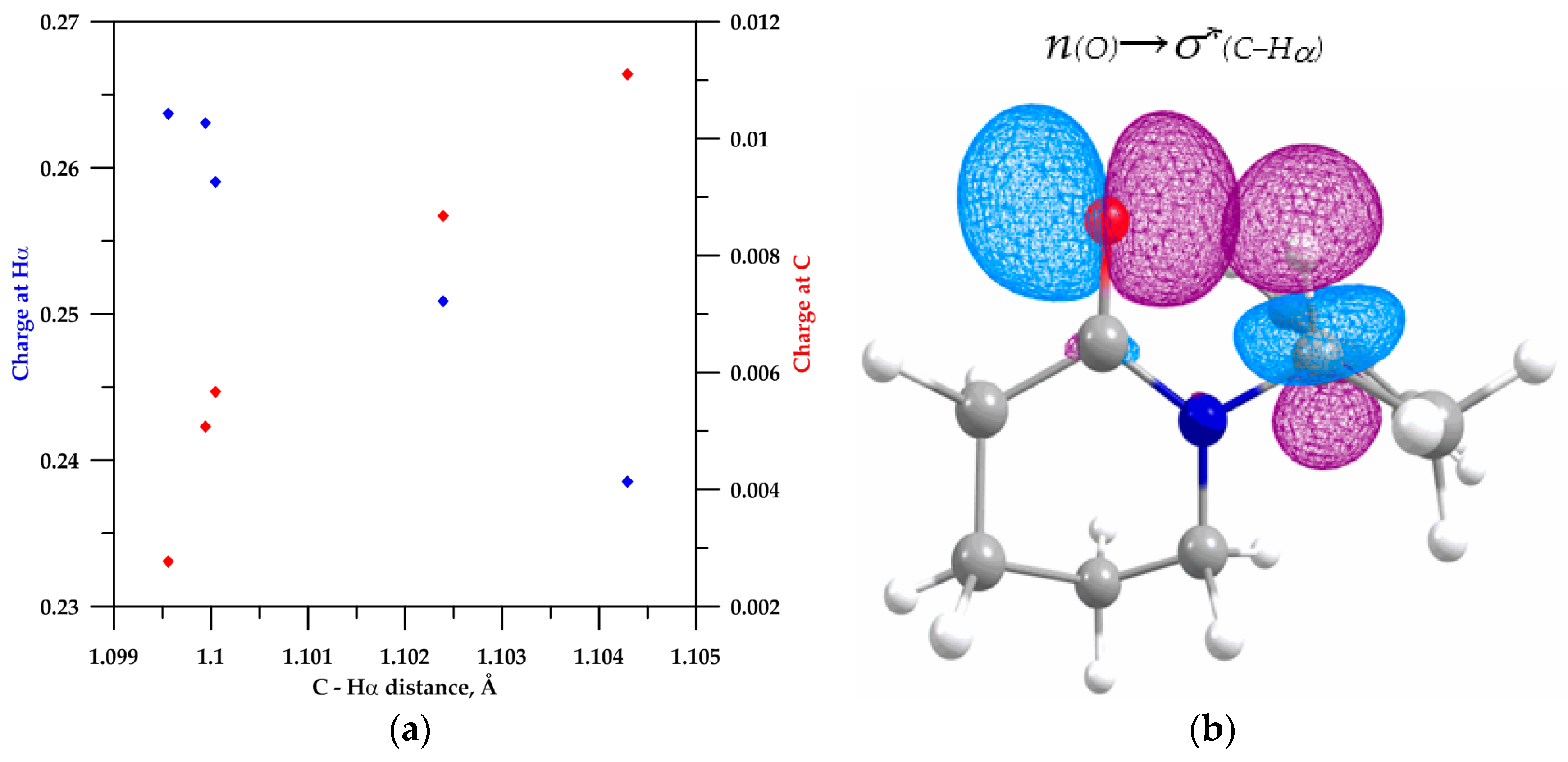
2.2. NBO Analysis
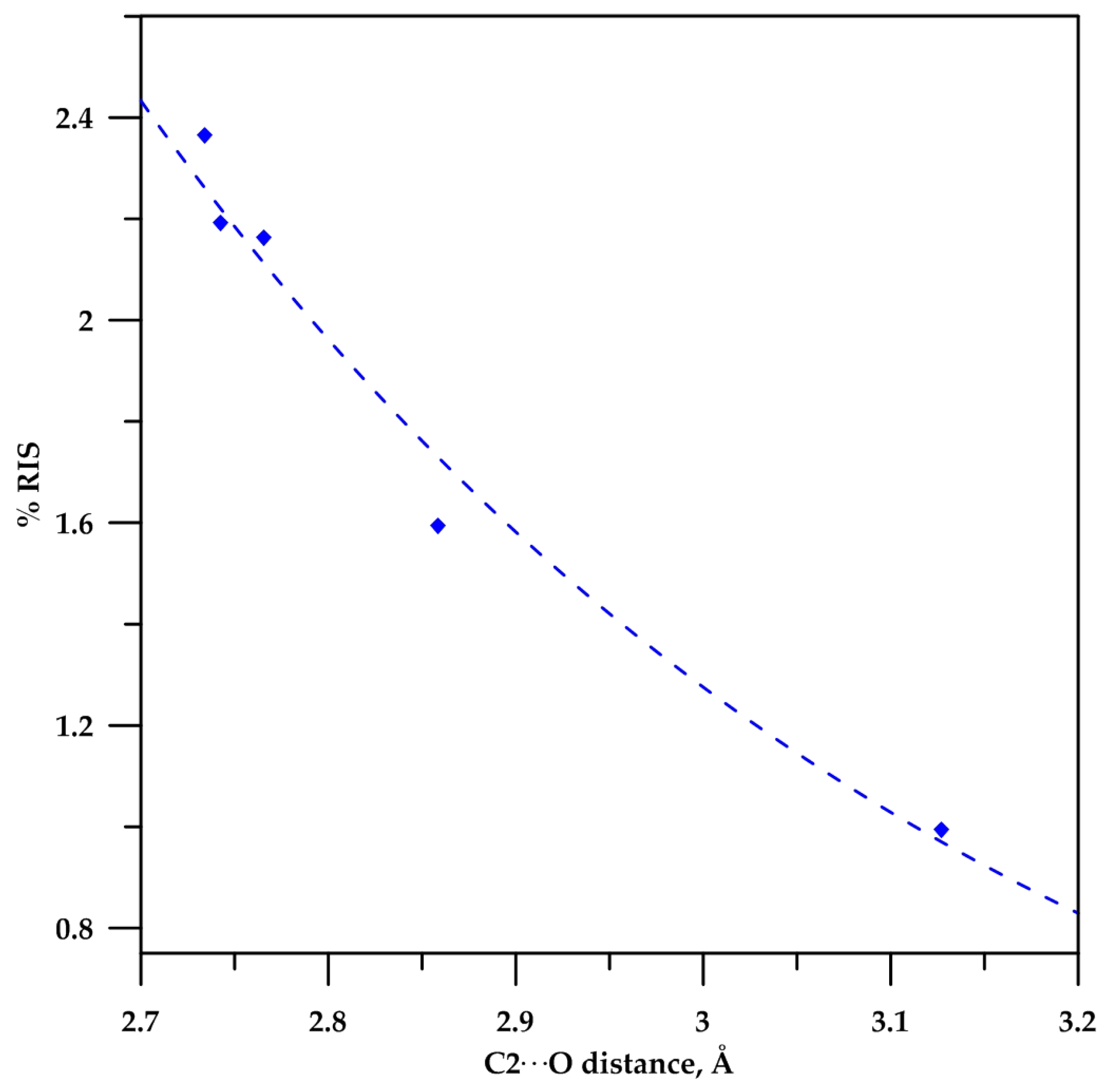
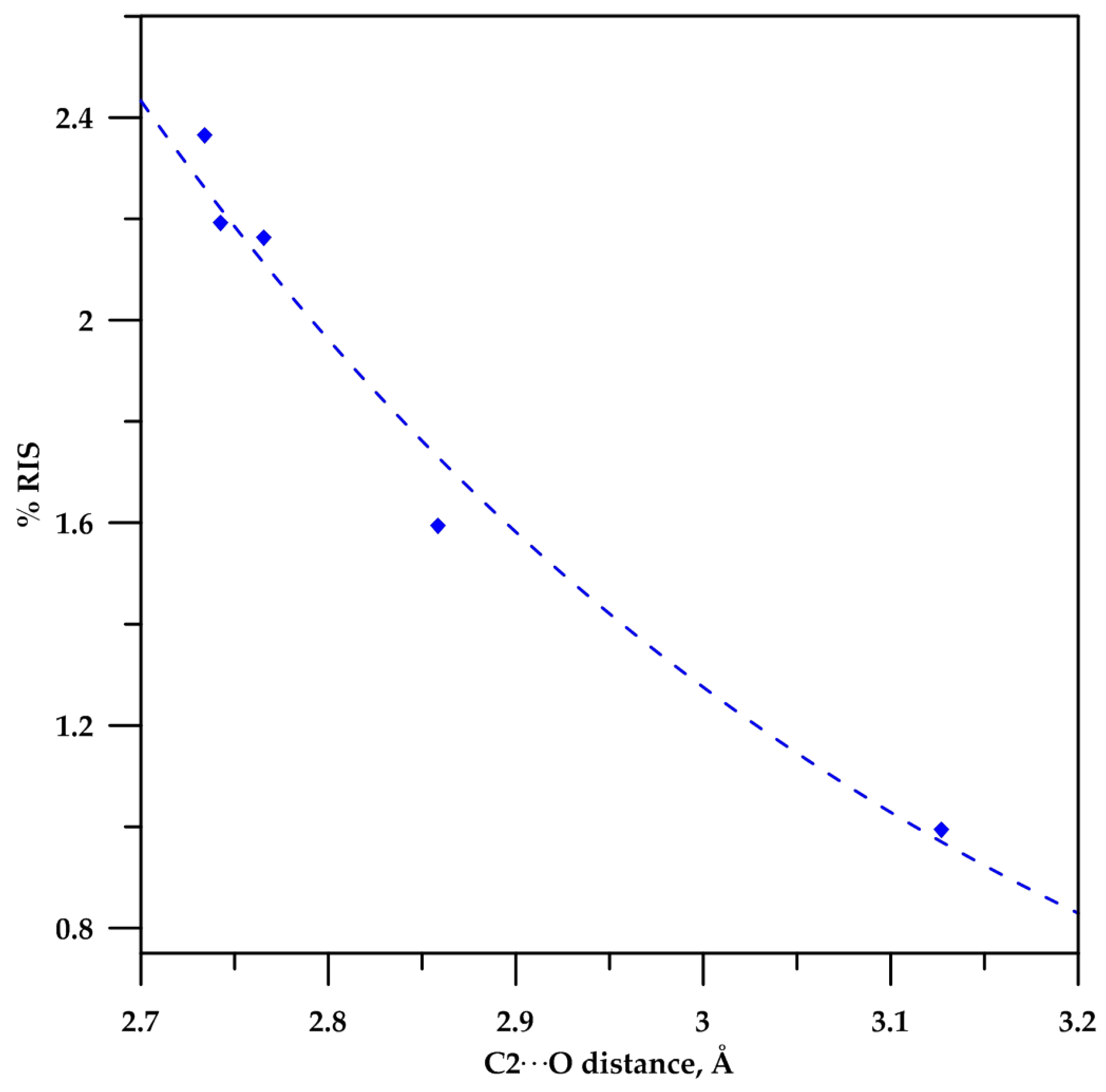
2.3. Analysis of the C−Hα Stretching
2.4. Analysis Based in Quantum Theory of Atoms in Molecules
4. Materials and Methods
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Desiraju, G.R. Crystal Gazing: Structure Prediction and Polymorphism. Science 1997, 278, 404–405. [Google Scholar] [CrossRef]
- Desiraju, G.R. The C−H···O Hydrogen Bond: Structural Implications and Supramolecular Design. Acc. Chem. Res. 1996, 29, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Steiner, T. Influence of C−H···O Interactions on the Conformation of Methyl Groups Quantified from Neutron Diffraction Data. J. Phys. Chem. A 2000, 104, 433–435. [Google Scholar] [CrossRef]
- Sheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Scheiner, S. Weak H-bonds. Comparisions of CH···O to NH···O proteins and PH···N to direct P···N interactions. Phys. Chem. Chem. Phys. 2011, 13, 13860–13872. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Analysis of Hydrogen Bonds in Crystals. Crystals 2016, 6, 59. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Subramanian, V.; Sathyamurthy, N. Hydrogen Bonding without Borders: An Atoms-in-Molecules Perspective. J. Phys. Chem. A 2006, 110, 3349–3351. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S.; Kar, T.; Gu, Y. Strength of the C-H···O Hydrogen Bond of Amino Acid Residues. J. Biol. Chem. 2001, 276, 9832–9837. [Google Scholar] [CrossRef] [PubMed]
- Shaw, N.; Cheng, C.; Tempel, W.; Chang, J.; Ng, J.; Wang, X.-Y.; Perrett, S.; Rose, J.; Rao, Z.; Wang, B.-C.; Liu, Z.-J. (NZ) CH···O Contacts assist crystallization of a ParB-like nuclease. BMC Struct. Biol. 2007, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Dissection of the Factors Affecting Formation of a CH···O H-Bond. A Case Study. Crystals 2015, 5, 327–345. [Google Scholar] [CrossRef]
- Jones, C.R.; Baruah, P.K.; Thompson, A.L.; Scheiner, S.; Smith, M.D. Can a C–H···O Interaction Be a Determinant of Conformation? J. Am. Chem. Soc. 2012, 134, 12064–12071. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Soria, V.; Sanchez, M.; Quintero, L.; Sartillo-Piscil, F. The 5-exo-trig radical cyclization reaction under reductive and oxidative conditions in the synthesis of optically pure GABA derivatives. Tetrahedron 2004, 60, 10809–10815. [Google Scholar] [CrossRef]
- Corey, E.J.; Rohde, J.J. The application of the formyl CH···O hydrogen bond postulate to the understanding of enantioselective reactions involving chiral boron lewis acids and aldehydes. Tetrahedron Lett. 1997, 38, 37–40. [Google Scholar] [CrossRef]
- Corey, E.J.; Rohde, J.J.; Fischer, A.; Azimioara, M.D. A hypothesis for conformational restriction in complexes of formyl compounds with boron Lewis acids. Experimental evidence for formyl CH···O and CH···F hydrogen bonds. Tetrahedron Lett. 1997, 38, 33–36. [Google Scholar] [CrossRef]
- Rodríguez-Soria, V.; Quintero, L.; Sartillo-Piscil, F. A novel stereoselective tin-free radical protocol for the enantioselective synthesis of pyrrolidinones and its application to the synthesis of biologically active GABA-derivatives. Tetrahedron 2008, 64, 2750–2754. [Google Scholar] [CrossRef]
- Rodríguez-Soria, V.; Quintero, L.; Sartillo-Piscil, F. Stereoselective 5-exo-trig radical cyclization in the enantioselective synthesis of Pregabalin. Tetrahedron Lett. 2007, 48, 4305–4308. [Google Scholar] [CrossRef]
- Sandoval-Lira, J.; Hernández-Pérez, J.M.; Sartillo-Piscil, F. Radical 1,4-phenyl migration favored by C-H···O interaction. A theoretical investigation. Tetrahedron Lett. 2012, 53, 6689–6693. [Google Scholar] [CrossRef]
- Fuentes, L.; Quintero, L.; Cordero-Vargas, A.; Eustaquio, C.; Terán, J.L.; Sartillo-Piscil, F. The amide bond rotation controlled by an unusual C–H···O hydrogen bonding that favors the 1,4-phenyl radical migration. Tetrahedron Lett. 2011, 52, 3630–3632. [Google Scholar] [CrossRef]
- Alkayar, Z.T.; Adams, H.; Coldham, I. Regiochemical and Stereochemical Studies of the Intramolecular Dipolar Cycloaddition of Nitrones Derived from Quaternary Aldehydes. Synlett 2016, 27, 447–449. [Google Scholar]
- Alabugin, I.V.; Gilmore, K. Finding the right path: Baldwin “Rules for Ring Closure” and stereoelectronic control of cyclizations. Chem. Commun. 2013, 49, 11246–11250. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Lira, J.; Fuentes, L.; Quintero, L.; Höpfl, H.; Hernández-Pérez, J.M.; Terán, J.L.; Sartillo-Piscil, F. The Stabilizing Role of the Intramolecular C-H···O Hydrogen Bond in Cyclic Amides Derived From α-Methylbenzylamine. J. Org. Chem. 2015, 80, 4481–4490. [Google Scholar] [CrossRef] [PubMed]
- Jaen, J.C.; Hart, A.C. (S)-α-Methylbenzylamine. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Hobza, P.; Havlas, Z. Blue-Shifting Hydrogen Bonds. Chem. Rev. 2000, 100, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Jorly, J.; Eluvathingal, D.J. Red-, Blue-, or No-Shift in Hydrogen Bonds: A Unified Explanation. J. Am. Chem. Soc. 2007, 129, 4620–4632. [Google Scholar]
- Alabugin, I.V.; Manoharan, M.; Peabody, S.; Weinhold, F. Electronic basis of improper hydrogen bonding: A subtle balance of hyperconjugation and rehybridization. J. Am. Chem. Soc. 2003, 125, 5973–5987. [Google Scholar] [CrossRef] [PubMed]
- Alabugin, I.V.; Bresch, S.; Manoharan, M. Hybridization Trends for Main Group Elements and Expanding the Bent’s Rule beyond Carbon: More than Electronegativity. J. Phys. Chem. A 2014, 118, 3663–3677. [Google Scholar] [CrossRef] [PubMed]
- Alabugin, I.V.; Bresch, S.; Passos dos Gomes, G. Orbital hybridization: A key electronic factor in control of structure and reactivity. J. Phys. Org. Chem. 2014, 28, 147–162. [Google Scholar] [CrossRef]
- Jyothish, J.; Jose, A.; Jemmis, E.D. Continuum in the X-Z---Y weak bonds: Z = main group elements. J. Comput. Chem. 2016, 37, 270–279. [Google Scholar]
- Bent, H.A. An Appraisal of Valence-bond Structures and Hybridization in Compounds of the First-row elements. Chem. Rev. 1961, 61, 275–311. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Schlegel, H.B. On the Physical Origin of Blue-Shifted Hydrogen Bonds. J. Am. Chem. Soc. 2002, 124, 9639–9647. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.M.; Liu, L.; Guo, Q.-X. Substituent effects on the blue-shifted hydrogen bonds. Chem. Phys. Lett. 2002, 365, 464–472. [Google Scholar] [CrossRef]
- Yang, Y. Coexistence of S–H···O and N–H···O Blue-shifted Hydrogen Bonds in a Small System: HSO···HNO. Chem. Lett. 2008, 37, 1070–1071. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near hartree-fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F.; Curtiss, L.A.; Pochatko, D.J. Natural bond orbital analysis of molecular interactions: Theoretical studies of binary complexes of HF, H2O, NH3, N2, O2, F2, CO and CO2 with HF, H2O, and NH3. J. Chem. Phys. 1986, 84, 5687–5705. [Google Scholar] [CrossRef]
- Chang, X.; Zhang, Y.; Weng, X.; Su, P.; Wu, W.; Mo, Y. Red-Shifting versus Blue-Shifting Hydrogen Bonds: Perspective from Ab Initio Valence Bond Theory. J. Phys. Chem. A 2016, 120, 2749–2756. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules, A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. A 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Solano-Altamirano, J.M.; Hernández-Pérez, J.M. DensToolKit: A comprehensive open-source package for analyzing the electron density and its derivative scalar and vector fields. Comput. Phys. Commun. 2015, 196, 362–371. [Google Scholar] [CrossRef]
- Sample Availability: Not available.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lactam | Φ, Open (°) | Φ, Closed (°) | ΔE (kcal/mol) |
|---|---|---|---|
| 1 | 166.71 | 33.415 | 1.20 |
| 2 | 163.10 | 28.003 | 0.34 |
| 3 | 156.24 | 20.410 | −2.19 |
| 4 | 166.56 | 9.837 | −2.11 |
| 5 | 156.39 | 8.575 | −4.04 |
| 6 | 157.02 | 3.847 | −2.90 |
| Lactam | O…H (Å) | C−H (Å) | C2...O (Å) | C−H...O (°) |
|---|---|---|---|---|
| 1 | 3.3600 | 1.1105 | 3.3501 | 79.97 |
| 2 | 2.7605 | 1.1042 | 3.1269 | 98.85 |
| 3 | 2.3449 | 1.1023 | 2.8585 | 106.37 |
| 4 | 2.1785 | 1.0999 | 2.7425 | 109.07 |
| 5 | 2.1933 | 1.1000 | 2.7655 | 109.71 |
| 6 | 2.1390 | 1.0995 | 2.7340 | 111.09 |
| Lactam | C−Hα (Å) | s-Character % at C2 | Polarization of C−Hα Bond | Charge (u.a.) | Population σ*(C−Hα) | E(2) (kcal/mol) | ||
|---|---|---|---|---|---|---|---|---|
| % C | % H | at C | at Hα | |||||
| 4b | 1.1046 | 20.99 | 60.43 | 39.57 | 0.01615 | 0.20563 | 0.01615 | - |
| 2 | 1.1042 | 22.05 | 62.010 | 37.990 | 0.01694 | 0.23853 | 0.01694 | 0.11 |
| 3 | 1.1023 | 22.49 | 62.750 | 37.350 | 0.01781 | 0.25087 | 0.01781 | 0.75 |
| 4 | 1.0999 | 22.87 | 63.390 | 36.610 | 0.01889 | 0.26308 | 0.01889 | 1.26 |
| 5 | 1.1000 | 22.75 | 63.190 | 36.810 | 0.01826 | 0.25905 | 0.01826 | 1.29 |
| 6 | 1.0995 | 22.76 | 63.440 | 36.560 | 0.01888 | 0.26374 | 0.01888 | 1.60 |
| Lactam | Conformers a | Conformers b | |
|---|---|---|---|
| ν (cm−1) | ν (cm−1) | %RIS | |
| 2 | 3105.26 | 3074.69 | 0.99 |
| 3 | 3123.34 | 3074.32 | 1.59 |
| 4 | 3145.59 | 3078.07 | 2.19 |
| 5 | 3145.03 | 3078.42 | 2.16 |
| 6 | 3147.57 | 3074.81 | 2.37 |
| Lactam | ρ (BCP) | ∇2ρ(r) (BCP) |
|---|---|---|
| 4 | 0.0226 | 0.0843 |
| 5 | 0.0219 | 0.0816 |
| 6 | 0.0241 | 0.0870 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mejía, S.; Hernández-Pérez, J.M.; Sandoval-Lira, J.; Sartillo-Piscil, F. Looking Inside the Intramolecular C−H∙∙∙O Hydrogen Bond in Lactams Derived from α-Methylbenzylamine. Molecules 2017, 22, 361. https://doi.org/10.3390/molecules22030361
Mejía S, Hernández-Pérez JM, Sandoval-Lira J, Sartillo-Piscil F. Looking Inside the Intramolecular C−H∙∙∙O Hydrogen Bond in Lactams Derived from α-Methylbenzylamine. Molecules. 2017; 22(3):361. https://doi.org/10.3390/molecules22030361
Chicago/Turabian StyleMejía, Sandra, Julio M. Hernández-Pérez, Jacinto Sandoval-Lira, and Fernando Sartillo-Piscil. 2017. "Looking Inside the Intramolecular C−H∙∙∙O Hydrogen Bond in Lactams Derived from α-Methylbenzylamine" Molecules 22, no. 3: 361. https://doi.org/10.3390/molecules22030361
APA StyleMejía, S., Hernández-Pérez, J. M., Sandoval-Lira, J., & Sartillo-Piscil, F. (2017). Looking Inside the Intramolecular C−H∙∙∙O Hydrogen Bond in Lactams Derived from α-Methylbenzylamine. Molecules, 22(3), 361. https://doi.org/10.3390/molecules22030361