1. Introduction
Coral reef is one of the biggest marine ecosystems. Because corals lack efficient physical protection in the heavily competitive environment, they mainly rely on chemical protection to survive. For the past two decades, it was found that a considerable part of the bioactive substances isolated from marine flora and fauna were actually generated from their symbiotic microorganisms [
1]. These findings attracted a great deal of attention in investigating the secondary metabolites from the microorganisms associated with algae and invertebrates, such as soft corals and sponges [
2,
3]. To date, a number of compounds including polyketides [
4], alkaloids [
5], lipids [
6], anthraquinones [
7], terpenes [
8] and peptides [
6] have been discovered from these commensal microorganisms. These compounds showed wide-ranging biological properties such as anti-fungal, cytotoxic, anti-feedant and anti-viral activities. For example, Dehai Li et al. obtained two unique fumiquinazoline alkaloids with anti-influenza virus A from a mangrove-derived fungus,
Neosartorya udagawae [
9].
To explore bioactive natural products, we have initiated a program to screen and discover unique molecules with cytotoxic activity from the marine-derived fungi related to invertebrates, such as soft corals and sea stars. In a previous study, the marine fungus
Neosartorya pseudofischeri manufactured a series of potent cytotoxic molecules, such as gliotoxin, acetylgliotoxin, and reduced gliotoxin [
10]. The fungal strain
Pseudallescheria boydii produced diverse chemical structures, and some of them showed notable cytotoxic activity against Sf9 cells from the fall armyworm
Spodoptera frugiperda. In a small-scale culture of
Dichotomomyces sp., we found that the EtOAc extract of the culture broth showed significant cytotoxic activity with an inhibition rate of 80% at 30 μg/mL against the human breast cancer cell line MDA-MB-435. A literature search revealed that the secondary metabolites of the genus
Dichotomomyces were abundant and displayed potent bioactivity. For instance, Henrik et al. obtained several indoloditerpenes with antagonistic activity that targeted GPR18 and cannabinoid receptors from
Dichotomomyces cejpii [
11]. Aiming to find novel chemicals with cytotoxic activity, we explored the secondary metabolites of
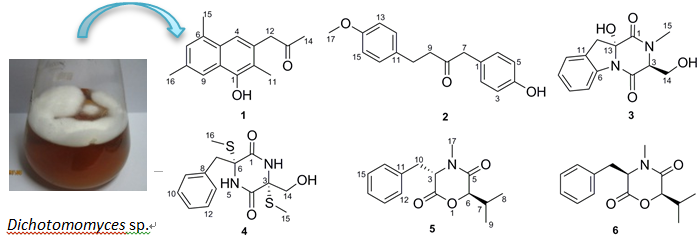
Dichotomomyces sp. and obtained two new molecules, dichotones A and B (
1 and
2), together with four known compounds, dichotocejpin C (
3), bis-
N-norgliovictin (
4), bassiatin (
5) and (3
R,6
R)-bassiatin (
6). Compound
6 displayed significant cytotoxicity against the human breast cancer cell line MDA-MB-435 and the human lung cancer cell line Calu3. Herein, we report the isolation, structure determination and bioactive evaluation of these compounds.
2. Results and Discussion
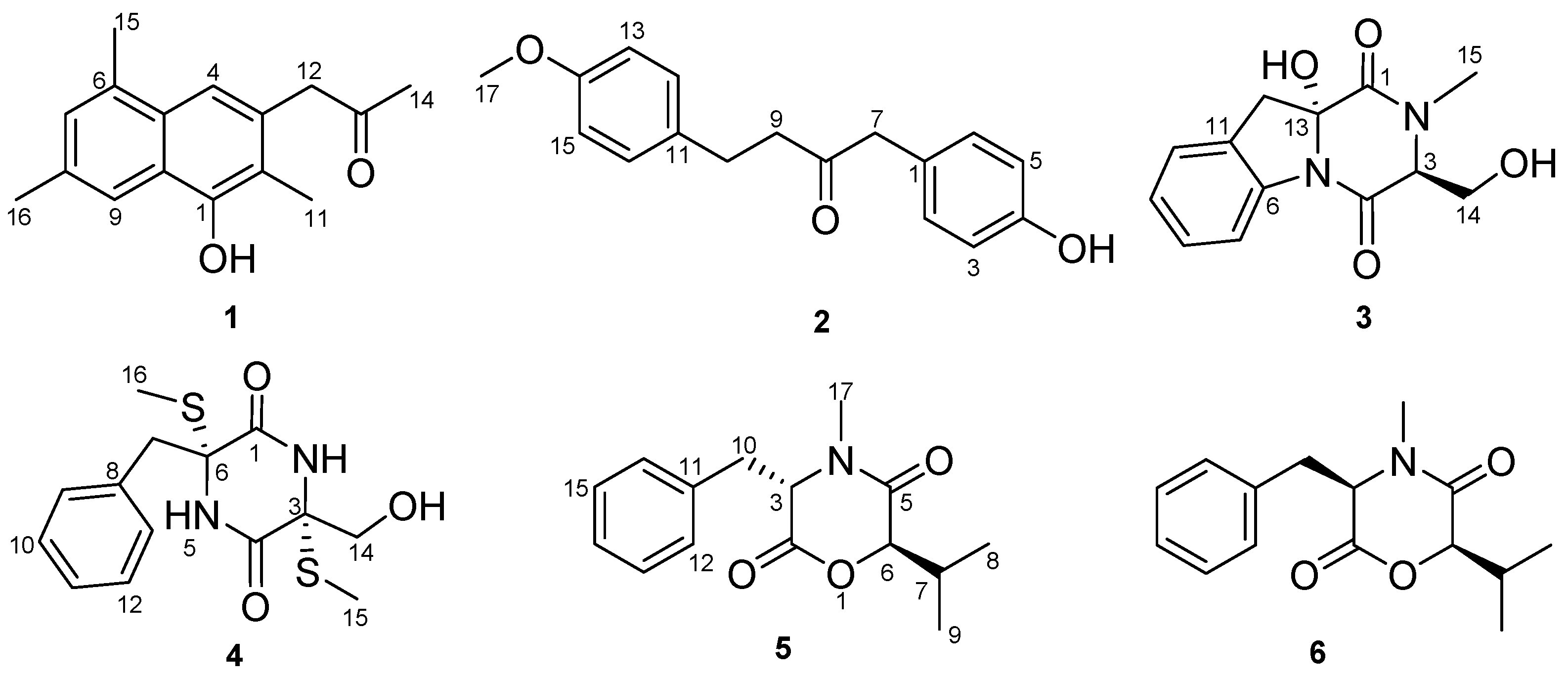
Dichotone A (
1) was obtained as a white amorphous powder. The HRESIMS (high resolution electrospray ionization mass spectroscopy) spectrum showed a strong quasi-molecular ion peak at
m/
z 241.1237 [M − H]
− (calcd. 241.1234), corresponding to the molecular formula C
16H
18O
2, requiring eight double equivalents. The
1H-NMR spectrum of dichotone A (
1) (
Table 1) displayed resonances for 18 protons, including four methyls (δ
H 2.16, 2.34, 2.41 and 2.50, each s), one methylene (δ
H 3.87, s), three aromatic proton signals (δ
H 7.39, 7.49 and 7.61, each s), and one exchangeable singlet proton signal at δ
H 4.87 for the hydroxyl moiety. The
13C NMR and DEPT spectra (
Table 1) showed signals for 16 carbons, including four methyls at δ
C 10.8, 17.1, 19.9 and 29.4, one methylene at δ
C 50.1, 10 aromatic carbons at δ
C 149.9, 132.0, 131.7, 131.5, 128.8, 128.6, 126.2, 125.6, 124.6 and 114.1, and one carbonyl carbon at δ
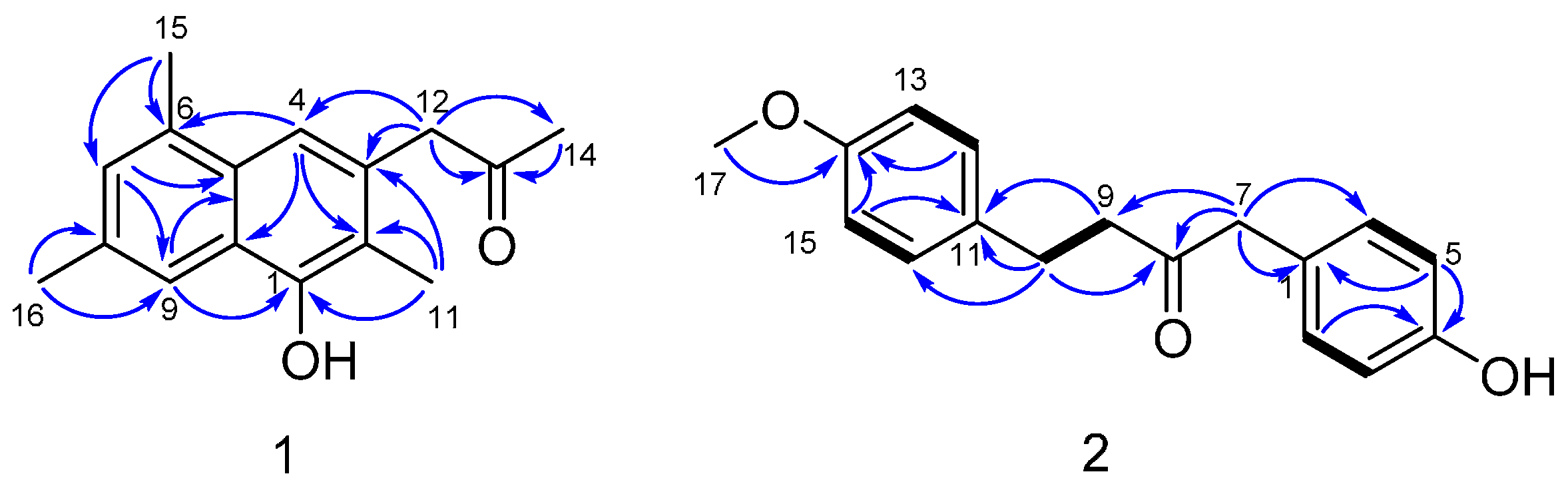
C 207.2. The existence of a naphthalene was deduced by the HMBC correlations (
Figure 2) from H-4 to C-2, C-10 and C-6, from H-7 to C-5 and C-9, and from H-9 to C-1 and C-5. Three methyls were substituted at C-2, C-6 and C-8, respectively, which was supported by the HMBC correlations from H
3-11 to C-1, C-2 and C-3, from H
3-15 to C-6 and C-7, and from H
3-16 to C-8 and C-9. The HMBC correlations from H
3-14 to C-13, from H
2-12 to C-13, C-14, C-3 and C-4 illustrated that a propan-2-one was linked to C-3. As showed in the HRESIMS spectrum, there was a hydroxyl substituted at C-3. The typical down-field shift of C-1 (δ
C 149.9) elaborated the substitution of a remaining hydroxyl at C-1. Thus, the structure of dichotone A (
1) was constructed as shown in
Figure 1.
Dichotone B (
2) was acquired as a white amorphous powder. It had the molecular formula C
17H
18O
3, which was established on the basis of the HRESIMS ion detected at
m/
z 269.1176 [M − H]
− (calcd. 269.1183). Analysis of the 1D NMR data and HSQC spectrum suggested the existence of 18 proton and 17 carbon signals, including one keto-carbonyl (δ
C 208.3), four couples of symmetric aromatic sp
2 methine groups (δ
C 114.0, δ
H 6.79 (d, 8.8); δ
C 115.7, δ
H 6.77 (d, 8.4); δ
C 129.4, δ
H 7.04 (d, 8.8); δ
C 130.8, δ
H 7.02 (d, 9.2) ), four sp
2 quaternary carbon atoms (δ
C 126.4, 133.1, 154.7, 158.0), three sp
3 methylene groups (δ
C 29.1, δ
H 2.80 (td, 6.8, 1.6); δ
C 43.7, δ
H 2.71 (td, 6.8, 1.6); δ
C 49.6, δ
H 3.57 (s)), one methoxy group (δ
C 55.4, δ
H 3.77 (s)), and one exchangeable singlet proton (δ
H 4.83, brs). Considering the nine degrees of unsaturation calculated from the molecular formula, there were two rings existing in the structure. The presence of two
para-benzene systems in
2 was deduced from characteristic
13C-NMR resonances (
Table 1; C-1 to C-6 and C-11 to C-16), along with the HMBC correlations from H-2 (H-6) to C-4, from H-5 (H-3) to C-1 and C-4, from H-15 (H-13) to C-11 and C-14, and from H-12 (H-16) to C-14 and the cross-peaks H-2/H-3 and H-12/H-13 in the COSY spectrum. The HMBC correlations from H
2-7 to C-8 and C-9, H
2-9 to C-8, H
2-10 to C-8 and the COSY correlations between H
2-9 and H
2-10 suggested the existence of a 1,4-disubstituted butan-2-one. Two
para-benzene systems were linked by the 1,4-disubstituted butan-2-one based on the HMBC correlations from H
2-7 to C-1 and C-6 (C-2), from H
2-9 to C-11, and from H
2-10 to C-11 and C-12 (C-16). A methoxy group was linked to C-14 by the evidence of the HMBC correlation from H
3-17 to C-14. A hydroxyl was substituted at C-4 because of the characteristic down-field shift of C-4 (δ
C 157.4), which was in accordance with the molecular formula C
17H
18O
3. Finally, the structure of
2 was established as shown in
Figure 1. More spectral data see
Supplementary.
Compounds
3–
6 were identified as dichotocejpin C [
12], bis-
N-norgliovictin [
10], bassiatin [
13] and (3
R,6
R)-bassiatin [
14], respectively, by comparing the data with the literature values.
The (3
R,6
R)-bassiatin (
6) and bassiatin (
5) are a pair of diastereomers. In the previous research, bassiatin (
5) was a platelet aggregation inhibitor [
13], while (3
R,6
R)-bassiatin (
6) could inhibit proliferation and induce apoptosis of several human cancer cells [
15,
16,
17]. Here, they were screened for their cytotoxic activities against the human breast cancer cell line MDA-MB-435 and the human lung cancer cell line Calu3. Interestingly, their cytotoxic activities were totally different. Compound
6 exhibited distinguished cytotoxic activities against the cell lines MDA-MB-435 and Calu3 with IC
50 values of 7.34 ± 0.20 and 14.54 ± 0.01 μM, respectively. However, compound
5 did not show any distinct activity with IC
50 values higher than 50μM. The cytotoxic activities of compounds
1–
4 were not evaluated in this assay due to the limited sample weight.
3. Materials and Methods
3.1. General Experimental Procedures
Silica gel (SiO2, 200−300 mesh, Qingdao Marine Chemical Inc., Qingdao, China) and Sephadex LH-20 (green herbs, Beijing, China) were used for column chromatography. Preparative HPLC was performed using a Shimadzu LC-20AT HPLC pump (Shimadzu Corporation, Nakagyo-ku, Kyoto, Japan) installed with an SPD-20A dual λ absorbance detector (Shimadzu Corporation, Nakagyo-ku, Kyoto, Japan) and a Shim-pack PRC-ODS HPLC column (250 mm × 20 mm, Shimadzu Corporation, Nakagyo-ku, Kyoto, Japan). 1D and 2D NMR spectra were recorded on Bruker Avance II 400 spectrometers (Bruker BioSpin AG, Industriestrasse 26, Fällanden, Switzerland) and a Varian Mercury-Plus 300 spectrometer (Varian Medical Systems In., Salt Lake City, UT, USA) in CDCl3 and acetone-d6. The chemical shifts are relative to the residual solvent signals (CDCl3: δH 7.260 and δC 77.160 and acetone-d6: δH 2.050 and δC 29.840). The high-resolution ESI-MS spectra was obtained with Shimadzu LCMS-IT-TOF (Shimadzu Corporation, Nakagyo-ku, Kyoto, Japan). UV spectra were recorded on a Shimadzu UV-Vis-NIR spectrophotometer (Shimadzu Corporation, Nakagyo-ku, Kyoto, Japan). IR spectra were recorded on a PerkinElmer Frontier FT-IR spectrophotometer (PerkinElmer Inc., Waltham, MA, USA).
3.2. Fungal Strain and Culture Method
The marine fungus Dichotomomyces sp. L-8 was obtained from the inner tissue of the soft coral Lobophytum crassum collected from Hainan Sanya National Coral Reef Reserve, China. This fungal strain was maintained in 15% (v/v) glycerol aqueous solution at −80 °C. A voucher specimen was deposited in the School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, China. Analysis of the ITS rDNA (GenBank KF999816) by BLAST database screening provided a 100% match to Dichotomomyces sp. (AB734448).
The marine fungus, Dichotomomyces sp., was cultured in the medium which contained glucose 10 g/L, peptone 5 g/L, yeast extract 2 g/L, sea salt 22 g/L, H2O 1 L and pH 7.5. Fungal mycelia were cut and transferred aseptically to 1000 mL Erlenmeyer flasks each containing 400 mL sterilized liquid medium. The flasks were incubated at 28 °C for 40 days.
3.3. Extraction and Isolation
Seventy liters of liquid culture were filtered through cheesecloth. The culture broth was extracted three times with EtOAc and then was concentrated by low-temperature rotary evaporation to afford a crude extract (20 g).
The extract (20 g) was chromatographed on a silica gel column (diameter: 8 cm, length: 70 cm, silica gel, 200 g) with a gradient of petroleum ether-EtOAc (100:0–0:100, v/v) followed by EtOAc–MeOH (100:0–0:100, v/v) to yield seven fractions (Fr. 1–Fr. 7). Fr. 3 was purified by silica gel column using a step gradient elution with CHCl2–acetone (10:0–0:10, v/v) to get 10 subfractions (Fr. 3-1–Fr. 3-10) after pooling the similar fractions as monitored by TLC. Fr. 3–5 was chromatographed on Sephadex LH-20 (MeOH), then purified with preparative HPLC (MeOH–H2O, 60:40, v/v) to obtain compound 5 (5 mg) and 6 (4 mg). Fraction 4 was chromatographed on ODS (MeOH–H2O, 20:80–100:0, v/v), then on Sephadex LH-20 with MeOH as the eluent followed by preparative HPLC (MeOH–H2O, 63:37, v/v) to afford compound 3 (0.9 mg). Meanwhile, compound 4 (0.9 mg) was crystallized from Fr. 4-4. Fraction 5 was separated as six subfractions (Fr. 5-1–Fr. 5-6) with ODS using a gradient elution MeOH–H2O (20:80–100:0, v/v). Fr. 5-3 and Fr. 5-4 were further purified by Sephadex LH-20 (MeOH) and then by preparative HPLC (MeOH–H2O, 56:44, v/v; MeOH–H2O, 59:41, v/v), respectively, to acquire compound 1 (0.7 mg) and 2 (0.9 mg).
3.4. Characterization of Compounds 1 and 2
Dichotone A (
1). White amorphous powder, UV (MeOH)
λmax (log ε): 235 (4.45), 201 (4.27); IR (KBr)
νmax 3392, 2965, 2926, 1714, 1670, 1262, 1155, 1094, 1048, 806 cm
−1.
1H and
13C-NMR data, see
Table 1; HRESIMS [M − H]
− m/
z 241.1237 (calcd. for C
16H
17O
2, 241.1234).
Dichotone B (
2). White amorphous powder, UV (MeOH)
λmax (log ε): 225 (3.95), 200 (4.22); IR (KBr)
νmax 3436, 2962, 2925, 1714, 1513, 1262, 1097, 1084, 1036, 804 cm
−1.
1H and
13C-NMR data, see
Table 1; HRESIMS [M − H]
− m/
z 269.1176 (calcd. for C
17H
17O
3, 269.1183).
3.5. Cytotoxicity Assay
In vitro cytotoxicities of compounds
5 and
6 were carried out with human cancer cells MDA-MB-435 and Calu3 and evaluated by means of the colorimetric MTT assay. MDA-MB-435 and calu3 were seeded into 96-well plates each 100 μL/well at a density of 1 × 10
4 cells per well and incubated at 37 °C for 24 h. Then the compounds were added to the cultures at various concentrations (0.125–50 μM). Then, 48 h later, 20 microliters MTT reagent (Genview, Houston, TX, USA) dissolved in phosphate-buffered saline (PBS) (pH 7.4) at a concentration of 5 mg·mL
−1 were added into each well, and the cells were incubated at 37 °C for additional 4 h. The MTT-formazan crystals were formed and then dissolved in 150 mL DMSO (Sangon Biotech, Shanghai, China). The absorbance was recorded at 570 nm with a reference wavelength of 630 nm using a microplate reader. The half maximal inhibitory concentration (IC
50) was calculated by Bliss’s software [
18], and the data was calculated by SPSS.
4. Conclusions
In summary, two new compounds, dichotones A and B (1 and 2), and four known compounds, dichotocejpin C (3), bis-N-norgliovictin (4), bassiatin (5) and (3R,6R)-bassiatin (6), were obtained from the fungus Dichotomomyces sp. The compound (3R,6R)-bassiatin (6) is the epimer of bassiatin (5) at C-3. Compound 6 displayed notable cytotoxic activities against the human breast cancer cell line MDA-MB-435 and the human lung cancer cell line Calu3; however, compound 5 showed no cytotoxicity, which indicated that the 3R-configuration is essential for the cytotoxicity of compound 6. It was supposed that only the 3R-configuration can specifically bind to the target molecule, such as protein kinase in signaling pathways. The absolute stereochemistry of natural products significantly influences their bioactivity due to the distinction in their recognition ability and binding force with the target organism. For example, the blocking action of the β receptor for S-(−)-propranolol was about 100-fold stronger than that of R-(+)-propranolol, while R-(+)-propranolol had an inhibitory effect on sodium channel. Therefore, the absolute configuration has become a critical issue in the drug development. Further investigation on the anticancer mechanism is in progress, which will facilitate clarifying the stereo-selectivity of (3R,6R)-bassiatin (6) on anticancer activity.






{kind=link}
{kind=link}
{kind=link}