
N-Pyrrylarylsulfones with High Therapeutic Potential


Abstract
:
1. Introduction
2. Pyrrolo[1,2-b]-s-triazolo[3,4-d][1,2,5]benzothiadiazepine 5,5-dioxide
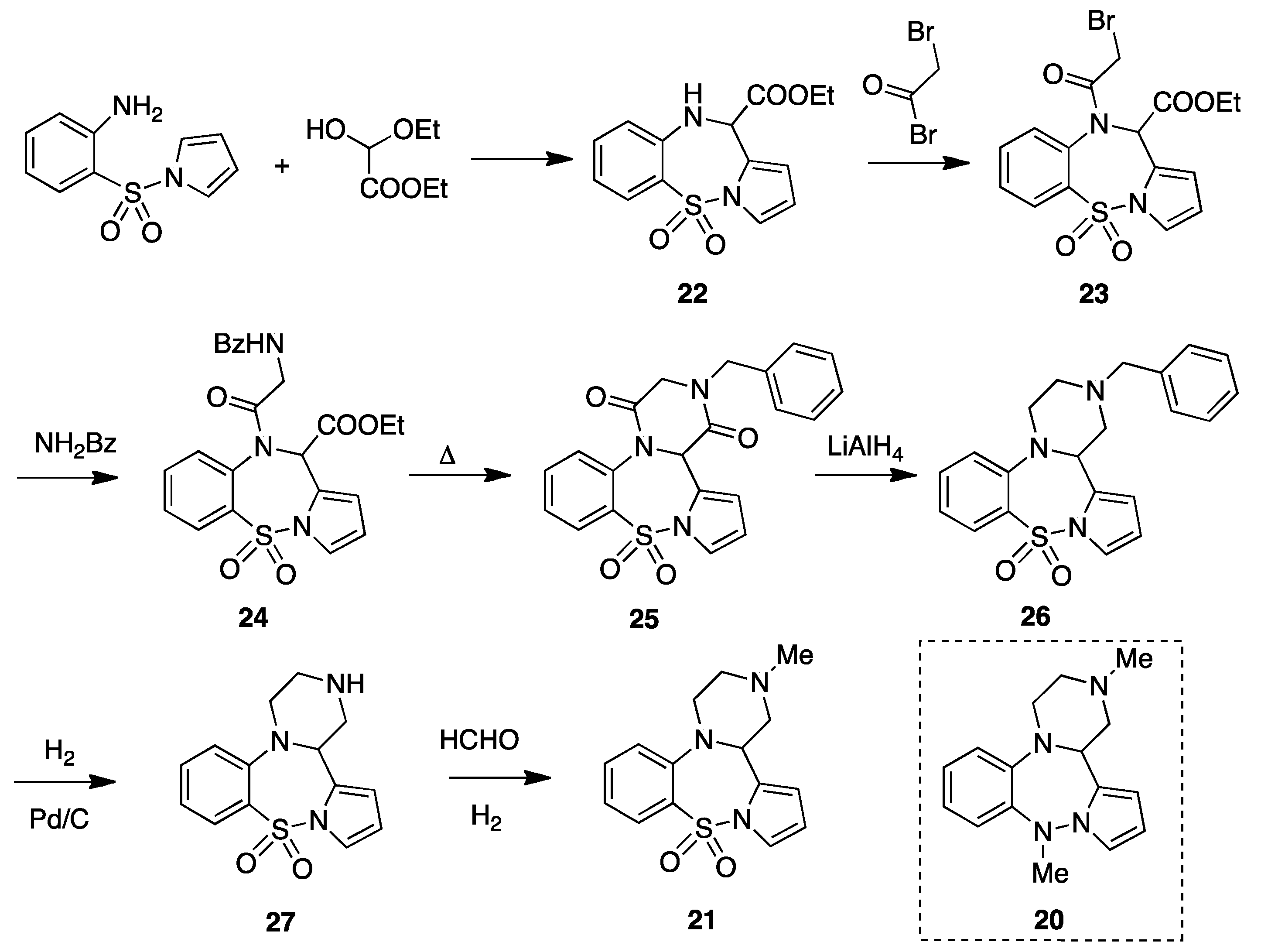
3. 2-Methyl-1,3,4,14b-tetrahydro-2H-pyrazino[2,1-d]pyrrolo[1,2-b][1,2,5]benzothiadia-zepine 10,10-dioxide
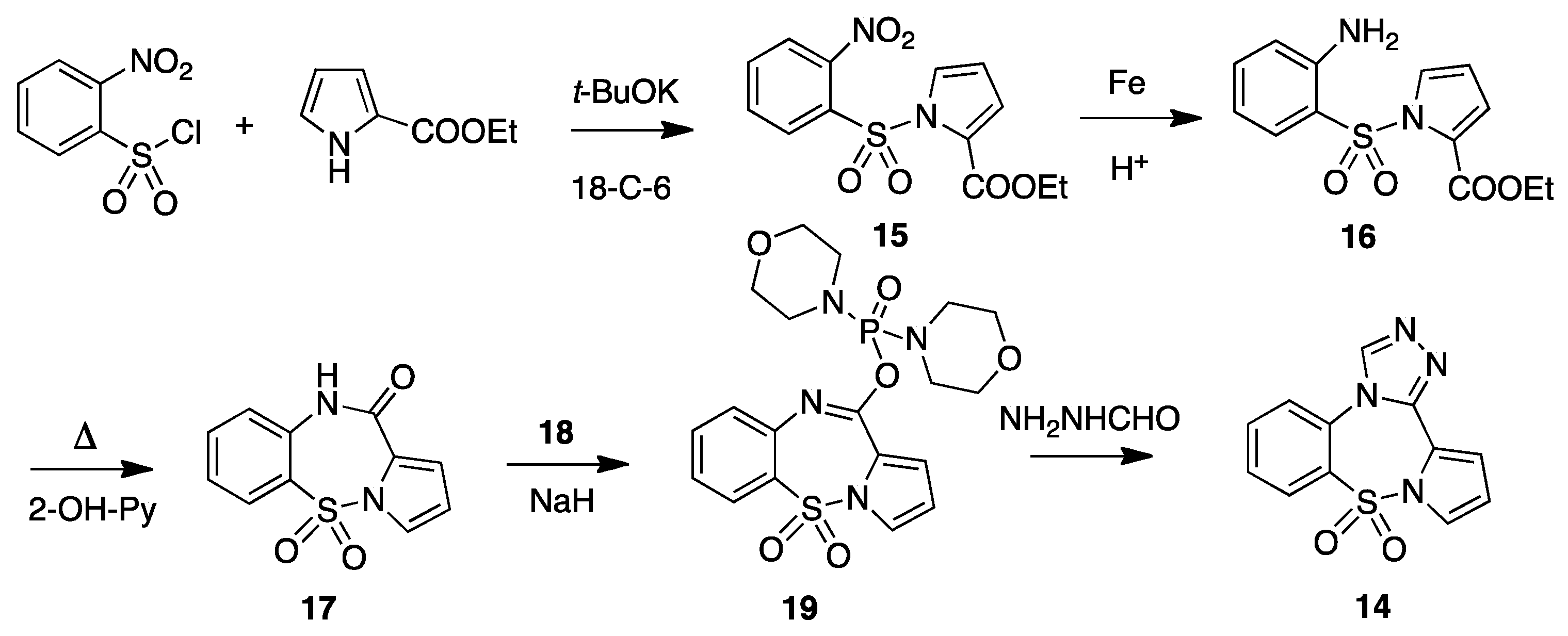
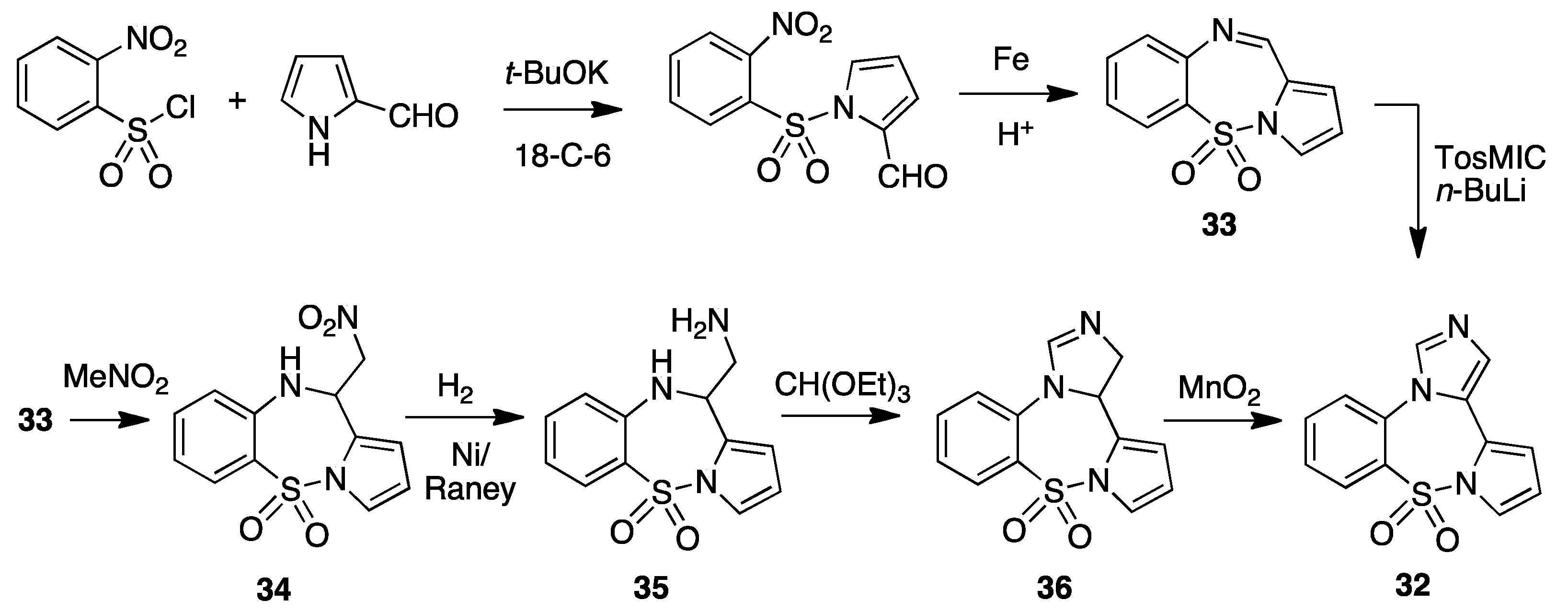
4. Imidazo[5,1-d]pyrrolo[1,2-b][1,2,5]benzothiadiazepine 9,9-dioxide
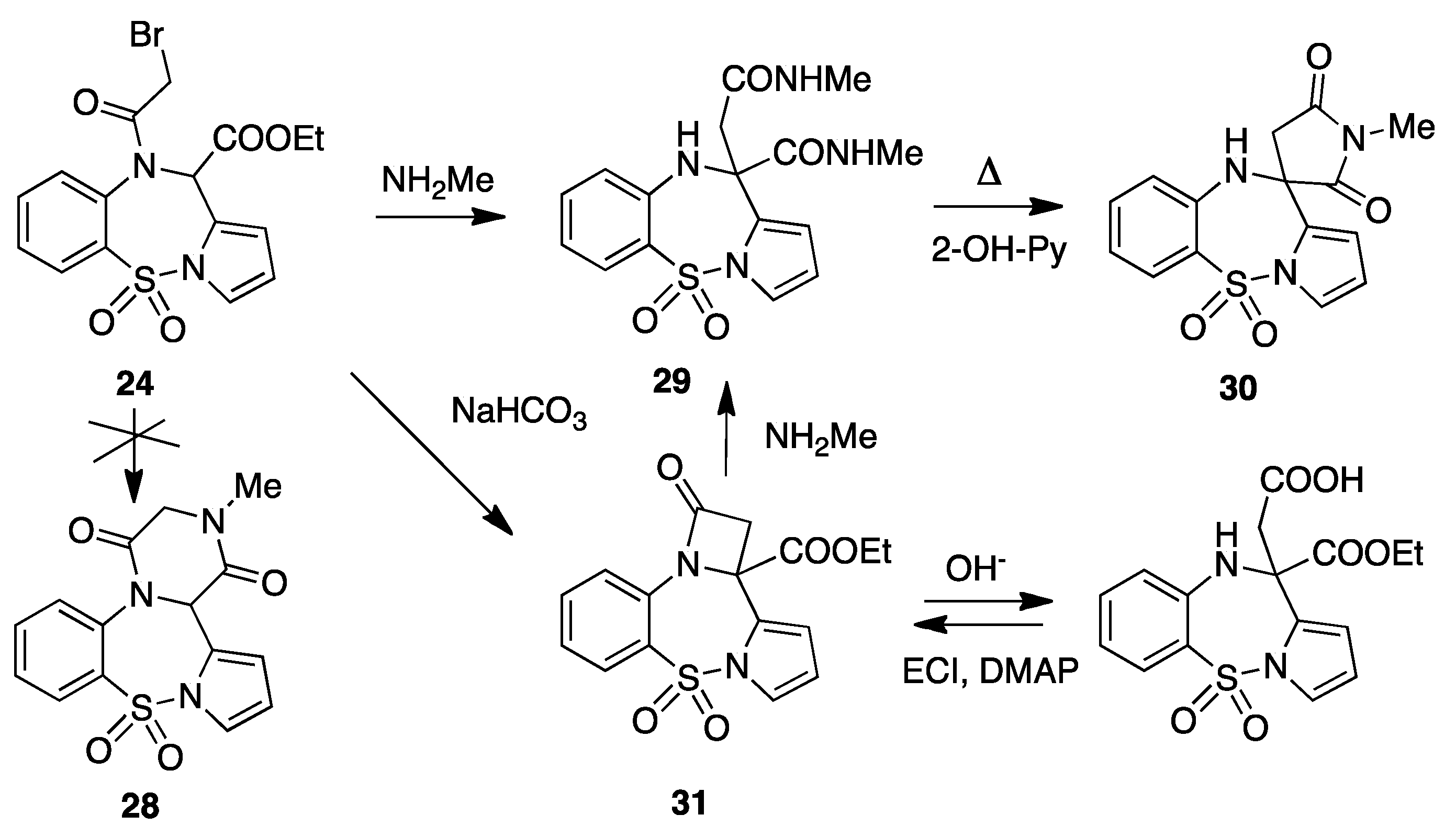
5. 5H-pyrrolo[1,2-b][1,2,5]benzothiadiazepin-11(10H)-one 5,5-dioxide
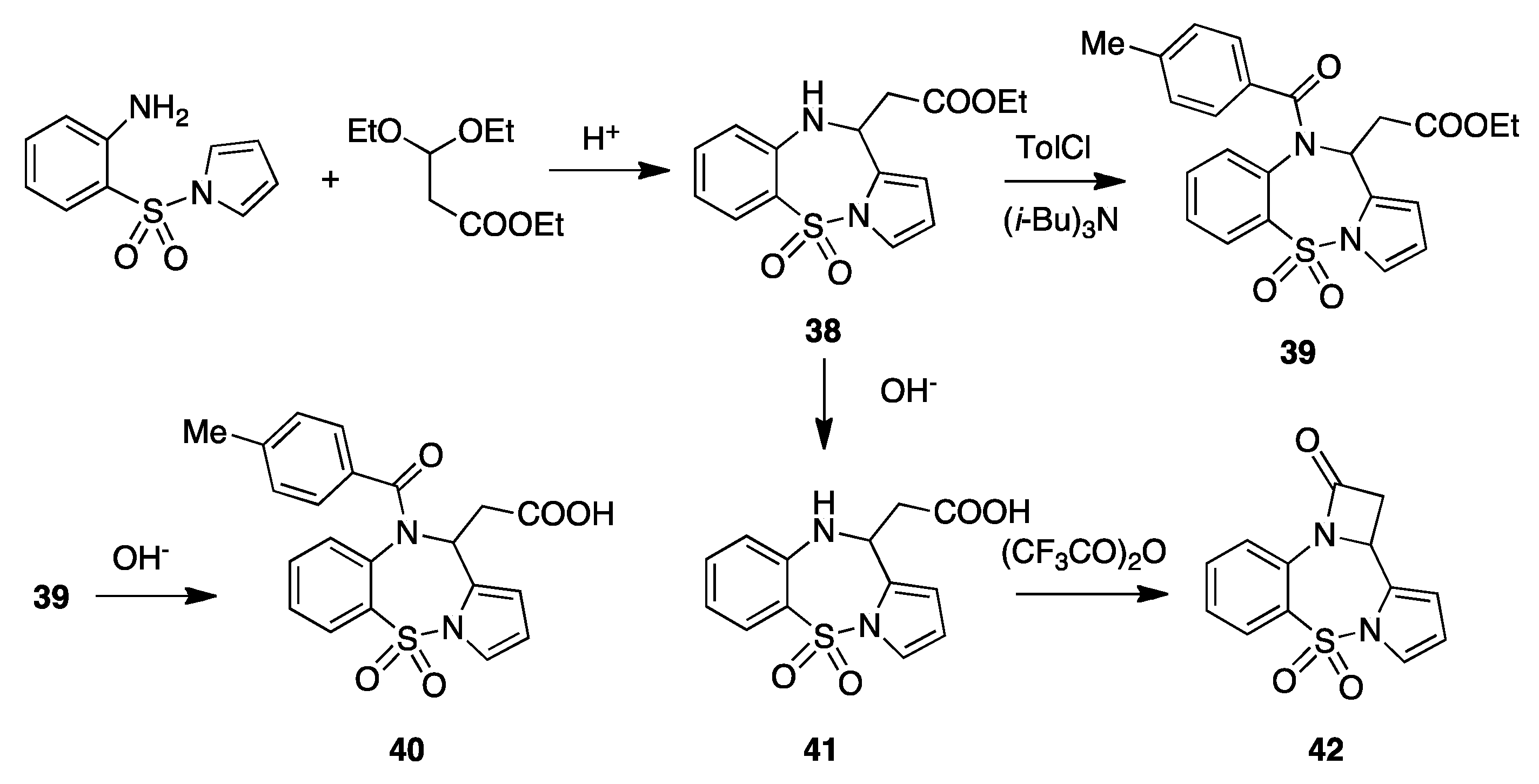
6. Pyrrolo[1,2-b][1,2,5]benzothiadiazepine Acetic Acid 5,5,-dioxide
7. PBTDs as Chronic Myelogenous Leukemia (CML) Agents
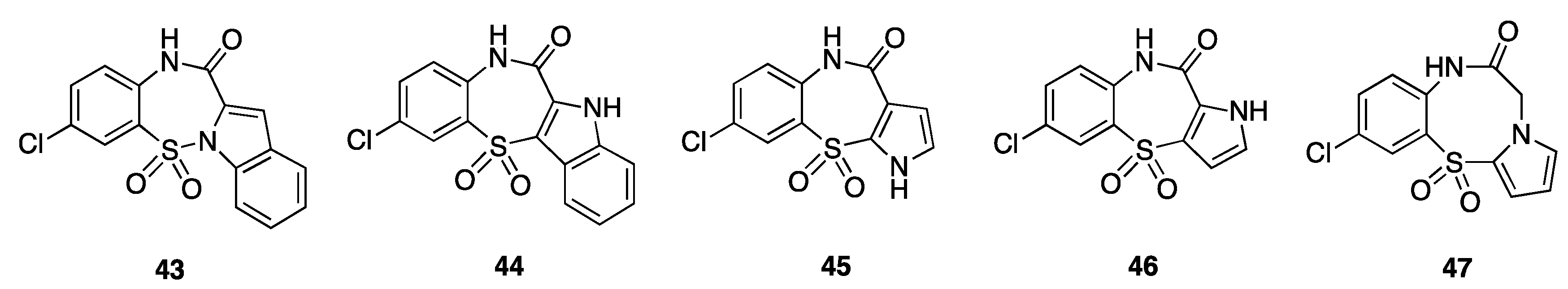
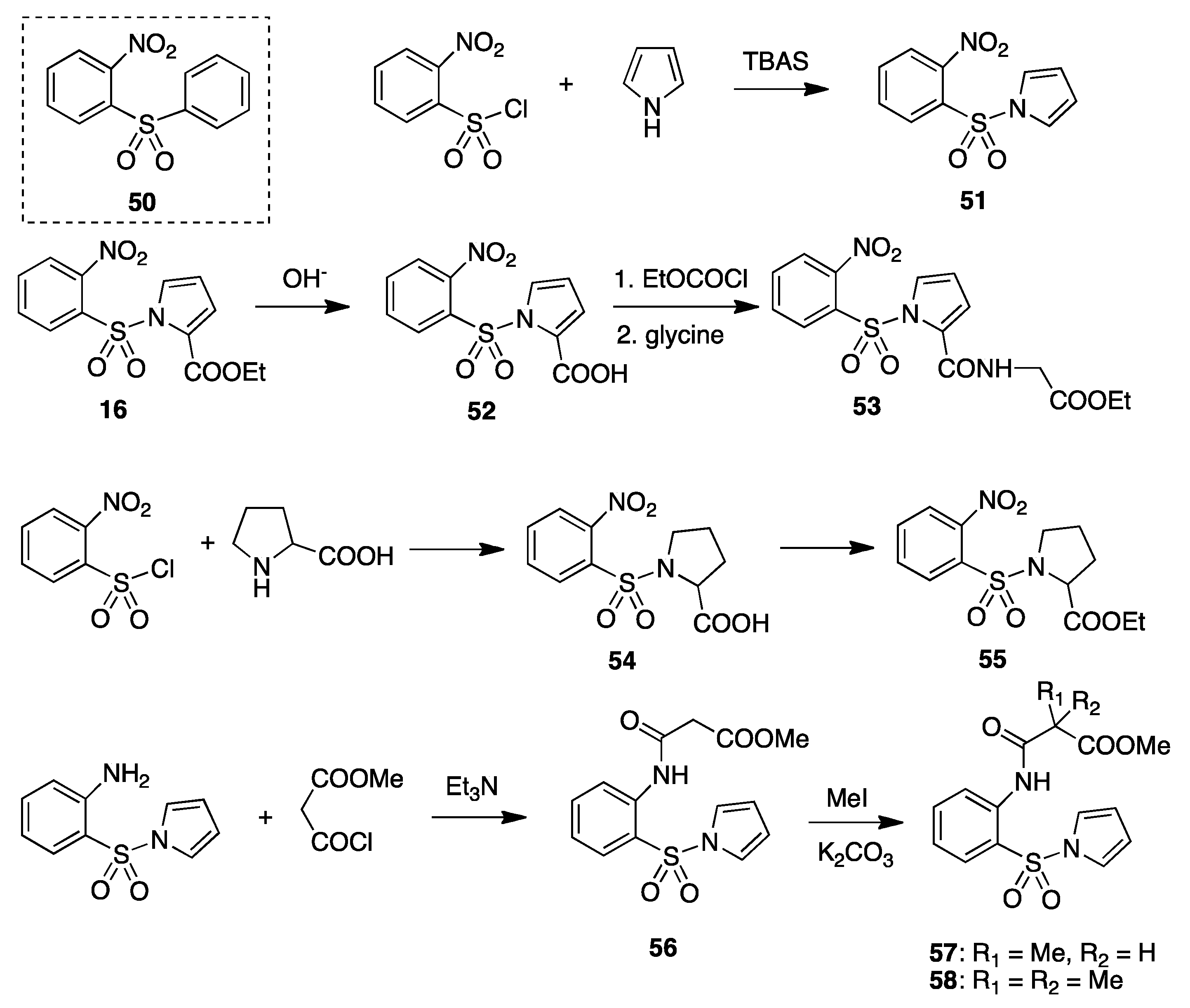
8. Pyrryl Aryl Sulfones
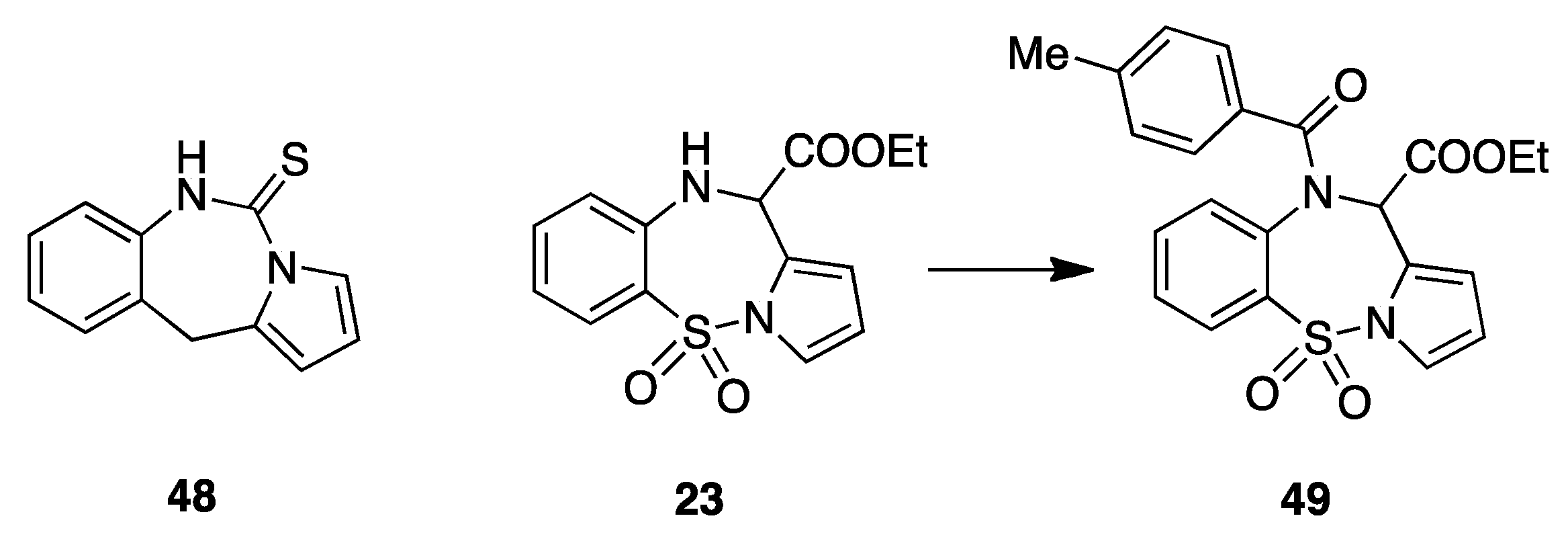
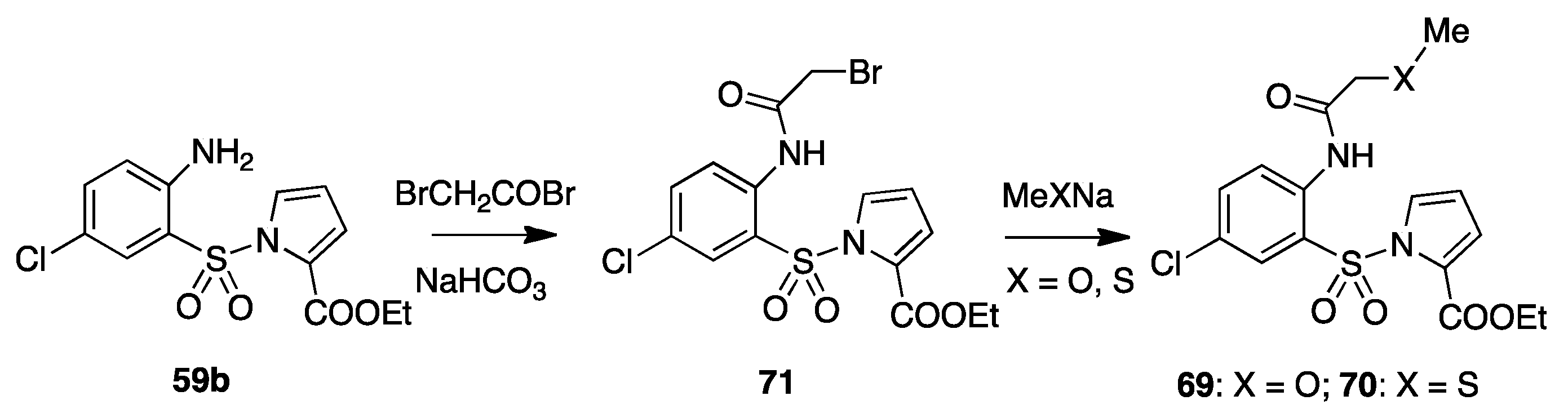
9. Acylamino Pyrryl Aryl Sulfones
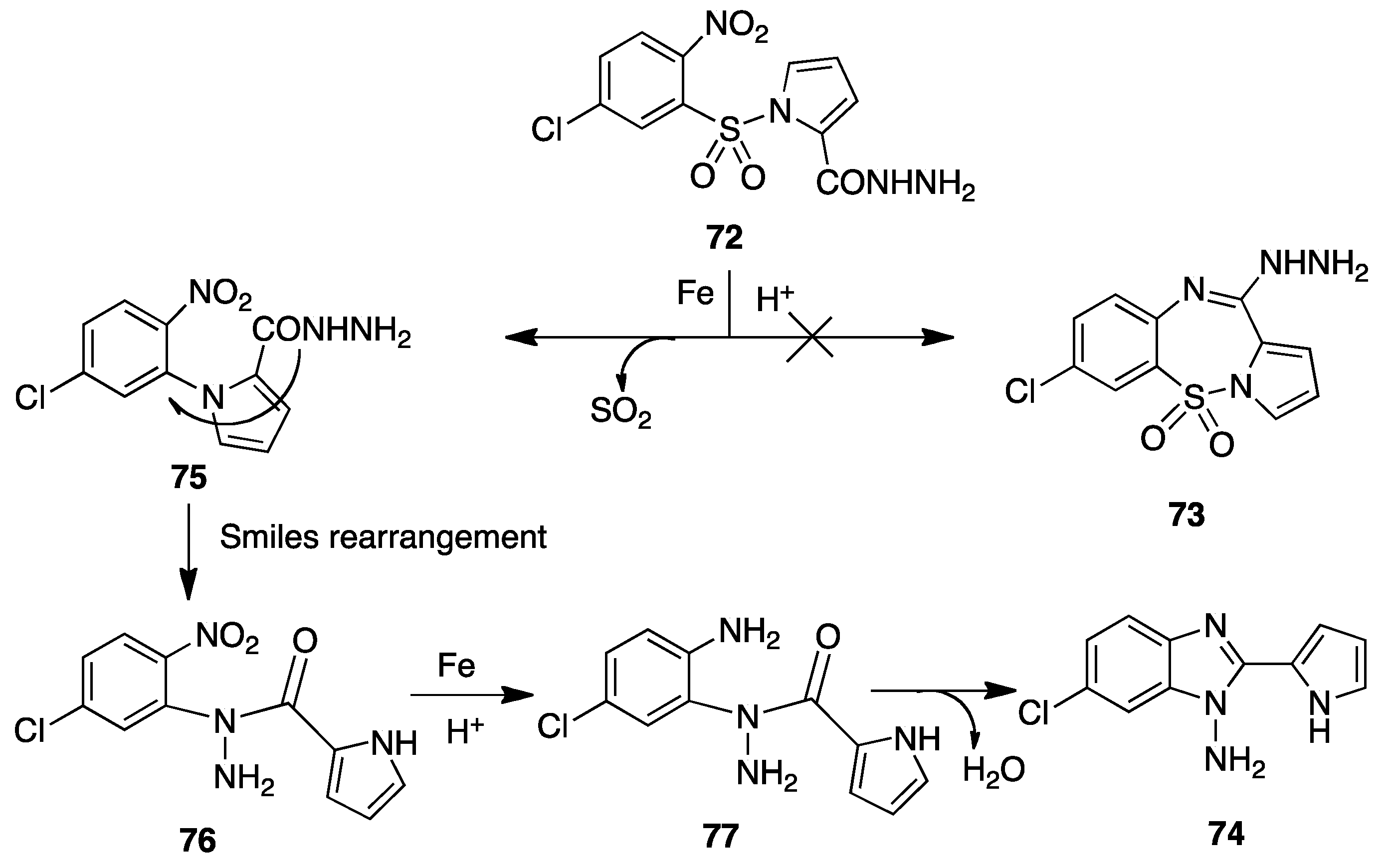
10. Smiles Rearrangement
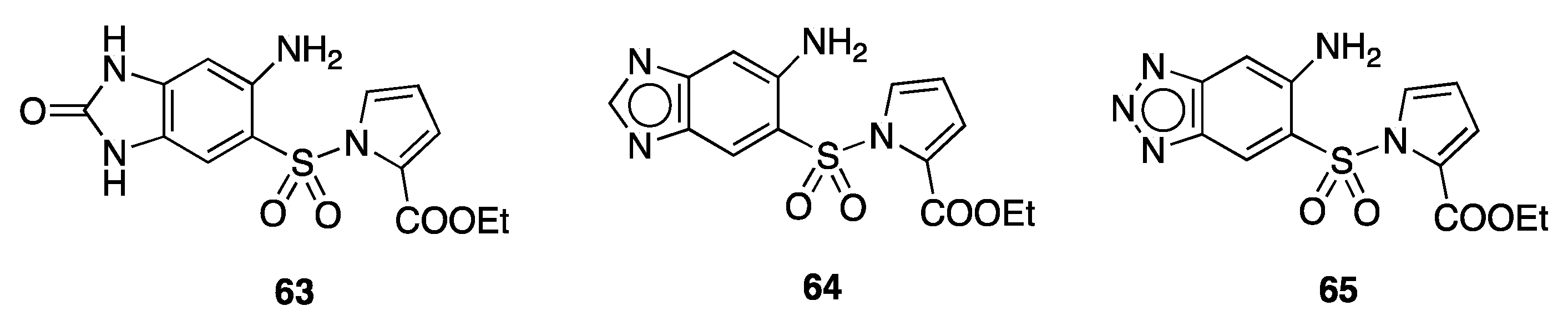
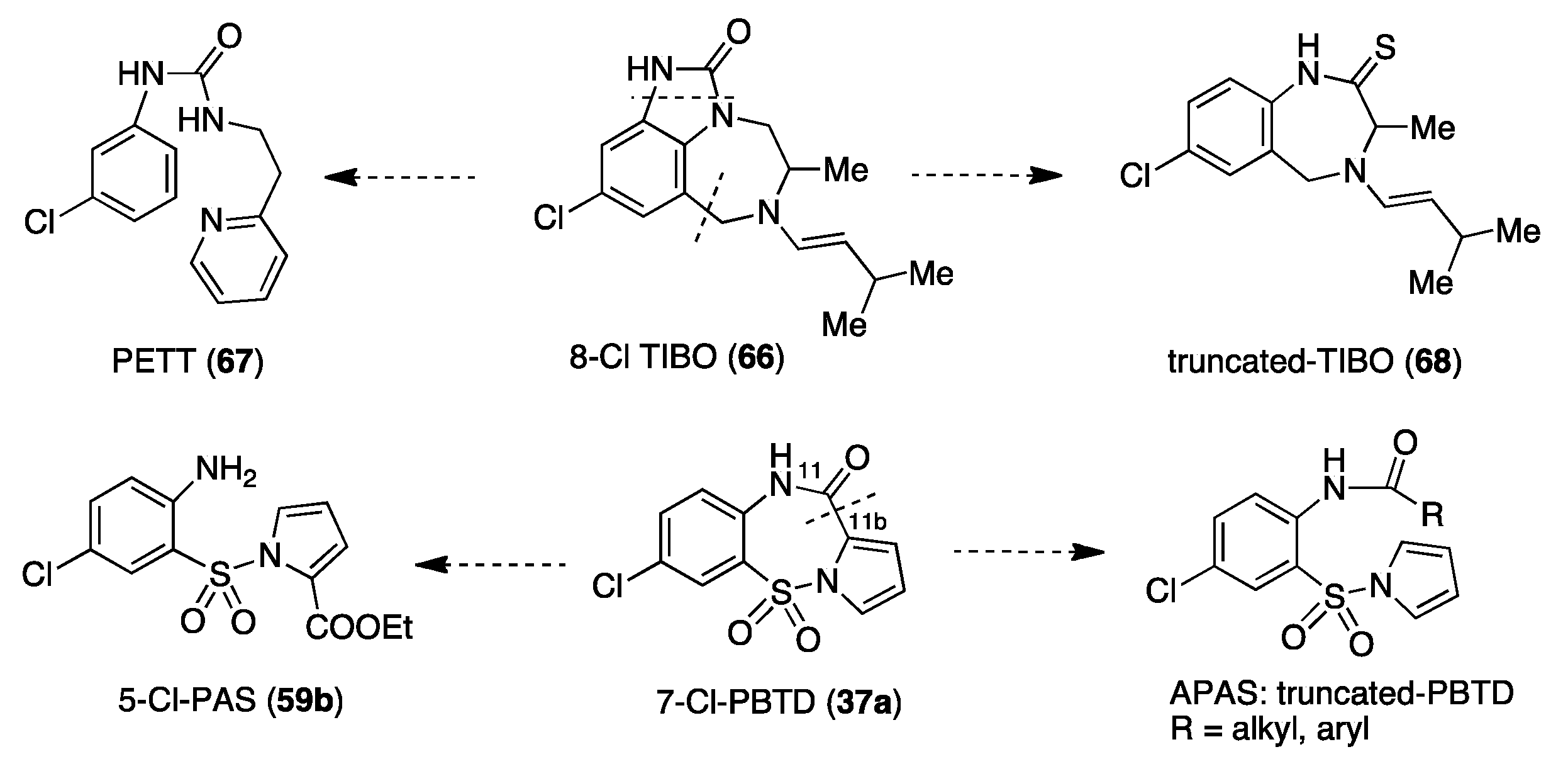
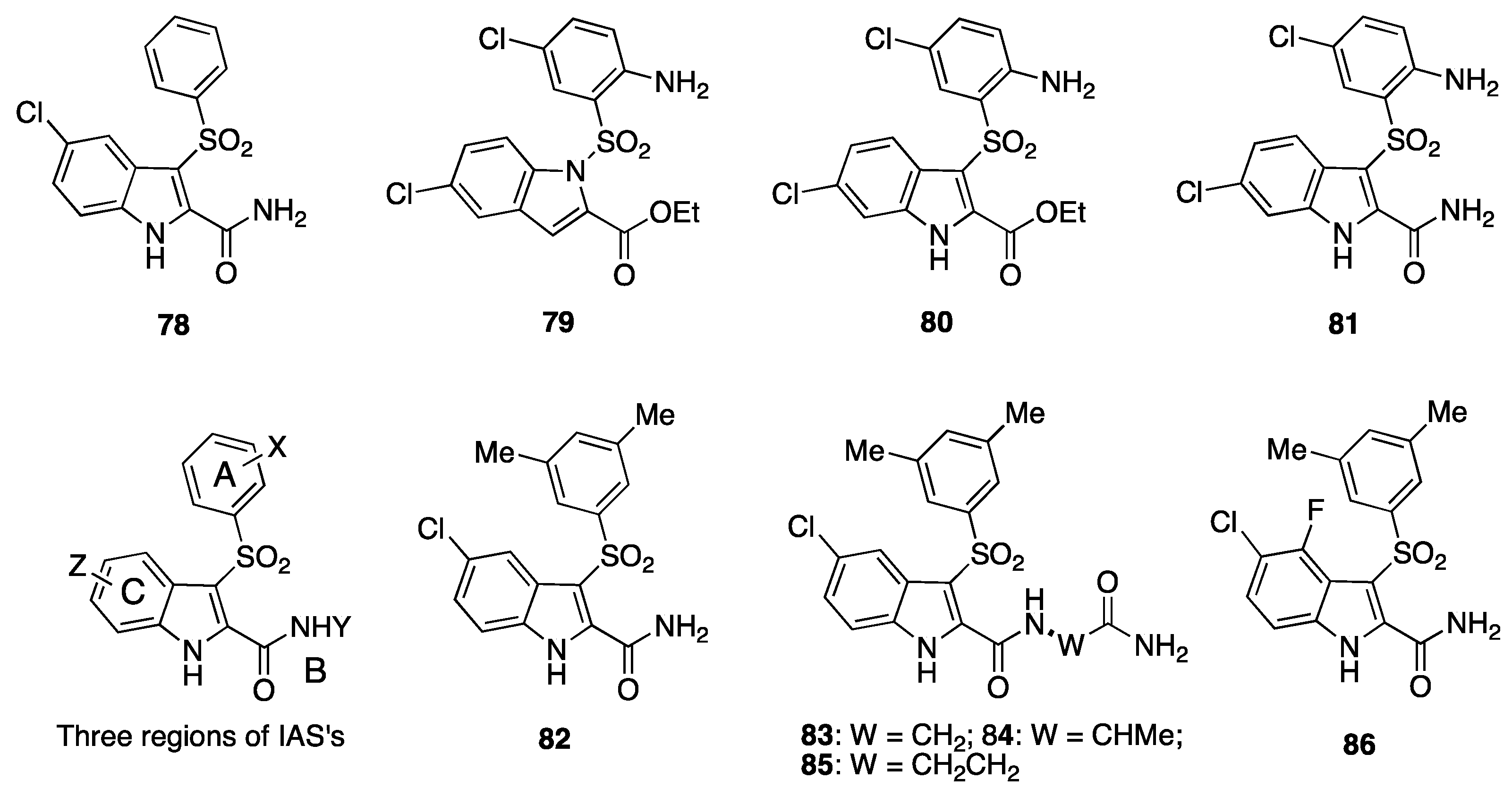
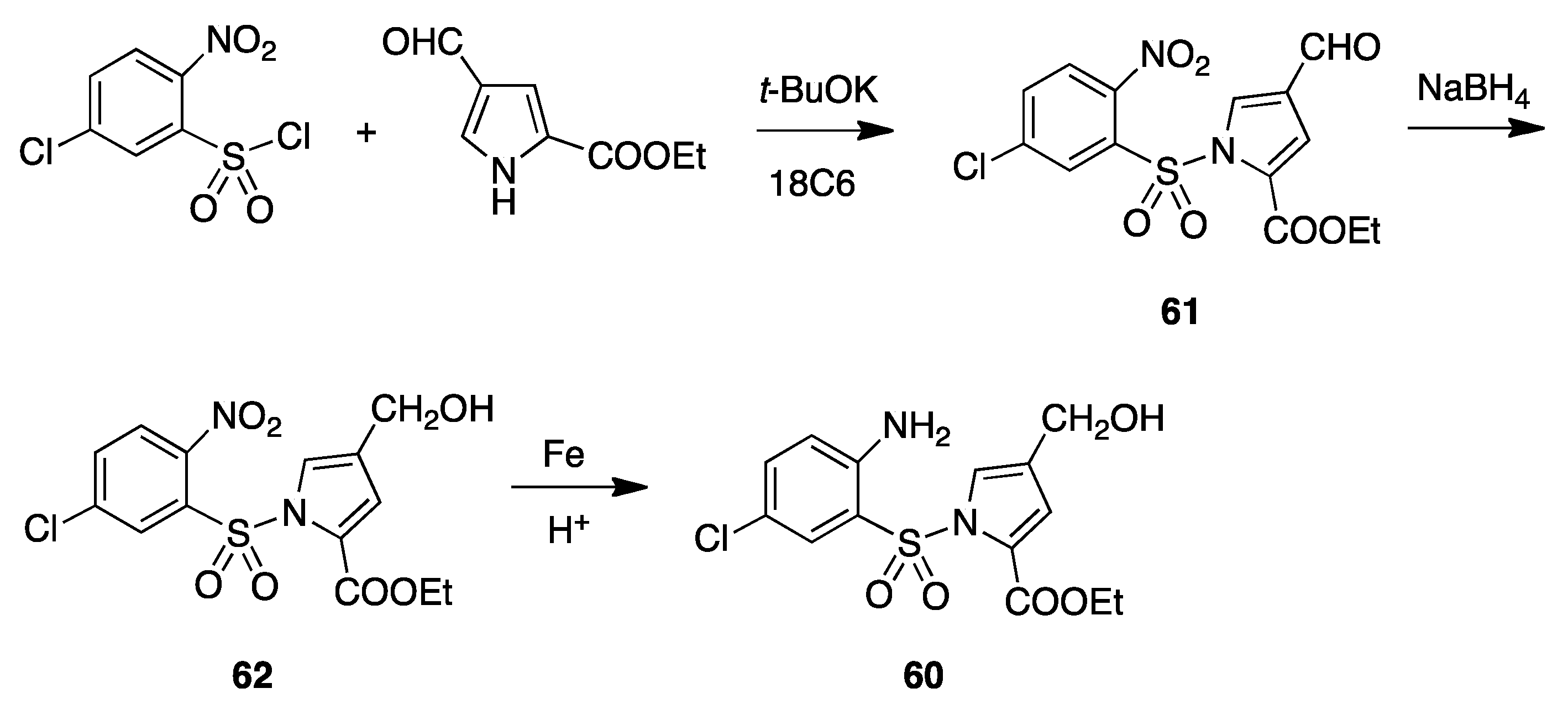
11. Structurally Related Compounds
12. Conclusions
Acknowledgments
Dedication
Conflicts of Interest
References
- Gerald, M.C. The Drug Book; Sterling Education: New York, NY, USA, 2013. [Google Scholar]
- Dogmagk, G. Ein Beitrag zur Chemotherapie der bakteriellen Infektionen. Deutsch. Med. Wochenschr. 1935, 61, 250–253. [Google Scholar] [CrossRef]
- Dogmagk, G. Eine neue Klasse von Desinfectionsmitteln. Deutsch. Med. Wochenschr. 1935, 61, 829–832. [Google Scholar] [CrossRef]
- Henry, R.J. The mode of action of sulfonamides. Bacteriol. Rev. 1943, 7, 175–262. [Google Scholar] [PubMed]
- Sulfamethoxazole. DrugBank. Available online: http://www.drugbank.ca (accessed on 17 August 2016).
- Fromm, E.; Wittmann, J. Derivate des p-nitrophenols. Ber. Deutsch. Chem. Ges. 1908, 41, 2264–2273. [Google Scholar] [CrossRef]
- Goulart, I.M.; Reis, A.C.; de Rezende, T.M.; Borges, A.S.; Ferreira, M.S.; Nishioka, S.A. Aplastic anaemia associated with multidrug therapy (dapsone, rifampicin and clofazimine) in a patient with lepromatous leprosy. Lepr. Rev. 2005, 76, 167–169. [Google Scholar] [PubMed]
- Brown, A.; Captain, B. 50 years of thiazides: Should thiazide diuretics be considered third-line hypertension treatment? Am. J. Ther. 2011, 18, e244–e254. [Google Scholar] [CrossRef] [PubMed]
- Fregly, M.J. Effect of chlorothiazide and hydrochlorothiazide on blood pressure and thyroid activity of hypertensive rats. Am. J. Cardiol. 1961, 8, 890–898. [Google Scholar] [CrossRef]
- Maxwell, R.A.; Eckhardt, S.B. Furosemide in “Drug Discovery”; The Humana Press Inc.: New York, NY, USA, 1990; pp. 67–77. [Google Scholar]
- Stumvoll, M.; Goldstein, B.J.; van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Tzoulaki, I.; Molokhia, M.; Curcin, V.; Little, M.P.; Millett, C.J.; Ng, A.; Hughes, R.I.; Khunti, K.; Wilkins, M.R.; Majeed, A.; et al. Risk of cardiovascular disease and all cause mortality among patients with type 2 diabetes prescribed oral antidiabetes drugs: Retrospective cohort study using UK general practice research database. BMJ 2009, 339, b4731. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, P.N.; Kandasamy, R.A.; Crotty, C.M.; Espie, G.S. Ethoxyzolamide Differentially Inhibits CO2 Uptake and Na+-Independent and Na+-Dependent HCO3− Uptake in the Cyanobacterium Synechococcus sp. UTEX 625. Plant Physiol. 1996, 112, 79–88. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Dancing with chemical formulae of antivirals: A personal account. Biochem. Pharmacol. 2013, 86, 711–725. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. Dancing with chemical formulae of antivirals: A panoramic view. Biochem. Pharmacol. 2013, 86, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Mehellou, Y.; De Clercq, E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go? J. Med. Chem. 2010, 53, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Vere Hodge, R.A. Meeting report: 28th International conference on antiviral research in Rome, Italy. Antivir. Res. 2015, 123, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-HIV drug discovery and development: Current innovations and future trends. J. Med. Chem. 2015, 59, 2849–2878. [Google Scholar] [CrossRef] [PubMed]
- López-Labrador, F.X. Hepatitis C Virus NS3/4A protease inhibitors. Recent Pat. Antiinfect. Drug Discov. 2008, 3, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Gerber, L.; Welzel, T.M.; Zeuzem, S. New therapeutic strategies in HCV: Polymerase inhibitors. Liver Int. 2013, 3 (Suppl. 1), 85–92. [Google Scholar] [CrossRef] [PubMed]
- Bandmann, H.J.; Breit, R. The mafenide story. Br. J. Dermatol. 1973, 89, 219–221. [Google Scholar] [CrossRef] [PubMed]
- McCormack, P.L. Celecoxib: A review of its use for symptomatic relief in the treatment of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis. Drugs 2011, 71, 2457–2489. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.; Gumber, D.; Abbot, A.; Dhimana, S.; Sharma, P. Pyrrole: A resourceful small molecule in key medicinal hetero-aromatics. RCS Adv. 2015, 5, 15233–15266. [Google Scholar] [CrossRef]
- Gordee, R.S.; Matthews, T.R. Systemic antifungal activity of pyrrolnitrin. Appl. Microbiol. 1969, 17, 690–694. [Google Scholar] [PubMed]
- Cordrey, L.J. Tolmetin sodium, a new anti-arthritis drug: Double-blind and long-term studies. J. Am. Geriatr. Soc. 1976, 24, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Rényi, L.; Larsson, L.G.; Berg, S.; Svensson, B.E.; Thorell, G.; Ross, S.B. Biochemical and behavioural effects of isamoltane, a beta-adrenoceptor antagonist with affinity for the 5-HT1B receptor of rat brain. Naunyn Schmiedebergs Arch. Pharmacol. 1991, 343, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Aarsand, A.K.; Petersen, P.H.; Sandberg, S. Estimation and application of biological variation of urinary-aminolevulinic acid and porphobilinogen in healthy individuals and in patients with acute intermittent porphyria. Clin. Chem. 2006, 52, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.D. The discovery and development of atorvastatin, a potent novel hypolipidemic agent. Prog. Med. Chem. 2002, 40, 1–22. [Google Scholar] [PubMed]
- Hopkins, C.R.; Lindsley, C.W.; Niswender, C.M. mGluR4-positive allosteric modulation as potential treatment for Parkinson’s disease. Future Med. Chem. 2009, 1, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Gogliotti, R.D.; Blobaum, A.L.; Morrison, R.M.; Daniels, J.S.; Salovich, J.M.; Cheung, Y.Y.; Rodriguez, A.L.; Loch, M.T.; Conn, P.J.; Lindsley, C.W.; et al. Discovery and characterization of a novel series of N-phenylsulfonyl-1H-pyrrole picolinamides as positive allosteric modulators of the metabotropic glutamate receptor 4 (mGlu4). Bioorg. Med. Chem. Lett. 2016, 26, 2984–2987. [Google Scholar] [CrossRef] [PubMed]
- Stefancich, G.; Silvestri, R. Research on nitrogen containing heterocyclic compounds. XVI. Synthesis of 1,3,4,14b-tetrahydro-2,10-dimethyl-2H,10H-pyrazino-[2,1-d]pyrrolo[1,2-b][1,2,5]benzotriazepine (1:1) maleate (10-methyl-10-azaaptazepine). J. Heterocycl. Chem. 1989, 26, 745–746. [Google Scholar] [CrossRef]
- Stefancich, G.; Artico, M.; Silvestri, R.; Pantaleoni, G.C.; Giorgi, R.; Palumbo, G. Research on psycotropic agents. III. Antidepressant activity and neuropsycobehavioural effects of new 5H-pyrrolo[1,2-b][1,2,5]benzotriazepine derivatives. Farmaco 1990, 45, 7–27. [Google Scholar] [PubMed]
- Stefancich, G.; Artico, M.; Silvestri, R.; Prosini, P.P.; Pantaleoni, G.C.; Giorgi, R.; Palumbo, G. Non-steroidal antiinflammatory agents. VII. Synthesis and antiinflammatory activity of 5-methyl-10,11-dihydro-5H-pyrrolo[1,2-b][1,2,5]-benzotriazepine-11-acetic acid and its 10-aroyl derivatives. Farmaco 1990, 45, 817–831. [Google Scholar] [PubMed]
- Artico, M.; Silvestri, R.; Stefancich, G. Heterocycles with a benzothiadiazepine moiety. 1. Synthesis of pyrrolo[1,2-b]-s-triazolo[3,4-d][1,2,5]benzothiadiazepine 5,5-dioxide. Synth. Commun. 1992, 22, 1433–1439. [Google Scholar] [CrossRef]
- Stefancich, G.; Silvestri, R.; Pagnozzi, E.; Artico, M. Heterocycles with a benzothiadiazepine moiety. 2. Synthesis of 2-methyl-1,3,4,14b-tetrahydro-2H-pyrazino[2,1-d]pyrrolo[1,2-b][1,2,5]benzothiadiazepine 10,10-dioxide (Tiaatpazepine). J. Heterocycl. Chem. 1994, 31, 867–869. [Google Scholar]
- Silvestri, R.; Pagnozzi, E.; Stefancich, G.; Artico, M. Heterocycles with a benzothiadiazepine moiety. 4. Synthesis of novel tetracyclic rings by intramolecular cyclization of 10-bromoacetyl-10,11-dihydro-11-ethoxycarbonylpyrrolo[1,2-b][1,2,5]benzothiadiazepine 5,5-dioxide and its derivatives. Synth. Commun. 1994, 24, 2685–2695. [Google Scholar] [CrossRef]
- Silvestri, R.; Artico, M.; La Regina, G.; di Pasquali, A.; de Martino, G.; la Torre, F.; Cirilli, R.; Cagnotto, A.; Mennini, T. Chiral resolution and binding study of 1,3,4,14b-tetrahydro-2,10-dimethyl-2H,10H-pyrazino[2,1-d]pyrrolo[1,2-b][1,2,5]benzotriazepine (10-methyl-10-azaaptazeine) and 2-methyl-1,3,4,14b-tetrahydro-2H-pyrazino-[2,1-d]pyrrolo[1,2-b][1,2,5]benzothiadiazepine 10,10-dioxide (tiaaptazepine). Farmaco 2005, 60, 931–937. [Google Scholar]
- Silvestri, R.; Artico, M.; Pagnozzi, E.; Stefancich, G. Heterocycles with a benzothiadiazepine moiety. 3. Synthesis of imidazo[5,1-d]pyrrolo[1,2-b][1,2,5]benzothiadiazepine 9,9-dioxide. J. Heterocycl. Chem. 1994, 31, 1033–1036. [Google Scholar] [CrossRef]
- Artico, M.; Silvestri, R.; Pagnozzi, E.; Stefancich, G.; Massa, S.; Loi, A.G.; Scano, P.; Corrias, S.; Spiga, M.G.; La Colla, P. 5H-Pyrrolo[1,2-b][1,2,5]benzothiadiazepines (PBTDs): A novel class of HIV-1-specific non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. 1996, 4, 837–885. [Google Scholar] [CrossRef]
- Ettorre, A.; Silvestri, R.; Artico, M.; Massa, S.; La Colla, P. Crystal structure of 7-chloro[1,2-b][1,2,5]benzothiadiazepin-10(11H)-one-5,5-dioxide, C11H7N2O3ClS. Z. Kristallogr. NCS 2001, 216, 57–58. [Google Scholar]
- Schäfer, W.; Friebe, W.G.; Leinert, H.; Mertens, A.; Poll, T.; von der Saal, W.; Zilch, H.; Nuber, B.; Ziegler, M.L. Non-nucleoside inhibitors of HIV-1 reverse transcriptase: Molecular modeling and X-ray structure investigations. J. Med. Chem. 1993, 36, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Artico, M.; Silvestri, R.; Pagnozzi, E.; Stefancich, G.; Massa, S.; La Colla, P. Synthesis and anti-HIV activity of 10,11-dihydropyrrolo[1,2-b][1,2,5]benzothiadiazepine-11-acetic acid 5,5-dioxide derivatives and related compounds. Farmaco 1996, 51, 425–430. [Google Scholar]
- Silvestri, R.; Artico, M.; Bruno, B.; Massa, S.; Novellino, E.; Greco, G.; Marongiu, M.E.; Pani, A.; de Montis, A.; La Colla, P. Synthesis and biological evaluation of 5H-indolo[3,2-b][1,5]benzothiazepine derivatives, designed as conformationally constrained analogues of the human immunodeficiency virus type 1 reverse transcriptase inhibitor L-737,126. Antivir. Chem. Chemother. 1998, 9, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Artico, M.; Stefancich, G.; Silvestri, R.; Massa, S.; Pagnozzi, E.; Loi, A.G.; Musu, D.; Doa, M.; Scano, P.; La Colla, P. Pyrrolobenzothiazepines: A new class of non-nucleoside HIV-1 reverse transcriptase inhibitors. Med. Chem. Res. 1994, 4, 283–290. [Google Scholar]
- Silvestri, R.; Pagnozzi, E.; Artico, M.; Stefancich, G.; Massa, S.; La Colla, P. Synthesis of 9H-pyrrolo[2,1-b][1,3,6]benzothiadiazocin-10(11H)-one 4,4-dioxide, potential anti-HIV agent. J. Heterocycl. Chem. 1995, 32, 683–685. [Google Scholar] [CrossRef]
- Thurston, D.E.; Bose, D.S. Synthesis of DNA-interactive pyrrolo-[2,1-c][1,4]benzodiazepines. Chem. Rev. 1994, 94, 433–465. [Google Scholar] [CrossRef]
- Silvestri, R.; Marfè, G.; Artico, M.; La Regina, G.; De Martino, G.; Lavecchia, A.; Novellino, E.; Morgante, E.; di Stefano, C.; Catalano, G.; et al. Pyrrolo[1,2-b][1,2,5]benzothiadiazepines (PBTDs): A new class of agents endowed with high apoptotic activity in chronic myelogenous leukemia K562 cells and in cells from patients at onset and imatinib-resistant. J. Med. Chem. 2006, 49, 5840–5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marfè, G.; di Stefano, C.; Silvestri, R.; Abruzzese, E.; Catalano, G.; Di Renzo, L.; Filomeni, G.; Giorda, E.; La Regina, G.; Morgante, E.; et al. Pyrrolo[1,2-b][1,2,5]benzothiadiazepines (PBTDs) induce apoptosis in chronic myelogenous leukemic K562 cells. BMC Cancer 2007, 7, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stefano, C.; Marfe, G.; Trawinska, M.M.; Sinibaldi-Salimei, P.; Silvestri, R.; Amadori, S.; Abruzzese, E. Pyrrolo[1,2-b][1,2,5]benzothiadiazepines (PBTDs) exert their anti-proliferative activity by interfering with Akt-mTOR signaling and bax:bcl-2 ratio modulation in cells from chronic myeloid leukemic patients. Cancer Sci. 2010, 101, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, R.; Marfè, G.; Di Stefano, C.; Sinibaldi Salimei, P.; De Martino, M.G.; La Regina, G.; Abruzzese, E.; Catalano, G. Derivati delle Benzodiazepine e loro uso in Campo Medico. Patent IT2006/000401, 3 August 2005. [Google Scholar]
- Silvestri, R.; Marfè, G.; Abruzzese, E.; Catalano, G.; Di Stefano, C.; Novellino, E.; Sinibaldi Salimei, P.; La Regina, G.; Lavecchia, A. Benzodiazepine Derivatives and Uses Thereof in Medical Field. Patent WO 2007/015280, 8 February 2007. [Google Scholar]
- McMahon, J.B.; Gulakowski, R.J.; Weislow, O.S.; Schultz, R.J.; Narayanan, V.L.; Clanton, D.J.; Pedemonte, R.; Wassmundt, F.W.; Buckheit, R.W., Jr.; Decker, W.D.; et al. Diarylsulfones, a new chemical class of nonnucleoside antiviral inhibitors of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 1993, 37, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Artico, M.; Silvestri, R.; Stefancich, G.; Massa, S.; Pagnozzi, E.; Musu, D.; Scintu, F.; Pinna, E.; Tinti, E.; La Colla, P. Synthesis of pyrryl aryl sulfones targeted at the HIV-1 reverse transcriptase. Arch. Pharm. (Weinheim) 1995, 328, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Langlois, N.; Andriamialisoa, R.Z. Synthesis of sulfonamide analogs of the pyrrolo[1,4]benzodiazepine antibiotic abbeymycin. Heterocycles 1989, 8, 1529–1536. [Google Scholar] [CrossRef]
- Artico, M.; Silvestri, R.; Massa, S.; Loi, A.G.; Corrias, S.; Piras, G.; La Colla, P. 2-Sulfonyl-4-chloroanilino moiety: A potent pharmacophore for the anti-human immunodeficiency virus type 1 activity of pyrrolyl aryl sulfones. J. Med. Chem. 1996, 39, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, R.; Artico, M.; Massa, S.; Stefancich, G.; Congeddu, E.; Putzolu, M.; La Colla, P. Sulfone derivatives with anti-HIV activity. Farmaco 1997, 52, 323–329. [Google Scholar] [PubMed]
- Pawels, R. Discovery of TIBO, a new family of HIV-1 specific reverse transcriptase inhibitors. In The Search for Antiviral Drugs; Adams, J., Merluzzi, V.J., Eds.; Birkhäuser: Boston, MA, USA, 1993; Chapter 4; pp. 71–104. [Google Scholar]
- De Clercq, E. Antiviral therapy for human immunodeficiency virus infections. Clin. Microbiol Rev. 1995, 8, 200–239. [Google Scholar] [PubMed]
- Baba, M.; Okamoto, M.; Makino, M.; Kimura, Y.; Ikeuchi, T.; Sakaguchi, T.; Okamoto, T. Potent and selective inhibition of human immunodeficiency virus type 1 transcription by piperazinyloxoquinoline derivatives. Antimicrob. Agents Chemother. 1997, 41, 1250–1255. [Google Scholar] [PubMed]
- Högberg, M.; Sahlberg, C.; Engelhardt, P.; Noréen, R.; Kangasmetsä, J.; Johansson, N.G.; Oberg, B.; Vrang, L.; Zhang, H.; Sahlberg, B.L.; et al. Urea-PETT compounds as a new class of HIV-1 reverse transcriptase inhibitors. 3. Synthesis and further structure-activity relationship studies of PETT analogues. J. Med. Chem. 1999, 20, 4150–4160. [Google Scholar] [CrossRef]
- Ren, J.; Esnouf, R.; Garman, E.; Somers, D.; Ross, C.; Kirby, I.; Keeling, J.; Darby, G.; Jones, Y.; Stuart, D.; et al. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Biol. 1995, 2, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Artico, M.; Silvestri, R.; Pagnozzi, E.; Bruno, B.; Novellino, E.; Greco, G.; Massa, S.; Ettorre, A.; Loi, A.G.; Scintu, F.; et al. Structure-based design, synthesis and biological evaluation of novel pyrrolyl aryl sulfones (PASs), HIV-1 non-nucleoside reverse transcriptase inhibitors active at nanomolar concentrations. J. Med. Chem. 2000, 43, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, R.; Artico, M.; La Regina, G.; De Martino, G.; La Colla, M.; Loddo, R.; La Colla, P. Anti-HIV-1 activity of pyrryl aryl sulfone (PAS) derivatives. Synthesis and SAR studies of novel esters and amides at the position 2 of the pyrrole nucleus. Farmaco 2004, 59, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, R.; Artico, M.; De Martino, G.; Novellino, E.; Greco, G.; Lavecchia, A.; Massa, S.; Loi, A.G.; Doratiotto, S.; La Colla, P. Computer-assisted design, synthesis and biological evaluation of novel pyrrolyl heteroaryl sulfones targeted at HIV-1 reverse transcriptase as non-nucleoside inhibitors. Bioorg. Med. Chem. 2000, 8, 2305–2309. [Google Scholar] [CrossRef]
- Cantrell, A.S.; Engelhardt, P.; Högberg, M.; Jaskunas, S.R.; Johansson, N.G.; Jordan, C.L.; Kangasmetsä, J.; Kinnick, M.D.; Lind, P.; Morin, J.M., Jr.; et al. Phenethylthiazolylthiourea (PETT) compounds as a new class of HIV-1 reverse transcriptase inhibitors. 2. Synthesis and further structure-activity relationship studies of PETT analogs. J. Med. Chem. 1996, 39, 4261–4274. [Google Scholar] [CrossRef] [PubMed]
- Breslin, H.J.; Kukla, M.J.; Kromis, T.; Cullis, H.; de Knaep, F.; Pauwels, R.; Andries, K.; De Clercq, E.; Janssen, M.A.; Janssen, P.A. Synthesis and anti-HIV activity of 1,3,4,5-tetrahydro-2H-1,4-benzodiazepin-2-one (TBO) derivatives. Truncated 4,5,6,7-tetrahydro-5-methylimidazo[4,5,1-jk][1,4]benzodiazepin-2(1H)-on es (TIBO) analogues. Bioorg. Med. Chem. 1999, 7, 2427–2436. [Google Scholar] [CrossRef]
- Pauwels, R.; Andries, K.; Desmyter, J.; Schols, D.; Kukla, M.J.; Breslin, H.J.; Raeymaeckers, A.; van Gelder, J.; Woestenborghs, R.; Heykants, J.; et al. Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature 1990, 343, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Ding, J.; Hsiou, Y.; Clark, A.D., Jr.; Moereels, H.; Koymans, L.; Andries, K.; Pauwels, R.; Janssen, P.A.; Boyer, P.L.; et al. Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RT and 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-resistant mutant. J. Mol. Biol. 1996, 26, 1085–1100. [Google Scholar] [CrossRef]
- Silvestri, R.; De Martino, G.; Artico, M.; La Regina, G.; Ragno, R.; Loddo, R.; La Colla, P.; Marongiu, M.E.; La Colla, M.; Pani, A. Anti-HIV-1 NNRT agents: Acylamino pyrryl aryl sulfones (APASs) as truncated analogues of tricyclic PBTDs. Med. Chem. Res. 2002, 11, 195–218. [Google Scholar]
- Chan, J.H.; Hong, J.S.; Hunter, R.N., 3rd; Orr, G.F.; Cowan, J.R.; Sherman, D.B.; Sparks, S.M.; Reitter, B.E.; Andrews, C.W., 3rd; Hazen, R.J.; et al. 2-Amino-6-arylsulfonylbenzonitriles as non-nucleoside reverse transcriptase inhibitors of HIV-1. J. Med. Chem. 2001, 44, 1866–1882. [Google Scholar] [CrossRef] [PubMed]
- Titmuss, S.J.; Keller, P.A.; Griffith, R. Docking experiments in the flexible non-nucleoside inhibitor binding pocket of HIV-1 reverse transcriptase. Bioorg. Med Chem. 1999, 7, 1163–1170. [Google Scholar] [CrossRef]
- Silvestri, R.; Pifferi, A.; De Martino, G.; Saturnino, C.; Artico, M. Reductive Smiles rearrangement of 1-[(5-chloro-2-nitrophenyl)sulfonyl]-1H-pyrrole-2-carbohydrazide to 1-amino-6-chloro-2-(1H-pyrrol-2-yl)benzimidazole. Heterocycles 2000, 53, 2163–2174. [Google Scholar]
- Skarżewski, J.; Skrowaczewska, Z. The smiles rearrangement: Mechanism of unusual acyl and 2,4-dinitrophenyl migrations in aryl acylamino ethers. Tetrahedron 1976, 32, 1221–1224. [Google Scholar] [CrossRef]
- Williams, T.A.; Ciccarone, T.M.; Saari, W.S.; Wai, J.S.; Greenlee, W.J.; Balani, S.K.; Goldman, M.E.; Theoharides, A.D. Indoles as Inhibitors of HIV Reverse Transcriptase. Patent Application EP 0 530 907 A1, 28 August 1992. [Google Scholar]
- Williams, T.A.; Ciccarone, T.M.; Greenlee, W.J.; Balani, S.K.; Goldman, M.E.; Hoffman, J.M., Jr.; Lumma, W.C.; Huff, J.R.; Rooney, C.S.; Sanderson, P.E.; Theoharides, A.D. Patent Application WO 94/19321 A1, 01 September 1994.
- Williams, T.M.; Ciccarone, T.M.; MacTough, S.C.; Rooney, C.S.; Balani, S.K.; Condra, J.H.; Emini, E.A.; Goldman, M.E.; Greenlee, W.J.; Kauffman, L.R.; et al. 5-Chloro-3-(phenylsulfonyl)indole-2-carboxamide: A novel, non-nucleoside inhibitor of HIV-1 reverse transcriptase. J. Med. Chem. 1993, 36, 1291–1294. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, R.; Artico, M. Indolyl aryl sulfones (IASs): Development of highly potent NNRTIs active against wt-HIV-1 and clinically relevant drug resistant mutants. Curr. Pharm. Des. 2005, 11, 3779–3806. [Google Scholar] [CrossRef]
- Silvestri, R.; De Martino, G.; La Regina, G.; Artico, M.; Massa, S.; Vargiu, L.; Mura, M.; Loi, A.G.; Marceddu, T.; La Colla, P. Novel indolyl aryl sulfones active against HIV-1 carrying NNRTI resistance mutations: Synthesis and SAR studies. J. Med. Chem. 2003, 46, 2482–2493. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, R.; Artico, M.; De Martino, G.; La Regina, G.; Loddo, R.; La Colla, M.; La Colla, P. Simple, short peptide derivatives of a sulfonylindolecarboxamide (L-737,126) active in vitro against HIV-1 wild-type and variants carrying non-nucleoside reverse transcriptase inhibitor resistance mutations. J. Med. Chem. 2004, 47, 3892–3896. [Google Scholar] [CrossRef] [PubMed]
- Piscitelli, F.; Coluccia, A.; Brancale, A.; La Regina, G.; Sansone, A.; Giordano, C.; Balzarini, J.; Maga, G.; Zanoli, S.; Samuele, A.; et al. Indolylarylsulfon bearing natural and unnatural amino acids. Discovery of potent inhibitors of HIV-1 non-nucleoside wild type and resistant mutant strains reverse transcriptase and coxsackie B4 virus. J. Med. Chem. 2009, 52, 1922–1934. [Google Scholar] [CrossRef] [PubMed]
- La Regina, G.; Coluccia, A.; Piscitelli, F.; Bergamini, A.; Sinistro, A.; Cavazza, A.; Maga, G.; Samuele, A.; Zanoli, S.; Novellino, E.; et al. Indolyl aryl sulfones as HIV-1 non-nucleoside reverse transcriptase inhibitors: Role of two halogen atoms at the indole ring in developing new analogues with improved antiviral activity. J. Med. Chem. 2007, 50, 5034–5038. [Google Scholar] [CrossRef] [PubMed]






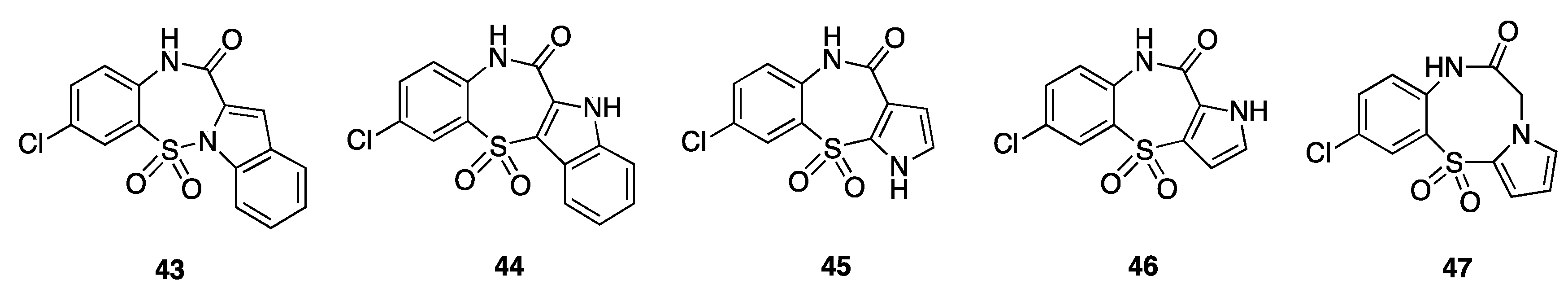









| Compound | R | HIV-1 IIIB | ||
|---|---|---|---|---|
| CC50 b (μM) | EC50 c (μM) | SI d | ||
| 37a | H | >300 | 1.0 | >300 |
| 37b | Me | >300 | 0.5 | >600 |
| 37c | Et | 283 | 2.4 | 118 |
| 37d | Propyl | 126 | 14 | 9 |
| 37e | Isopropyl | >300 | Nd e | - |
| 37f | Allyl | >300 | 3.7 | >81 |
| 37g | Crotyl | >300 | 4.1 | >73 |
| 37h | Dimethylallyl | >300 | 129 | >2 |
| Patient | Sex | Age | Source a | % of Apoptosis | |||
|---|---|---|---|---|---|---|---|
| 23 | 49 | ||||||
| 24 h | 48 h | 24 h | 48 h | ||||
| 1 | M | 45 | PB a | 64 | 70 | 77 | 85 |
| 2 | M | 60 | BM b | 50 | 70 | 70 | 85 |
| 3 | F | 73 | PB | 65 | 79 | 66 | 82 |
| 4 | M | 83 | BM | 50 | 70 | 50 | 75 |
| 5 | F | 46 | PB | 50 | 70 | 60 | 80 |
| 6 | F | 27 | PB | 50 | 70 | 60 | 80 |
| 7 | F | 45 | PB | 60 | 80 | 64 | 80 |
| 8 | M | 35 | PB | 60 | 80 | 65 | 85 |
| 9 | M | 66 | PB | 60 | 80 | 60 | 80 |
| 10 | F | 38 | PB | 50 | 70 | 70 | 80 |
| 11 | F | 65 | PB | 52 | 71 | 55 | 78 |
| 12 | F | 27 | PB | 52 | 73 | 55 | 78 |
| Patient | Sex | Age | Source a | Percent Apoptosis | |||
|---|---|---|---|---|---|---|---|
| 23 | 49 | ||||||
| 24 h | 48 h | 24 h | 48 h | ||||
| 13 | M | 38 | PB a | 60 | 80 | 40 | 60 |
| 14 | F | 70 | PB | 55 | 78 | 50 | 70 |
| Compound | HIV-1 IIIB | ||
|---|---|---|---|
| CC50 b (μM) | EC50 c (μM) | SI d | |
| 16 | >308 | 15.08 | >20 |
| 51 | 36.55 | >36.55 | - |
| 52 | >337.5 | >337.5 | - |
| 53 | >262.2 | >262.2 | - |
| 54 | >333 | >333 | - |
| 55 | 255 | >255 | - |
| 56 | >370 | 63 | >5.8 |
| 57 | >279 | >279 | - |
| 58 | >285 | >285 | - |
| NPPS | - | 1.4 | - |
| Compound | R1 | R2 | HIV-1 IIIB | ||
|---|---|---|---|---|---|
| CC50 b (μM) | EC50 c (μM) | SI d | |||
| 59a | 2-NH2-5-Cl | 2-COOMe | >300 | 0.18 | >2140 |
| 59b | 2-NH2-5-Cl | 2-COOEt | >300 | 0.14 | >2140 |
| 59c | 2-NO2 | 2-COOEt | >300 | 15 | >20 |
| 59d | 2-Cl | 2-COOEt | 141 | 25 | 5 |
| 59e | 2-NH2-5-Cl | 2-COOCH2CHC=CH2 | 100 | 0.40 | 250 |
| 59f | 2-NHCHO-5-Cl | 2-COOEt | >300 | 1.0 | >300 |
| 59g | 2-NHCOMe-5-Cl | 2-COOEt | ≥300 | 1.0 | ≥300 |
| 59h | 2-NHCOOEt | 2-COOEt | >300 | 1.0 | >300 |
| NVP e | >10000 | 0.60 | >167 | ||
| Compound | IC50 ± SD (μM) a | ||
|---|---|---|---|
| WT IIIB | Y181C | L100I | |
| 59a | 0.45 ± 0.09 | 6.9 ± 2.3 | 7.4 ± 1.2 |
| 59b | 0.40 ± 0.05 | 7.5 ± 1.4 | 8.5 ± 1.0 |
| 59c | 0.40 ± 0.14 | 5.0 ± 1.5 | 10 ± 3.1 |
| 59d | 0.27 ± 0.10 | 8.0 ± 2.0 | 14 ± 1.2 |
| 59e | 0.90 ± 0.12 | 14 ± 2.5 | >20 |
| 59f | >20 | >20 | >20 |
| 59g | >20 | >20 | >20 |
| 59h | >20 | >20 | >20 |
| NVP | 0.60 ± 0.1 | >20 | 3.5 ± 0.18 |
| Compound | CC50 b (μM) | HIV-1 IIIB | IC50 e (μM) | |
|---|---|---|---|---|
| EC50 c (μM) | SI d | |||
| 60 | 240 | 0.042 | 5333 | 0.05 |
| NVP f | >200 | 0.35 | >571 | 0.64 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Famiglini, V.; Castellano, S.; Silvestri, R. N-Pyrrylarylsulfones with High Therapeutic Potential. Molecules 2017, 22, 434. https://doi.org/10.3390/molecules22030434
Famiglini V, Castellano S, Silvestri R. N-Pyrrylarylsulfones with High Therapeutic Potential. Molecules. 2017; 22(3):434. https://doi.org/10.3390/molecules22030434
Chicago/Turabian StyleFamiglini, Valeria, Sabrina Castellano, and Romano Silvestri. 2017. "N-Pyrrylarylsulfones with High Therapeutic Potential" Molecules 22, no. 3: 434. https://doi.org/10.3390/molecules22030434
APA StyleFamiglini, V., Castellano, S., & Silvestri, R. (2017). N-Pyrrylarylsulfones with High Therapeutic Potential. Molecules, 22(3), 434. https://doi.org/10.3390/molecules22030434