
A Direct Method for ?-Selective Glycosylation with an N-Acetylglucosamine Donor Armed by a 4-O-TBDMS Protecting Group


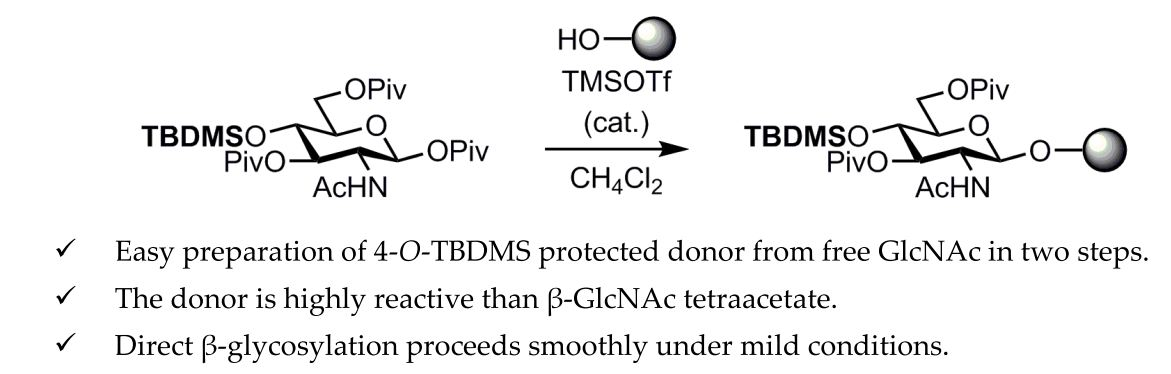
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
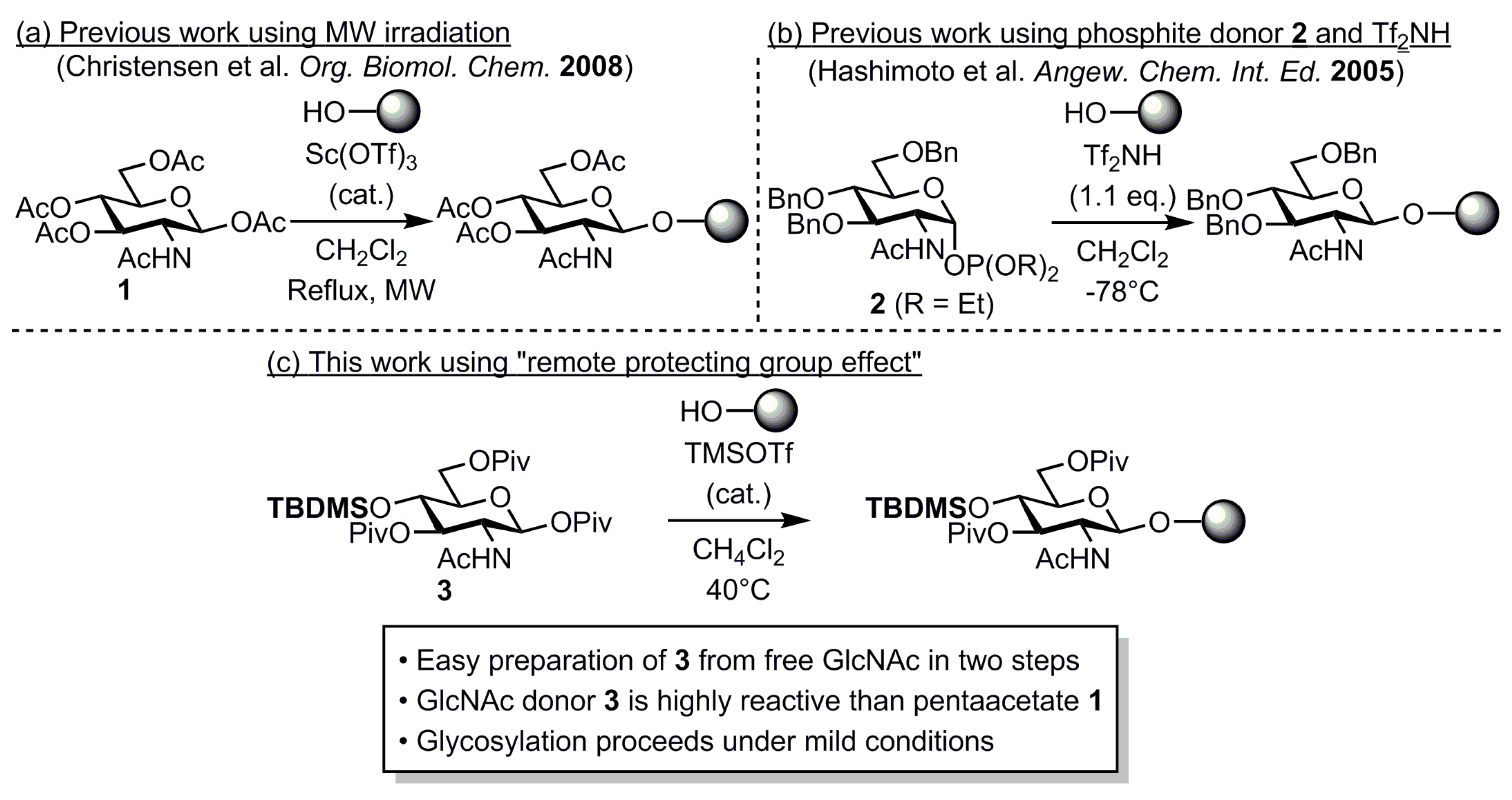
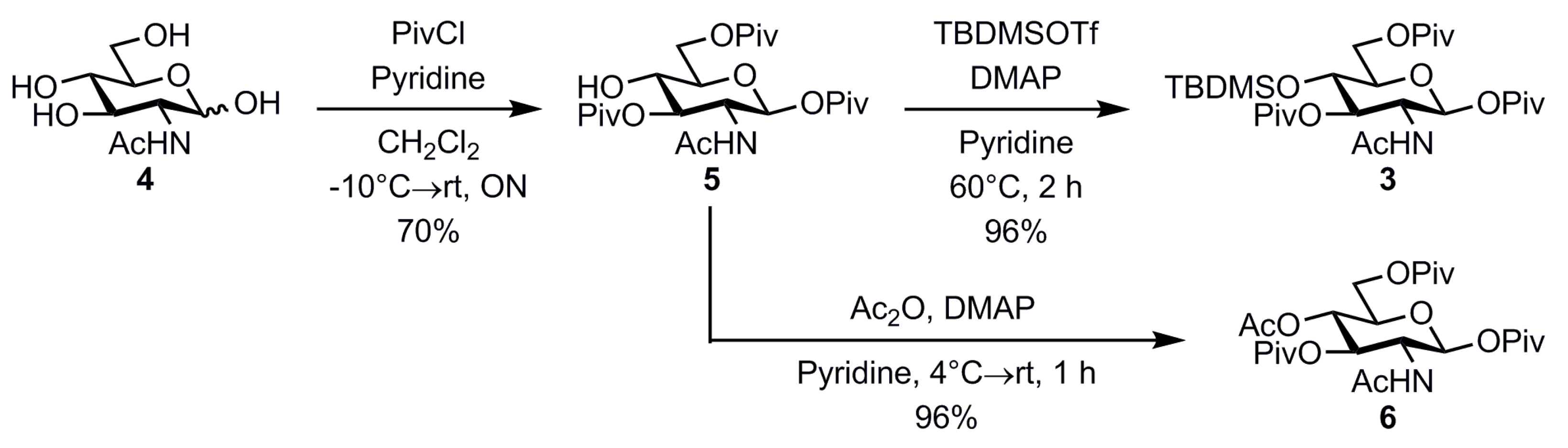
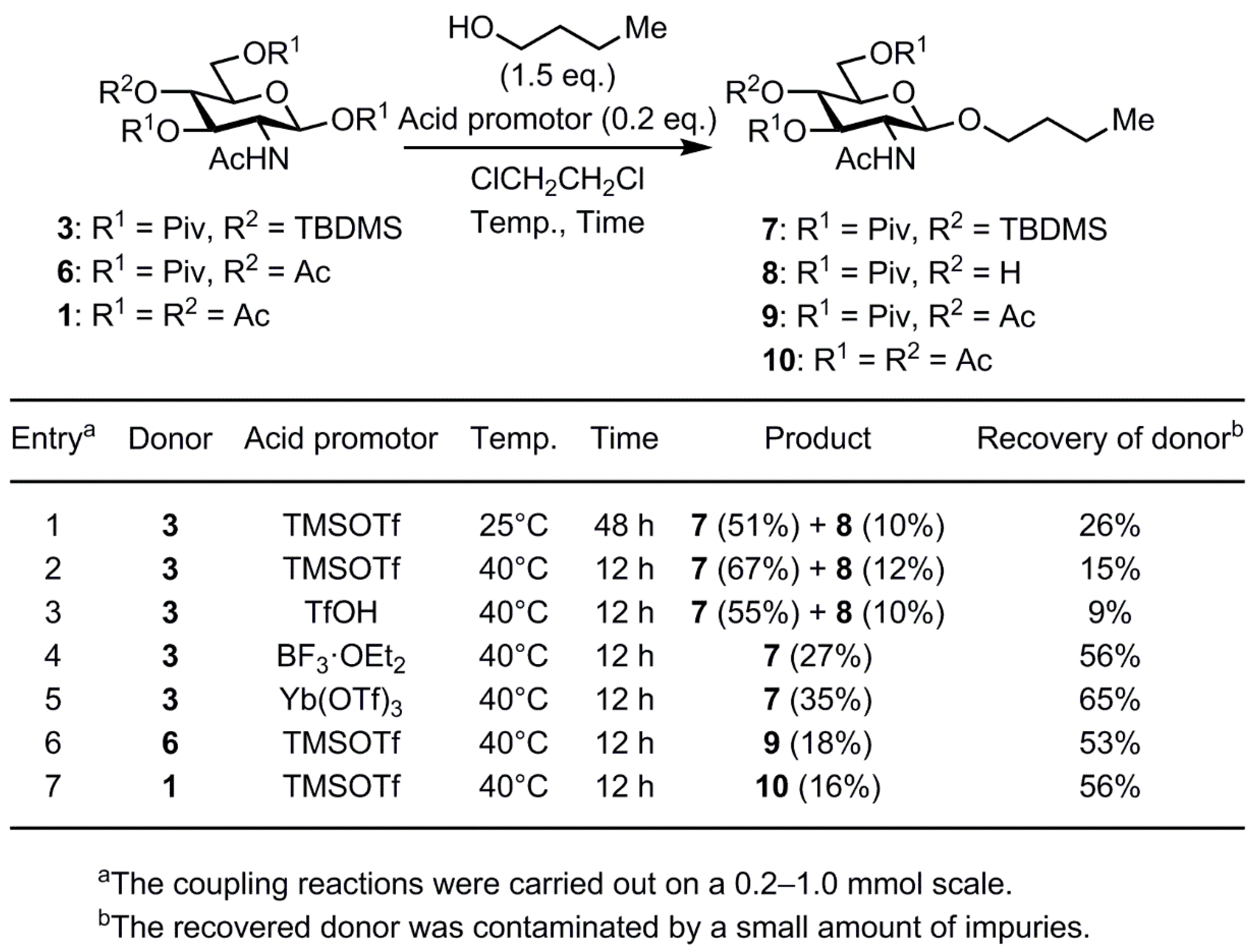
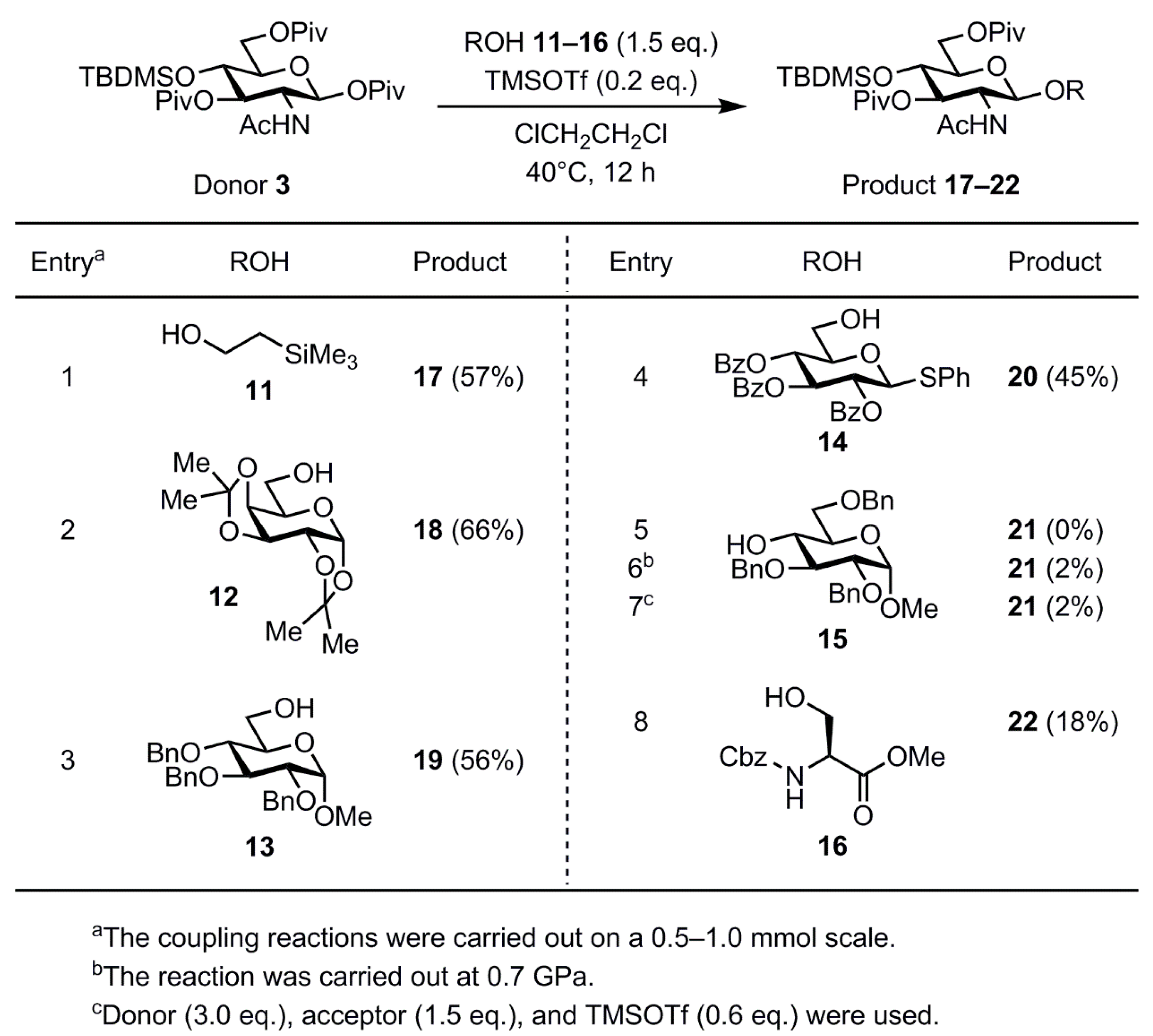
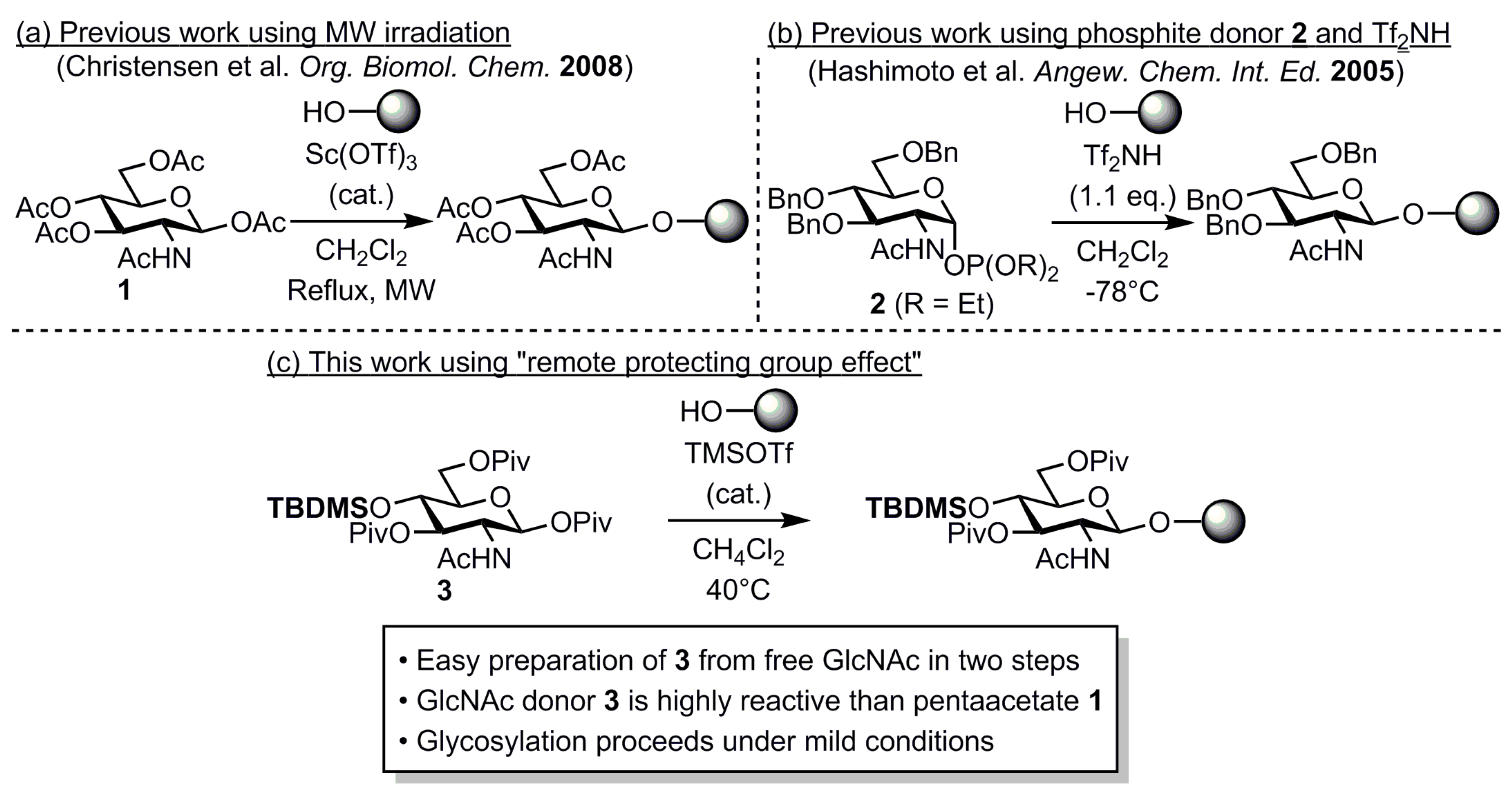
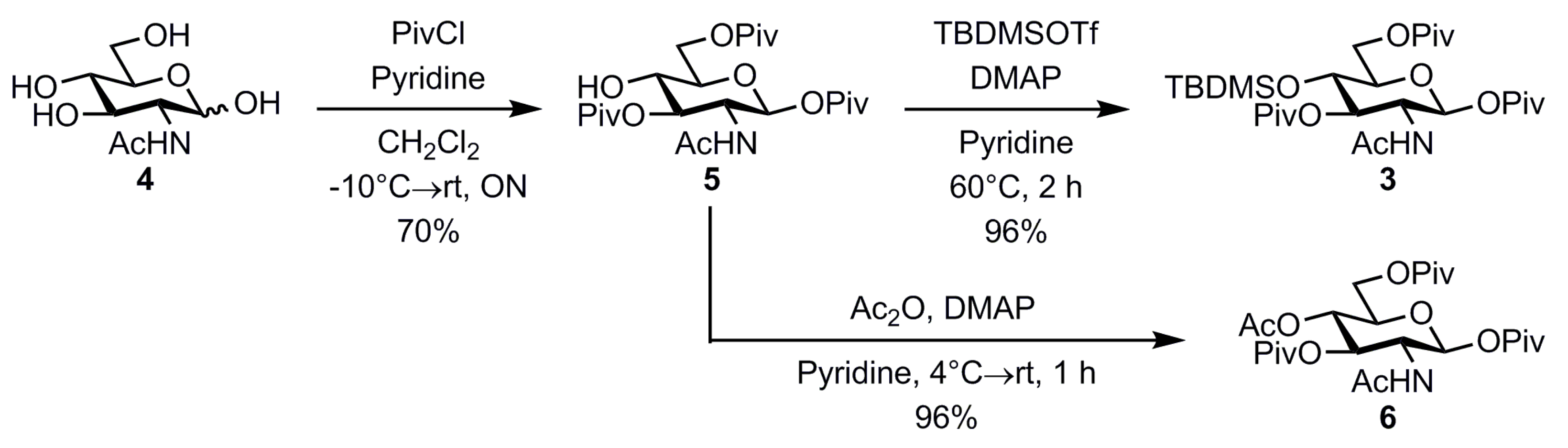
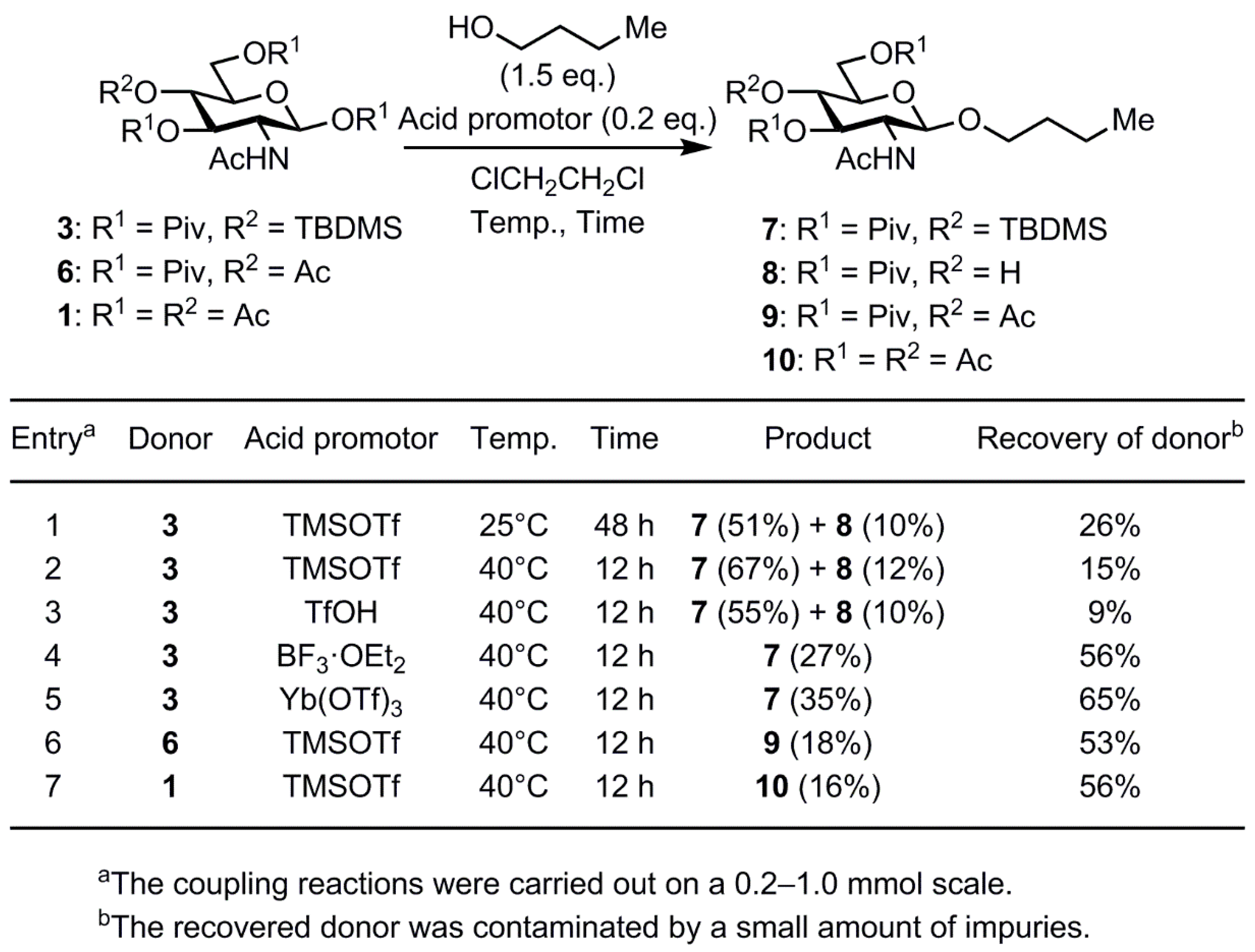
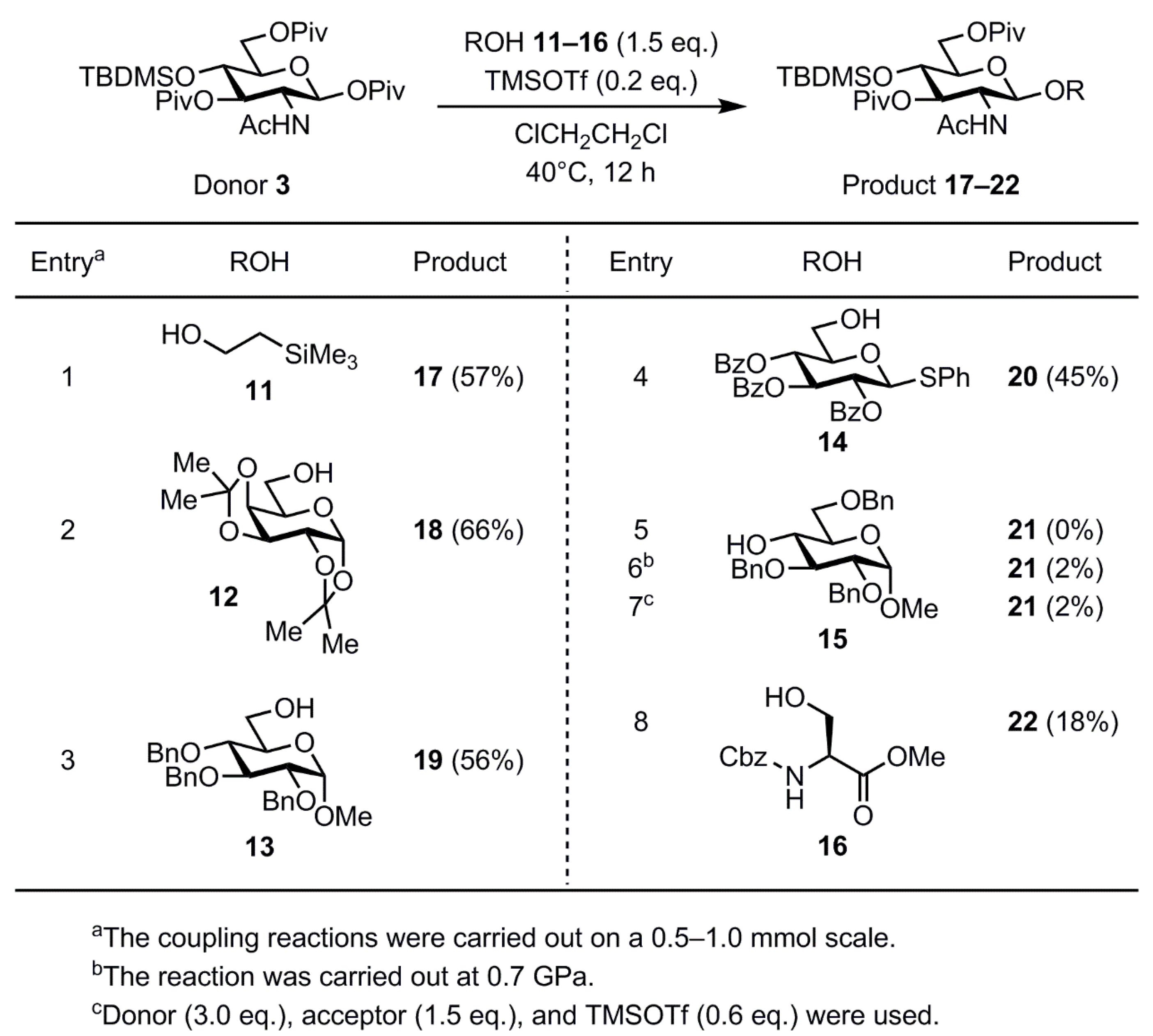
2. Results and Discussion
3. Experimental Section
3.1. General Methods
3.2. General Experimental Procedure and Physical Data for All New Compounds
3.3. General Procedure for the Glycosylation with GlcNAc Donors
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dwek, R.A. Glycobiology: Toward understanding the function of sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef] [PubMed]
- Sytkowski, A.J. Erythropoietin; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Usuki, H.; Nitoda, T.; Ichikawa, M.; Yamaji, N.; Iwashita, T.; Komura, H.; Kanzaki, H. TMG-chitotriomycin, an Enzyme Inhibitor Specific for Insert and Fungal β-N-Acetylglucosaminidases, Produced by Actinomycete Streptomyces anulatus NBRC 13369. J. Am. Chem. Soc. 2008, 130, 4146–4152. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, S.; Maki, M.; Kintaka, K.; Imada, A.; Asai, M. Isolation and characterization of bulgecins, new bacterial metabolites with bulge-inducing activity. J. Antibiot. 1985, 38, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, R.U.; Takada, T.; Chung, B.Y. Synthesis of 2-amino-2-deoxy-β-d-glucopyranosides. Properties and use of 2-deoxy-2-phthalimidoglycosyl halides. ACS Symp. Ser. 1976, 39, 90–115. [Google Scholar]
- Imoto, M.; Yoshimura, H.; Sakaguchi, N.; Kusumoto, S.; Shiba, T. Total synthesis of Escherichia coli lipid A. Tetrahedron Lett. 1985, 26, 1545–1548. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Matsubara, K. Stereoselective synthesis of 2-amino-deoxy-β-d-glucopyranosides and galactopyranosides by using a catalytic amount of tin(II) trifluoromethanesulfonate. Chem. Lett. 1992, 21, 1755–1758. [Google Scholar] [CrossRef]
- Krag, J.; Christiansen, M.S.; Petersen, J.G.; Jensen, H.H. Direct chemical glycosylation with pentenyl- and thioglycoside donors of N-acetylglucosamine. Carbohydr. Res. 2010, 345, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Marqvorsen, M.H.S.; Pedersen, M.J.; Rasmussen, M.R.; Kristensen, S.M.; Dahl-Lassen, R.; Jensen, H.H. Why Is Direct Glycosylation with N-Acetylglucosamine Donors Such a Poor Reaction and What Can Be Done about It? J. Org. Chem. 2017, 82, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Stévenin, A.; Boyer, F.-D.; Beau, J.-M. Highly selective formation of β-glycosides of N-acetylglucosamine using catalytic iron(III) triflate. Eur. J. Org. Chem. 2012, 2012, 1699–1702. [Google Scholar] [CrossRef]
- Christensen, H.; Christiansen, M.S.; Petersen, J.; Jesen, H.H. Direct formation of β-glycosides of N-acetyl glycosamines mediated by rare earth metal triflates. Org. Biomol. Chem. 2008, 6, 3276–3283. [Google Scholar] [CrossRef] [PubMed]
- Arihara, R.; Nakamura, S.; Hashimoto, S. Direct and stereoselective synthesis of 2-acetamido-2-deoxy-β-d-glycopyranosides by using the phosphite method. Angew. Chem. Int. Ed. 2005, 44, 2245–2249. [Google Scholar] [CrossRef] [PubMed]
- Mydock, L.K.; Demchenko, A.V. Superarming the S-benzoxazolyl glycosyl donors by simple 2-O-benzoyl-3,4,6-tri-O-benzyl protection. Org. Lett. 2008, 10, 2103–2106. [Google Scholar] [CrossRef] [PubMed]
- Tomono, S.; Kusumi, S.; Takahashi, D.; Toshima, K. Armed-disarmed effect of remote protecting groups on the glycosylation reaction of 2,3-dideoxyglycosyl donors. Tetrahedron Lett. 2011, 52, 2399–2403. [Google Scholar] [CrossRef]
- Feng, J.; Ling, C.-C. An efficient conversion of N-acetyl-d-glucosamine to N-acetyl-d-galactosamine and derivatives. Carbohydr. Res. 2010, 345, 2450–2457. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, R.U.; Kondo, T. Benzyl trifluoromethanesulfonate. Preparation of tri-O-acetyl-2-O-benzyl-α-d-galactopyranosyl bromide from 1,3,4,6-tetra-O-acetyl-α-d-galactopyranose. Carbohydr. Res. 1974, 35, C4–C6. [Google Scholar]
- Yamada, K.; Fujita, H.; Kunishima, M. A novel acid-catalyzed O-benzylating reagent with the smallest unit of imidate structure. Org. Lett. 2012, 14, 5026–5029. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.M.; Nordstrøm, L.U.; Bols, M. “Super armed” glycosyl donors: Conformational arming of thioglycosides by silylation. J. Am. Chem. Soc. 2007, 129, 9222–9235. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gildersleeve, J.C. Mechanistic studies and methods to prevent aglycon transfer of thioglycosides. J. Am. Chem. Soc. 2006, 128, 11612–11619. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Gama, Y.; Yasumoto, M.; Ishigami, Y. Glycosylation reaction under high pressure. Tetrahedron Lett. 1990, 31, 6549–6552. [Google Scholar] [CrossRef]
- Sample Availability: Not Available.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, H.; Hamaya, Y.; Kotsuki, H. A Direct Method for ?-Selective Glycosylation with an N-Acetylglucosamine Donor Armed by a 4-O-TBDMS Protecting Group. Molecules 2017, 22, 429. https://doi.org/10.3390/molecules22030429
Tanaka H, Hamaya Y, Kotsuki H. A Direct Method for ?-Selective Glycosylation with an N-Acetylglucosamine Donor Armed by a 4-O-TBDMS Protecting Group. Molecules. 2017; 22(3):429. https://doi.org/10.3390/molecules22030429
Chicago/Turabian StyleTanaka, Hidenori, Yu Hamaya, and Hiyoshizo Kotsuki. 2017. "A Direct Method for ?-Selective Glycosylation with an N-Acetylglucosamine Donor Armed by a 4-O-TBDMS Protecting Group" Molecules 22, no. 3: 429. https://doi.org/10.3390/molecules22030429
APA StyleTanaka, H., Hamaya, Y., & Kotsuki, H. (2017). A Direct Method for ?-Selective Glycosylation with an N-Acetylglucosamine Donor Armed by a 4-O-TBDMS Protecting Group. Molecules, 22(3), 429. https://doi.org/10.3390/molecules22030429