
An Efficient One-Pot Protocol for the Synthesis of Substituted 3,4-Dihydropyrimidin-2(1H)-ones Using Metallophthalocyanines (MPcs) as Potent Heterogeneous Catalysts: Synthesis, Characterization, Aggregation and Antimicrobial Activity



Abstract
:1. Introduction
2. Results
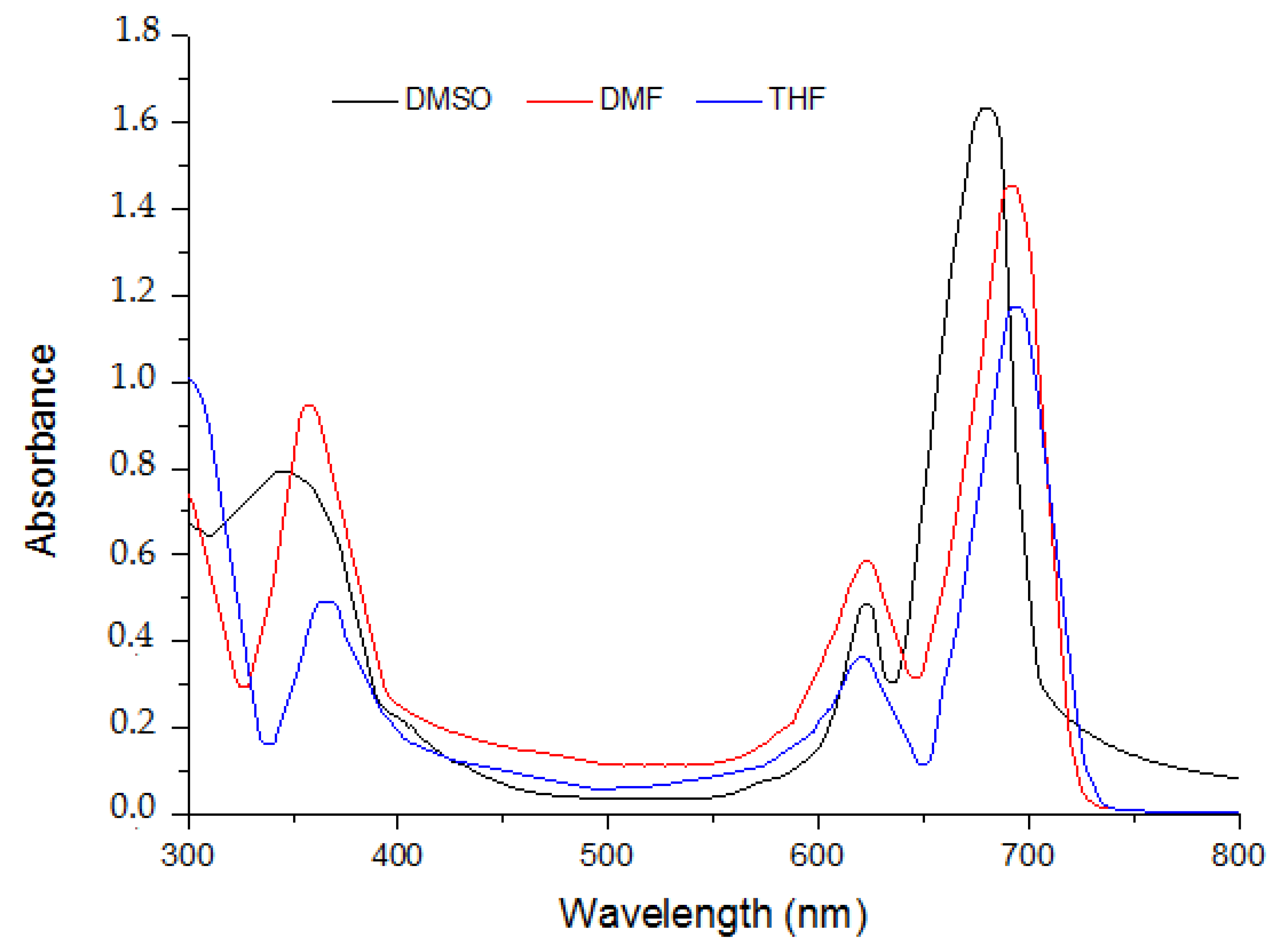
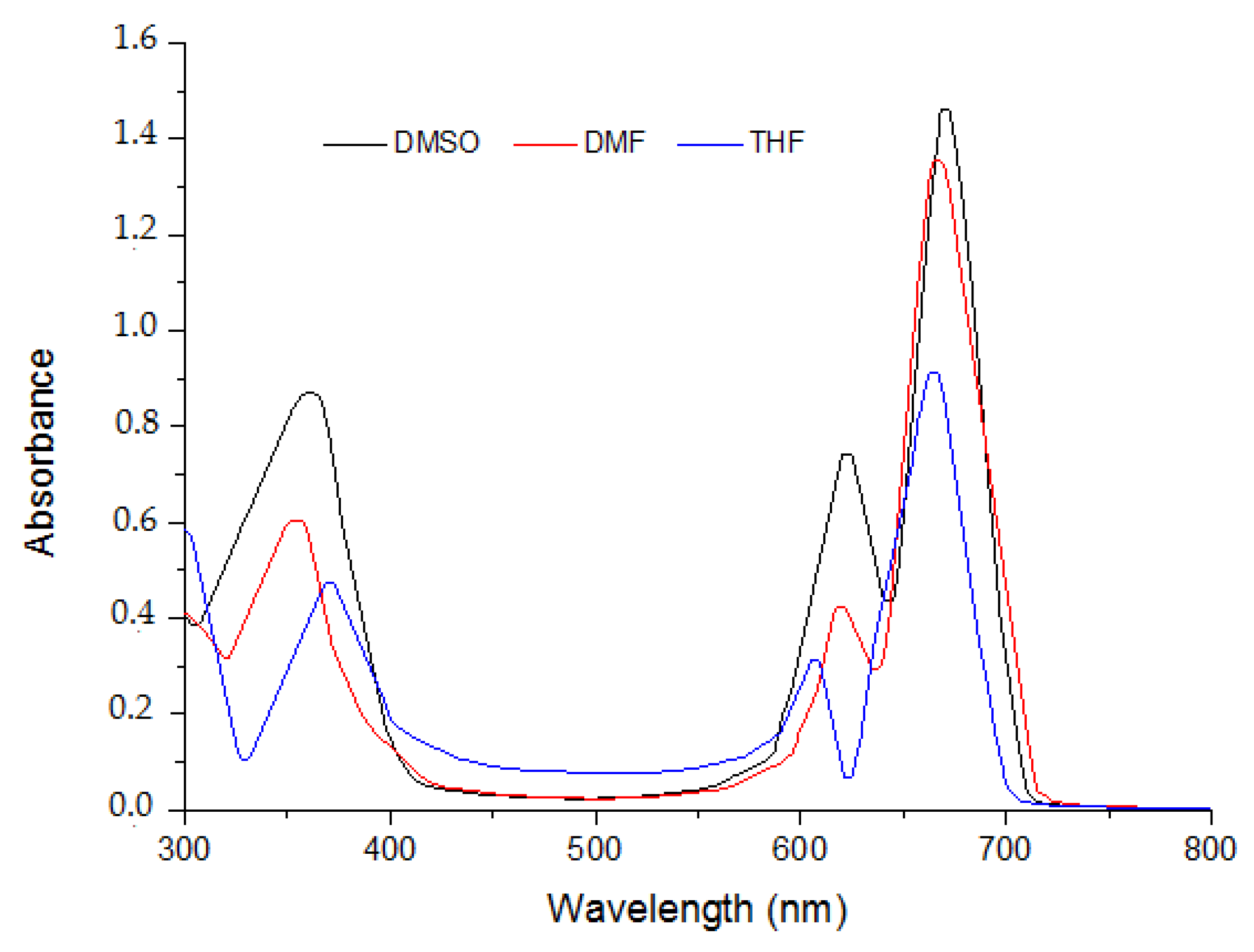
2.1. Ground State Electronic Absorption and Aggregation Properties
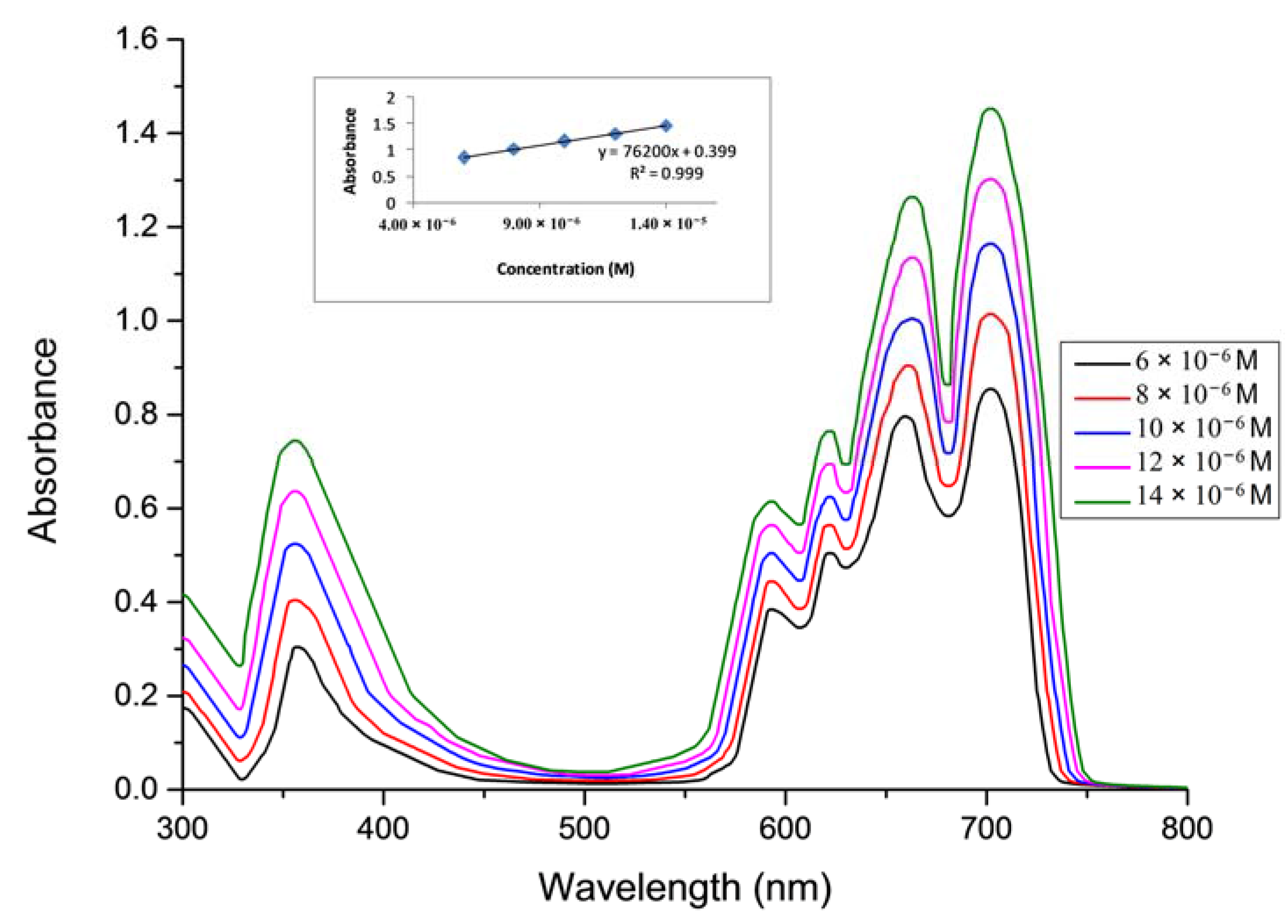
2.2. Aggregations Studies
2.3. Catalytic Eficiency of Metallophthalocyanines 5–7
2.4. Recycling Performance of the Catalysts 5–7
2.5. Antimicrobial Activity
3. Experimental Section
3.1. General Information
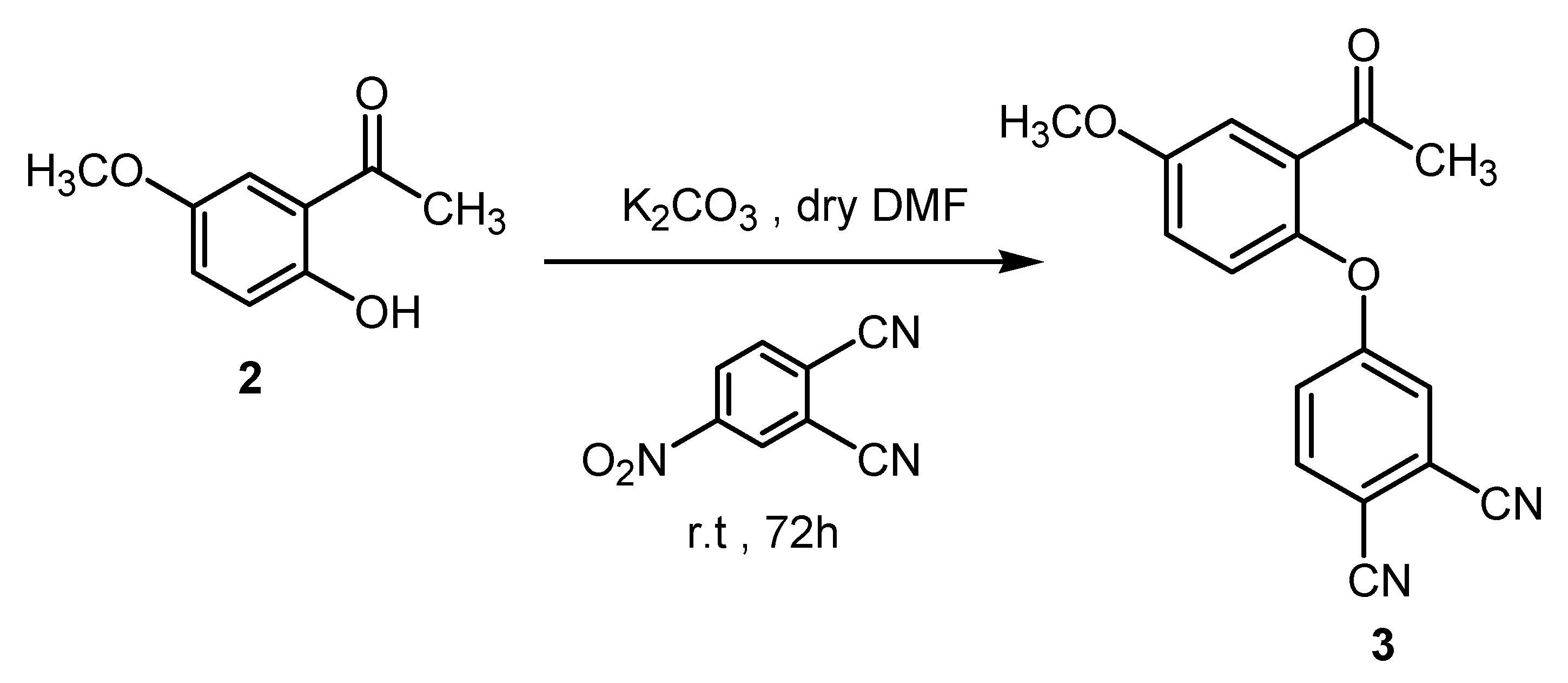
3.2. Synthesis of 4-(2-Acetyl-4-methoxyphenoxy)phthalonitrile (3)
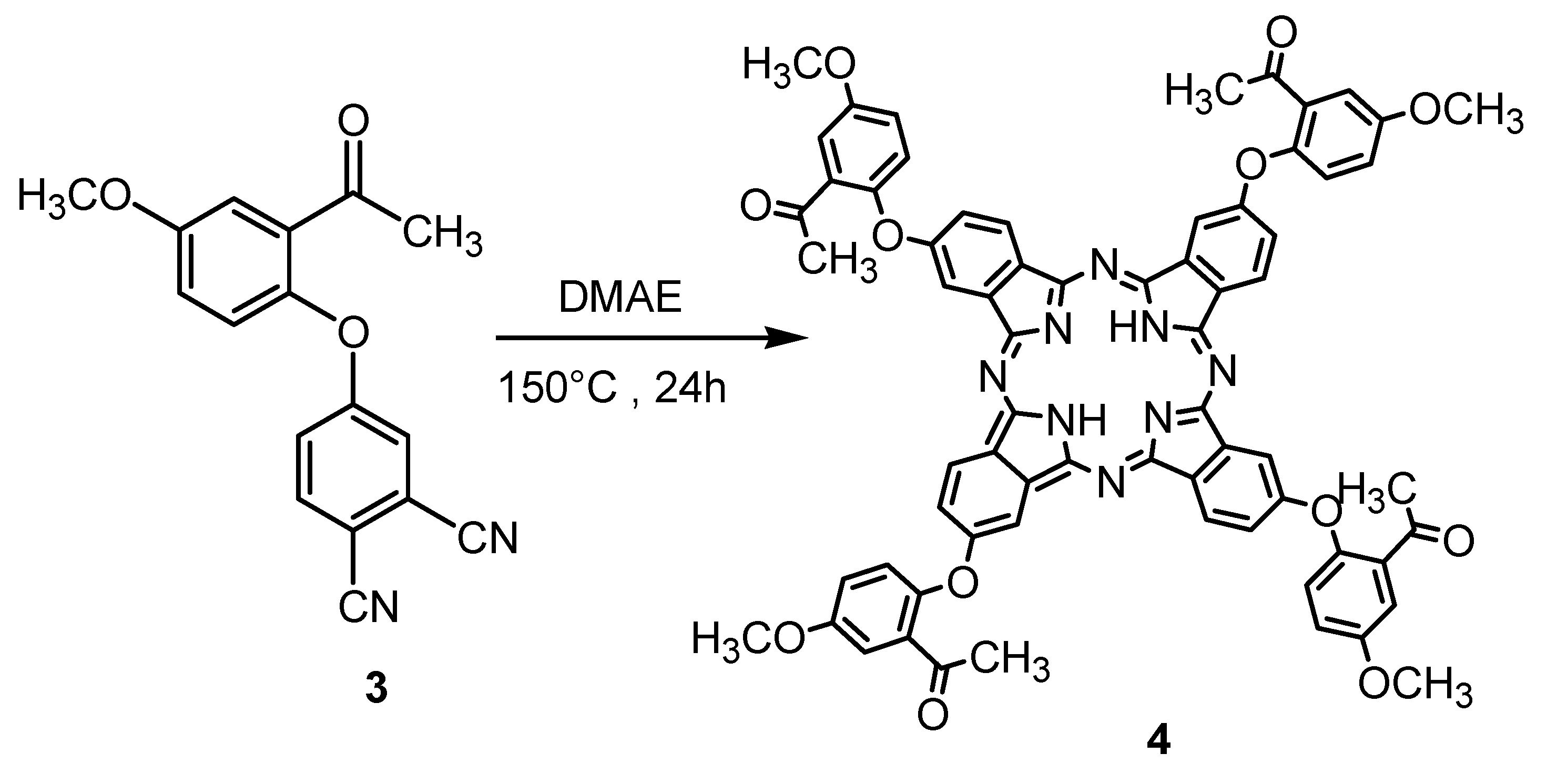
3.3. Synthesis of Metal-Free 4
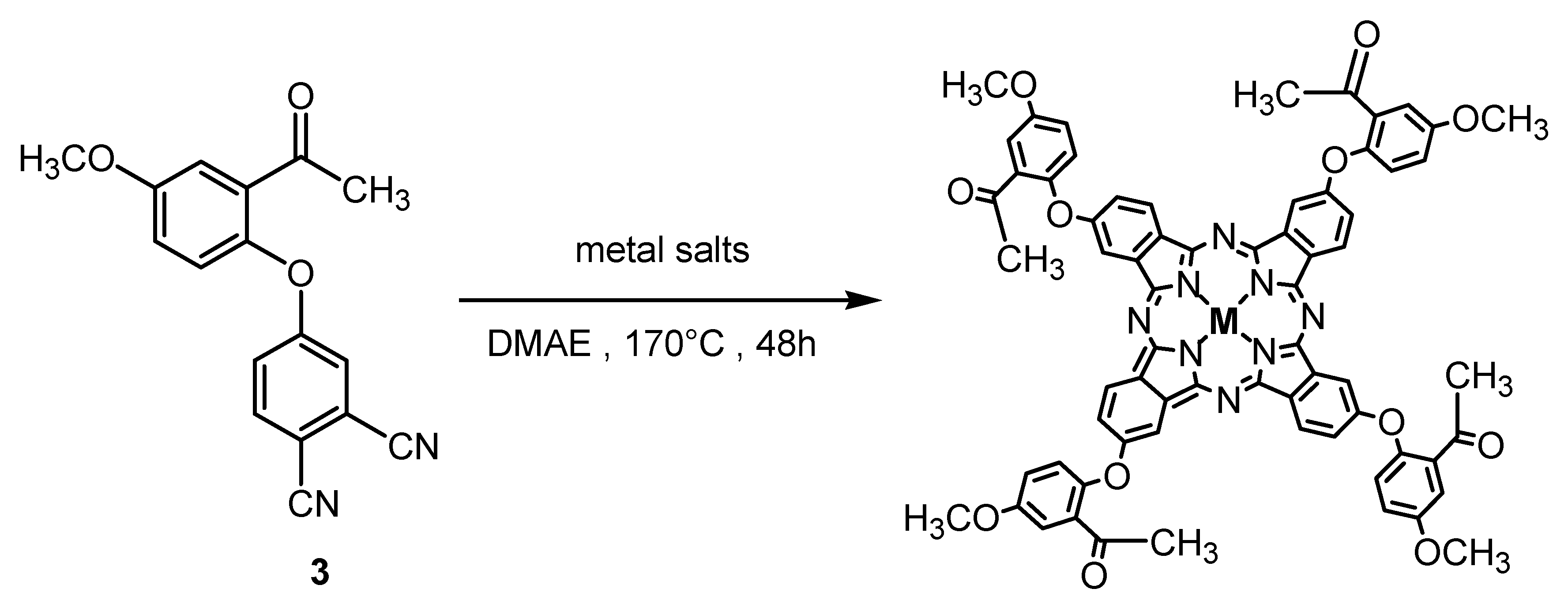
3.4. General Procedure for the Synthesis of Metallophthalocyanines 5–7
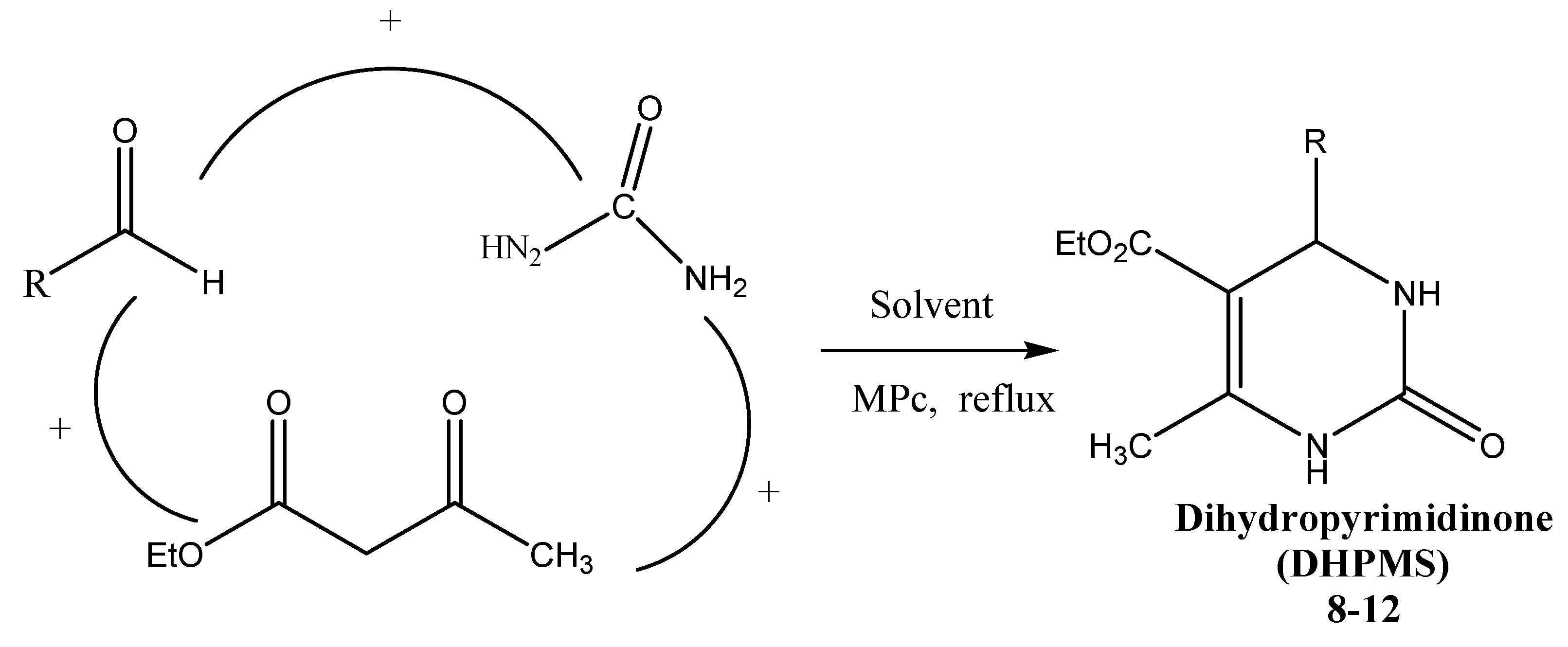
3.5. General Procedure for Preparation of Compounds 8–12
3.6. Antimicrobial Activities
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kleidernigg, O.P.; Kappe, C.O. Separation of enantiomers of 4-aryldihydropyrimidines by direct enantioselective HPLC. A critical comparison of chiral stationary phases. Tetrahedron Asymmetry 1997, 8, 2057–2067. [Google Scholar] [CrossRef]
- Biginelli, P. Synthesis of 3,4-dihydropyrimidin-2(1H)-ones was discovered in 1893 by Pietro Biginelli. Gazz. Chim. Ital. 1893, 23, 360–416. [Google Scholar]
- Kappe, C.O. 100 years of the biginelli dihydropyrimidine synthesis. Tetrahedron 1993, 49, 6937–6963. [Google Scholar] [CrossRef]
- Kappe, C.O.; Fabian, W.M.F.; Semones, M.A. Conformational analysis of 4-aryl-dihydropyrimidine calcium channel modulators. A comparison of ab initio, semiempirical and X-ray crystallographic studies. Tetrahedron 1997, 53, 2803–2816. [Google Scholar] [CrossRef]
- Atwal, K.S.; Rovnyak, G.C.; O’Reilly, B.C.; Schwartz, J. Substituted 1,4-dihydropyrimidines. III: Synthesis of selectively functionalized 2-hetero-1,4-dihydropyrimidines. J. Org. Chem. 1989, 54, 5898–5907. [Google Scholar] [CrossRef]
- Wipf, P.; Cunningham, A.A. Solid phase protocol of the biginelli dihydropyrimidine synthesis suitable for combinatorial chemistry. Tetrahedron Lett. 1995, 36, 7819–7822. [Google Scholar] [CrossRef]
- Kappe, C.O.; Uray, G.; Roschger, P.; Lindner, W.; Kratky, C.; Keller, W. Synthesis and Reactions and Resolution of a of Biginelli Compounds -5.1 Facile Preparation Stable 5-Dihydropyrimidinecarboxylic Acid. Tetrahedron 1992, 48, 5473–5480. [Google Scholar] [CrossRef]
- Kappe, C.O. Recent Advances in the Biginelli dihydropyrimidine synthesis. New Tricks from an old dog. Acc. Chem. Res. 2000, 33, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.O. Biologically active dihydropyrimidones of the Biginelli-type—A literature survey. Eur. J. Med. Chem. 2000, 35, 1043–1052. [Google Scholar] [CrossRef]
- Biginelli, P. The Biginelli Reaction. Gazz. Chim. Ital. 1893, 23, 360–413. [Google Scholar]
- Bruno, P.; Zhang, W. Synthesis of diverse dihydropyrimidine-related scaffolds by fluorous benzaldehyde-based Biginelli reaction and post-condensation modifications. Beilstein J. Org. Chem. 2011, 7, 1294–1298. [Google Scholar]
- Shobha, D.; Chari, M.A.; Mano, A.; Selvan, S.T.; Mukkanti, K.; Vinu, A. Synthesis of 3,4-dihydropyrimidin-2-ones (DHPMs) using mesoporous aluminosilicate (AlKIT-5) catalyst with cage type pore structure. Tetrahedron 2009, 65, 10608–10611. [Google Scholar] [CrossRef]
- O’Reilly, B.C.; Atwal, K.S. Synthesis of substituted 1,2,3,4-Tetrahydro-6-methyl-2-oxo-5-pyrimidinecarboxylic acid esters: The Biginelli condensation revisited. Heterocycles 1987, 26, 1185–1188. [Google Scholar] [CrossRef]
- Clark, J.H. Catalysis of Organic Reactions by Supported Reagents; VCH Publishers: New York, NY, USA, 1994; pp. 35–68. [Google Scholar]
- Sheldon, R.A.; Van Bekkum, H. Catalysis through Heterogeneous Catalysis; Wiely-VCH Publishers: Weinheim, Germany, 2002. [Google Scholar]
- Pérollier, C.; Sorokin, A.B. Preparation of α,β-acetylenic ketones by catalytic heterogeneous oxidation of alkynes. Chem. Commun. 2002, 1548–1549. [Google Scholar] [CrossRef]
- Meunier, B.; Sorokin, A. Oxidation of pollutants catalyzed by metallophthalocyanines. Acc. Chem. Res. 1997, 30, 470–476. [Google Scholar] [CrossRef]
- Sorokin, A.; De Suzzoni-Dezard, S.; Poullain, D.; Noël, J.P.; Meunier, B. CO2 as the ultimate degradation product in the H2O2 oxidation of 2,4,6-trichlorophenol catalyzed by iron tetrasulfophthalocyanine. J. Am. Chem. Soc. 1996, 118, 7410–7411. [Google Scholar] [CrossRef]
- Grootboom, N.; Nyokong, T. Iron perchlorophthalocyanine and tetrasulfophthalocyanine catalyzed oxidation of cyclohexane using hydrogen peroxide, chloroperoxybenzoic acid andtert-butylhydroperoxide as oxidants. J. Mol. Catal. A Chem. 2002, 179, 113–123. [Google Scholar] [CrossRef]
- Jain, S.L.; Joseph, J.K.; Singhal, S.; Sain, B. Metallophthalocyanines (MPcs) as efficient heterogeneous catalysts for Biginelli condensation: Application and comparison in catalytic activity of different MPcs for one pot synthesis of 3,4-dihydropyrimidin-2-(1H)-ones. J. Mol. Catal. A Chem. 2007, 268, 134–138. [Google Scholar] [CrossRef]
- Bayrak, R.; Akçay, H.T.; Beriş, F.Ş.; Şahin, E.; Bayrak, H.; Demirbaş, Ü. Synthesis, aggregation and spectroscopic studies of novel water soluble metal free, zinc, copper and magnesium phthalocyanines and investigation of their anti-bacterial properties. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 133, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Rawdha, M.; Wissal, E.; Olfa, N.; Antonio, R.; Abdullah, S.; Lasaad, B.; Naceur, H. One-pot three-component Biginelli-type reaction to synthesize 3,4-dihydropyrimidine-2-(1H)-ones catalyzed by Co phthalocyanines: Synthesis, characterization, aggregation behavior and antibacterial activity. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 167, 165–174. [Google Scholar]
- Rosenmund, K.W.; Lohfert, H. Über Synthese von Polyphenol-Ketonen. Eur. J. Inorg. Chem. 1928, 61, 2601–2607. [Google Scholar] [CrossRef]
- Zhao, W.; Sun, J.; Xiang, H.; Zeng, Y.Y.; Li, X.B.; Xiao, H.; Chen, D.Y.; Ma, R.L. Synthesis and biological evaluation of new flavonoid fatty acid esters with anti-adipogenic and enhancing glucose consumption activities. Bioorg. Med. Chem. 2011, 19, 3192–3203. [Google Scholar] [CrossRef] [PubMed]
- Yenilmez, H.Y.; Okur, A.İ.; Gül, A. Peripherally tetra-palladated phthalocyanines. J. Organomet. Chem. 2007, 692, 940–945. [Google Scholar] [CrossRef]
- Değirmencioğlu, İ.; Atalay, E.; Er, M.; Köysal, Y.; Işık, Ş.; Serbest, K. Novel phthalocyanines containing substituted salicyclic hydrazone-1,3-thiazole moieties: Microwave-assisted synthesis, spectroscopic characterization, X-ray structure and thermal characterization. Dyes Pigments 2010, 84, 69–78. [Google Scholar] [CrossRef]
- Chauke, V.; Durmuş, M.; Nyokong, T. Photochemistry, photophysics and nonlinear optical parameters of phenoxy and tert-butylphenoxy substituted indium (III) phthalocyanines. J. Photochem. Photobiol. A Chem. 2007, 192, 179–187. [Google Scholar] [CrossRef]
- Bayrak, R.; Akçay, H.T.; Durmuş, M.; Değirmencioğlu, İ. Synthesis, photophysical and photochemical properties of highly soluble phthalocyanines substituted with four 3,5-dimethylpyrazole-1-methoxy groups. J. Organomet. Chem. 2011, 696, 3807–3815. [Google Scholar] [CrossRef]
- Wires, T.M. Synthesis and supramolecular chemistry of novel liquid crystalline crown ether-substituted phthalocyanines: Toward molecular wires and molecular ionoelectronics. J. Am. Chem. Soc. 1995, 117, 9957–9965. [Google Scholar]
- Wu, W.T.; Wu, W.H.; Ji, S.M.; Guo, H.M.; Wang, X.; Zhao, J.Z. The synthesis of 5,10,15,20-tetraarylporphyrins and their platinum (II) complexes as luminescent oxygen sensing materials. Dyes Pigment. 2011, 89, 199–207. [Google Scholar] [CrossRef]
- Koçan, H.; Burat, A.K. Synthesis and characterization of [7-(trifluoromethyl) quinolin-4-yl] oxy-substituted phthalocyanines. Monatsh. Chem. 2013, 144, 171–177. [Google Scholar] [CrossRef]
- Hojatollah, K.; Esmat, T.K.; Tayebeh, J. An efficient synthesis of 3,4-dihydropyrimidin-2(1H)-ones catalyzed by molten [Et3NH][HSO4]. Arab. J. Chem. 2012, 5, 485–488. [Google Scholar]
- Pasha, M.A.; Ramachandra, N.S.; Jayashankara, V.P. One pot synthesis of 3,4-dihydropyrimidin-2(1H)-ones/-thiones catalysed by zinc chloride: An improved procedure for the Biginelli reaction using microwaves under solvent free condition. Indian J. Chem. 2005, 44, 823–826. [Google Scholar]
- Guven, K.; Yucel, E.; Etintas, C. Antimicrobial activities of fruits of Crataegus and Pyrus species. Pharm. Biol. 2006, 44, 79–83. [Google Scholar] [CrossRef]
- National Committee for Clinical Laboratory Standard. Referece Method for Broth Dilution Antifungal Susceptibility Testing of Conidium Forming Filamentous Fungi; Proposed standard M38-P; National Committee for Clinical Laboratory Standards: Wayne, PA, USA, 1998. [Google Scholar]
- Medyouni, R.; Mtibaa, A.C.; Mellouli, L.; Romerosa, A.; Hamdi, N. Convenient synthesis of novel unmetalled and metallophthalocyanines bearing coumarin derivatives: Synthesis, characterization, aggregation behaviors and antimicrobial activity. J. Incl. Phenom. Macrocycl. Chem. 2016, 86, 201–210. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |





| M | Co | Zn | Cu |
|---|---|---|---|
| Compounds | 5 | 6 | 7 |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | Pcs | Bandes (Q), λmax (nm) (log ε) | Bandes (B), λmax (nm) (log ε) |
|---|---|---|---|
| DMF | 5 | 615 (4.591); 697 (5.135) | 340 (5.033) |
| 6 | 636 (4.632); 697 (5.064) | 343 (4.834) | |
| 7 | 614 (4.436); 675 (4.968) | 332 (4.645) | |
| DMSO | 5 | 622 (4.462); 708 (5.054) | 334 (4.728) |
| 6 | 631 (4.337); 701 (5.004) | 340 (4.526) | |
| 7 | 614 (4.246); 693 (4.885) | 351 (4.403) |
| Entry | Substrate | MPc | Reaction Time (h) | Solvent | Catalyst (mol %) | Yield a (%) |
|---|---|---|---|---|---|---|
| 1 | benzaldehyde | 5 | 1.5 | DMC | 2 | 98 |
| 2 | benzaldehyde | 6 | 3 | DMC | 2 | 90 |
| 3 | benzaldehyde | 7 | 2 | DMC | 2 | 85 |
| 4 | 3-methoxy benzaldehyde | 5 | 2.5 | EtOH | 2 | 80 |
| 5 | 3-methoxy benzaldehyde | 5 | 3 | THF | 2 | 75 |
| 6 | 3-methoxy benzaldehyde | 5 | 3.5 | CH3COCH3 | 5 | 98 |
| 7 | 3-methoxy benzaldehyde | 5 | 1 | DMC | 10 | 98 |
| 8 | 3-methoxy benzaldehyde | 5 | 4 | DMC | - | trace |
| Substrate | MPc | DHPM | Time (h) | Yield a (%) |
|---|---|---|---|---|
| Benzaldehyde | Co(II)Pc | 8 | 3 | 78 |
| 3-Methoxybenzaldehyde | Co(II)Pc | 9 | 4 | 92 |
| 4-Methylbenzaldehyde | Co(II)Pc | 10 | 2 | 96 |
| 4-Bromobenzaldehyde | Co(II)Pc | 11 | 2 | 92 |
| 4-Nitrobenzaldehyde | Co(II)Pc | 12 | 3 | 85 |
| Entry | Catalyst (mol) | Reaction Time (h) | Yield a (%) |
|---|---|---|---|
| 1 | 2 | 1 | 98 |
| 2 | Cycle1 | 1 | 90 |
| 3 | Cycle2 | 1 | 85 |
| 4 | Cycle3 | 1 | 95 |
| Indicator Microorganism | Compounds | MIC (mg/mL) |
|---|---|---|
| Micrococcus luteus LB 14110 | 8 | 10 |
| 9 | 1.25 | |
| 10 | 4 | |
| 11 | 1.5 | |
| 12 | 8 | |
| Staphylococcus aureus ATCC 6538 | 8 | 5 |
| 9 | 2.5 | |
| 10 | 5 | |
| 11 | 4 | |
| 12 | - | |
| Listeria monocytogenes ATCC 19117 | 8 | 5 |
| 9 | 0.625 | |
| 10 | - | |
| 11 | 5 | |
| 12 | 3.5 | |
| Salmonella Typhimurium ATCC 14028 | 8 | 10 |
| 9 | 5 | |
| 10 | - | |
| 11 | 5 | |
| 12 | 8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdi, N.; Medyouni, R.; Bilel, H.; Mansour, L.; Romerosa, A. An Efficient One-Pot Protocol for the Synthesis of Substituted 3,4-Dihydropyrimidin-2(1H)-ones Using Metallophthalocyanines (MPcs) as Potent Heterogeneous Catalysts: Synthesis, Characterization, Aggregation and Antimicrobial Activity. Molecules 2017, 22, 605. https://doi.org/10.3390/molecules22040605
Hamdi N, Medyouni R, Bilel H, Mansour L, Romerosa A. An Efficient One-Pot Protocol for the Synthesis of Substituted 3,4-Dihydropyrimidin-2(1H)-ones Using Metallophthalocyanines (MPcs) as Potent Heterogeneous Catalysts: Synthesis, Characterization, Aggregation and Antimicrobial Activity. Molecules. 2017; 22(4):605. https://doi.org/10.3390/molecules22040605
Chicago/Turabian StyleHamdi, Naceur, Rawdha Medyouni, Hallouma Bilel, Lamjed Mansour, and Antonio Romerosa. 2017. "An Efficient One-Pot Protocol for the Synthesis of Substituted 3,4-Dihydropyrimidin-2(1H)-ones Using Metallophthalocyanines (MPcs) as Potent Heterogeneous Catalysts: Synthesis, Characterization, Aggregation and Antimicrobial Activity" Molecules 22, no. 4: 605. https://doi.org/10.3390/molecules22040605
APA StyleHamdi, N., Medyouni, R., Bilel, H., Mansour, L., & Romerosa, A. (2017). An Efficient One-Pot Protocol for the Synthesis of Substituted 3,4-Dihydropyrimidin-2(1H)-ones Using Metallophthalocyanines (MPcs) as Potent Heterogeneous Catalysts: Synthesis, Characterization, Aggregation and Antimicrobial Activity. Molecules, 22(4), 605. https://doi.org/10.3390/molecules22040605