
Chroman-4-One Derivatives Targeting Pteridine Reductase 1 and Showing Anti-Parasitic Activity

 ,
,  ,
,  , ,
, , 
 , ,
, ,  , and
, and 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Inhibitory Activity towards PTR1
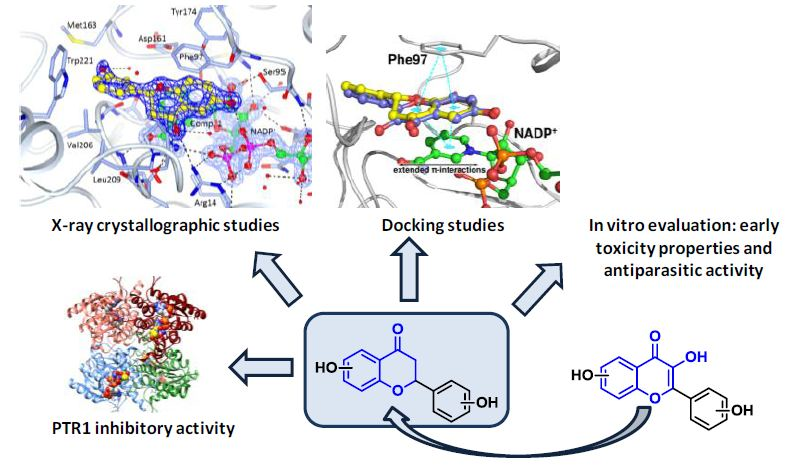
2.2. Crystallographic Structures of LmPTR1 and TbPTR1 in Complex with Flavanones
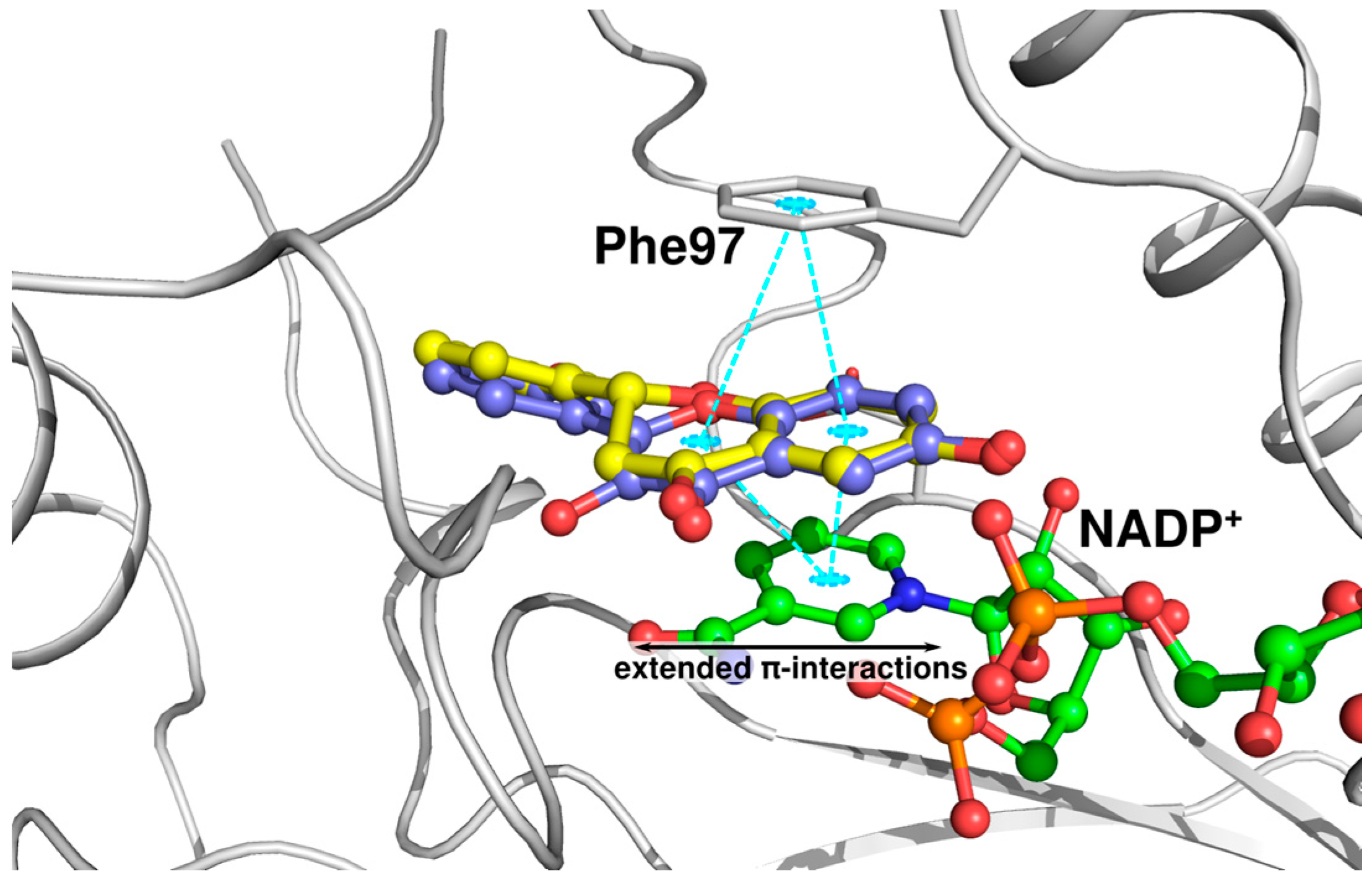
2.3. Docking Studies and Crystal Structure Comparison
2.4. Biological Profile of Compounds 1–3
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Compounds 1–3
3.3. Computational Methodology
3.4. Protein Purification and Kinetic Profile Characterization
3.5. TbPTR1 and LmPTR1 Enzyme Assay
3.6. Crystallization
3.7. Data Collection, Structure Solution and Refinement
3.8. In Vitro Evaluation of Activity against L. infantum Intramacrophage Amastigotes
3.9. In Vitro Evaluation of Activity against T. brucei
3.10. Cytotoxicity Assessment against THP-1 Macrophages
3.11. hERG Assay
3.12. Cytochrome P450 1A2, 2C9, 2C19, 2D6 and 3A4 Assays
3.13. Cytotoxicity Assay against A549 and W1-38 Cells
3.14. Aurora B Kinase Assay
3.15. Assessment of Mitochondrial Toxicity
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cooper, D.R.; Porebski, P.J.; Chruszcz, M.; Minor, W. X-ray crystallography: Assessment and validation of protein-small molecule complexes for drug discovery. Expert Opin. Drug Discov. 2011, 6, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Castillo, E.; Dea-Ayuela, M.A.; Bolás-Fernández, F.; Rangel, M.; González-Rosende, M.E. The kinetoplastid chemotherapy revisited: Current drugs, recent advances and future perspectives. Curr. Med. Chem. 2010, 17, 4027–4051. [Google Scholar] [CrossRef] [PubMed]
- Machado-Silva, A.; Guimarães, P.P.; Tavares, C.A.; Sinisterra, R.D. New perspectives for leishmaniasis chemotherapy over current anti-leishmanial drugs: A patent landscape. Expert Opin. Ther. Pat. 2015, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Barret, M.P.; Vincent, I.M.; Burchmore, R.J.; Kazibwe, A.J.; Matovu, E. Drug resistance in human African trypanosomiasis. Future Microbiol. 2011, 6, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Chatelain, E.; Ioset, J.R. Drug discovery and development for neglected diseases: The DNDi model. Drug Des. Dev. Ther. 2011, 5, 175–181. [Google Scholar] [PubMed]
- Gilbert, I.H. Drug discovery for neglected diseases: Molecular target-based and phenotypic approaches. J. Med. Chem. 2013, 56, 7719–7726. [Google Scholar] [CrossRef] [PubMed]
- Borsari, C.; Santarem, N.; Torrado, J.; Olías, A.I.; Corral, M.J.; Baptista, C.; Gul, S.; Wolf, M.; Kuzikov, M.; Ellinger, B.; et al. Methoxylated 2′-hydroxychalcones as antiparasitic hit compounds. Eur. J. Med. Chem. 2017, 126, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.E. Exploring the folate pathway in Plasmodium falciparum. Acta Trop. 2005, 94, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, I.H. Inhibitors of dihydrofolate reductase in Leishmania and trypanosomes. Biochim. Biophys. Acta 2002, 1587, 249–257. [Google Scholar] [CrossRef]
- Dawson, A.; Gibellini, F.; Sienkiewicz, N.; Tulloch, L.B.; Fyfe, P.K.; McLuskey, K.; Fairlamb, A.H.; Hunter, W.N. Structure and reactivity of Trypanosoma brucei pteridine reductase: Inhibition by the archetypal antifolate methotrexate. Mol. Microbiol. 2006, 61, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Guerrieri, D.; Ferrari, S.; Costi, M.P.; Michels, P.A. Biochemical effects of riluzole on Leishmania parasites. Exp. Parasitol. 2013, 133, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Barrack, K.L.; Tulloch, L.B.; Burke, L.A.; Fyfe, P.K.; Hunter, W.N. Structure of recombinant Leishmania donovani pteridine reductase reveals a disordered active site. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Morandi, F.; Motiejunas, D.; Nerini, E.; Henrich, S.; Luciani, R.; Venturelli, A.; Lazzari, S.; Calò, S.; Gupta, S.; et al. Virtual screening identification of non folate compounds, including a CNS drug, as antiparasitic agents inhibiting pteridine reductase. J. Med. Chem. 2011, 54, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Borsari, C.; Luciani, R.; Pozzi, C.; Pöhner, I.; Henrich, S.; Trande, M.; Cordeiro-da-Silva, A.; Santarém, N.; Baptista, C.; Tait, A.; et al. Profiling of flavonol derivatives for the development of anti-trypanosomatidic drugs. J. Med. Chem. 2016, 59, 7589–7616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sienkiewicz, N.; Ong, H.B.; Fairlamb, A.H. Trypanosoma brucei pteridine reductase 1 is essential for survival in vitro and for virulence in mice. Mol. Microbiol. 2010, 77, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Cavazzuti, A.; Paglietti, G.; Hunter, W.N.; Gamarro, F.; Piras, S.; Loriga, M.; Allecca, S.; Corona, P.; McLuskey, K.; Tulloch, L.; et al. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. USA 2008, 105, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalaf, A.I.; Huggan, J.K.; Suckling, C.J.; Gibson, C.L.; Stewart, K.; Giordani, F.; Barrett, M.P.; Wong, P.E.; Barrack, K.L.; Hunter, W.N. Structure-based design and synthesis of antiparasitic pyrrolopyrimidines targeting pteridine reductase 1. J. Med. Chem. 2014, 57, 6479–6494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tulloch, L.B.; Martini, V.P.; Iulek, J.; Huggan, J.K.; Lee, J.H.; Gibson, C.L.; Smith, T.K.; Suckling, C.J.; Hunter, W.N. Structure-based design of pteridine reductase inhibitors targeting African sleeping sickness and the leishmaniases. J. Med. Chem. 2010, 53, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Mpamhanga, C.P.; Spinks, D.; Tulloch, L.B.; Shanks, E.J.; Robinson, D.A.; Collie, I.T.; Fairlamb, A.H.; Wyatt, P.G.; Frearson, J.A.; Hunter, W.N.; et al. One scaffold, three binding modes: Novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J. Med. Chem. 2009, 52, 4454–4465. [Google Scholar] [CrossRef] [PubMed]
- Drinkwater, N.; Vu, H.; Lovell, K.M.; Criscione, K.R.; Collins, B.M.; Prisinzano, T.E.; Poulsen, S.A.; McLeish, M.J.; Grunewald, G.L.; Martin, J.L. Fragment-based screening by X-ray crystallography, MS and isothermal titration calorimetry to identify PNMT (phenylethanolamine N-methyltransferase) inhibitors. Biochem. J. 2010, 431, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, L.; Luciani, R.; Borsari, C.; Ferrari, S.; Costi, M.P.; Cruciani, G.; Spyrakis, F. Comparing Drug Images and Repurposing Drugs with BioGPS and FLAPdock: The Thymidylate Synthase Case. ChemMedChem 2016, 11, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J. Mol. Biol. 2005, 352, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Gourley, D.G.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Crystallization of recombinant Leishmania major pteridine reductase 1 (PTR1). Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 1608–1610. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2015-4: Maestro, Version 10.4; Schrödinger, LLC: New York, NY, USA, 2015.
- Sanschagrin, P.C.; Kuhn, L.A. Cluster Analysis of Consensus Water Sites in Thrombin and Trypsin Shows Conservation between Serine Proteases and Contributions to Ligand Specificity. Prot. Sci. 1998, 7, 2054–2064. [Google Scholar] [CrossRef] [PubMed]
- Small-Molecule Drug Discovery Suite 2015-4: Glide, Version 6.9; Schrödinger, LLC: New York, NY, USA, 2015.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 1.7.4.4; Schrödinger, LLC: New York, NY, USA, 2015.
- Benvenuti, M.; Mangani, S. Crystallization of soluble proteins in vapor diffusion for X-ray crystallography. Nat. Protoc. 2007, 2, 1633–1651. [Google Scholar] [CrossRef] [PubMed]
- Leslie, A.G.W. The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 48–57. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. An approach to multi-copy search in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 1622–1624. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.B.; Winn, M.D.; Ballard, C.C.; Ashton, A.W.; Patel, P.; Potterton, E.A.; McNicholas, S.J.; Cowtan, K.D.; Emsley, P. The new CCP4 Coordinate Library as a toolkit for the design of coordinate-related applications in protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2250–2255. [Google Scholar] [CrossRef] [PubMed]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Perrakis, A.; Morris, R.; Lamzin, V.S. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999, 6, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Moss, D.S.; Thornton, J.M. Main-chain bond lengths and bond angles in protein structures. J. Mol. Biol. 1993, 231, 1049–1067. [Google Scholar] [CrossRef] [PubMed]
- Potterton, L.; McNicholas, S.; Krissinel, E.; Gruber, J.; Cowtan, K.; Emsley, P.; Murshudov, G.N.; Cohen, S.; Perrakis, A.; Noble, M. Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2288–2294. [Google Scholar] [CrossRef] [PubMed]
- Siqueira-Neto, J.L.; Moon, S.; Jang, J.; Yang, G.; Lee, C.; Moon, H.K.; Chatelain, E.; Genovesio, A.; Cechetto, J.; Freitas-Junior, L.H. An Image-Based High-Content Screening Assay for Compounds Targeting Intracellular Leishmania donovani Amastigotes in Human Macrophages. PLoS Negl. Trop. Dis. 2012, 6, e1671. [Google Scholar] [CrossRef] [PubMed]
- Bowling, T.; Mercer, L.; Don, R.; Jacobs, R.; Nare, B. Application of a resazurin-based high-throughput screening assay for the identification and progression of new treatments for human African trypanosomiasis. Int. J. Parasitol. Drugs Resist. 2012, 2, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Piper, D.R.; Duff, S.R.; Eliason, H.C.; Frazee, W.J.; Frey, E.A.; Fuerstenau-Sharp, M.; Jachec, C.; Marks, B.D.; Pollok, B.A.; Shekhani, M.S.; et al. Development of the predictor HERG fluorescence polarization assay using a membrane protein enrichment approach. Assay Drug. Dev. Technol. 2008, 6, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Cali, J.J.; Ma, D.; Sobol, M.; Simpson, D.J.; Frackman, S.; Good, T.D.; Daily, W.J.; Liu, D. Luminogenic cytochrome P450 assays. Expert Opin. Drug Metab. Toxicol. 2006, 2, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Xue, K.; Gu, J.J.; Zhang, Q.; Mavis, C.; Hernandez-Ilizaliturri, F.J.; Czuczman, M.S.; Guo, Y. Vorinostat, a histonedeacetylase (HDAC) inhibitor, promotes cell cycle arrest and re-sensitizes rituximab- and chemo-resistant lymphoma cells to chemotherapy agents. J. Cancer Res. Clin. Oncol. 2016, 142, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Tanega, C.; Shen, M.; Mott, B.T.; Thomas, C.J.; MacArthur, R.; Inglese, J.; Auld, D.S. Comparison of bioluminescent kinase assays using substrate depletion and product formation. Assay Drug Dev. Technol. 2009, 7, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Pendergrass, W.; Wolf, N.; Poot, M. Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry A 2004, 61, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 TbPTR1 (μM) | IC50 LmPTR1 (μM) |
|---|---|---|
| 1 | 31.0 | 57.0 |
| 2 | 133.0 | 35.0 |
| 3 | 82.0 | 36.0 |
| 1A | 4.3 | 12.5 |
| 2A | NI at 50 μM | NI at 50 μM |
| 3A | 38.0 | 35.0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pisa, F.; Landi, G.; Dello Iacono, L.; Pozzi, C.; Borsari, C.; Ferrari, S.; Santucci, M.; Santarem, N.; Cordeiro-da-Silva, A.; Moraes, C.B.; et al. Chroman-4-One Derivatives Targeting Pteridine Reductase 1 and Showing Anti-Parasitic Activity. Molecules 2017, 22, 426. https://doi.org/10.3390/molecules22030426
Di Pisa F, Landi G, Dello Iacono L, Pozzi C, Borsari C, Ferrari S, Santucci M, Santarem N, Cordeiro-da-Silva A, Moraes CB, et al. Chroman-4-One Derivatives Targeting Pteridine Reductase 1 and Showing Anti-Parasitic Activity. Molecules. 2017; 22(3):426. https://doi.org/10.3390/molecules22030426
Chicago/Turabian StyleDi Pisa, Flavio, Giacomo Landi, Lucia Dello Iacono, Cecilia Pozzi, Chiara Borsari, Stefania Ferrari, Matteo Santucci, Nuno Santarem, Anabela Cordeiro-da-Silva, Carolina B. Moraes, and et al. 2017. "Chroman-4-One Derivatives Targeting Pteridine Reductase 1 and Showing Anti-Parasitic Activity" Molecules 22, no. 3: 426. https://doi.org/10.3390/molecules22030426
APA StyleDi Pisa, F., Landi, G., Dello Iacono, L., Pozzi, C., Borsari, C., Ferrari, S., Santucci, M., Santarem, N., Cordeiro-da-Silva, A., Moraes, C. B., Alcantara, L. M., Fontana, V., Freitas-Junior, L. H., Gul, S., Kuzikov, M., Behrens, B., Pöhner, I., Wade, R. C., Costi, M. P., & Mangani, S. (2017). Chroman-4-One Derivatives Targeting Pteridine Reductase 1 and Showing Anti-Parasitic Activity. Molecules, 22(3), 426. https://doi.org/10.3390/molecules22030426