
Recent Advances in Cyanamide Chemistry: Synthesis and Applications


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
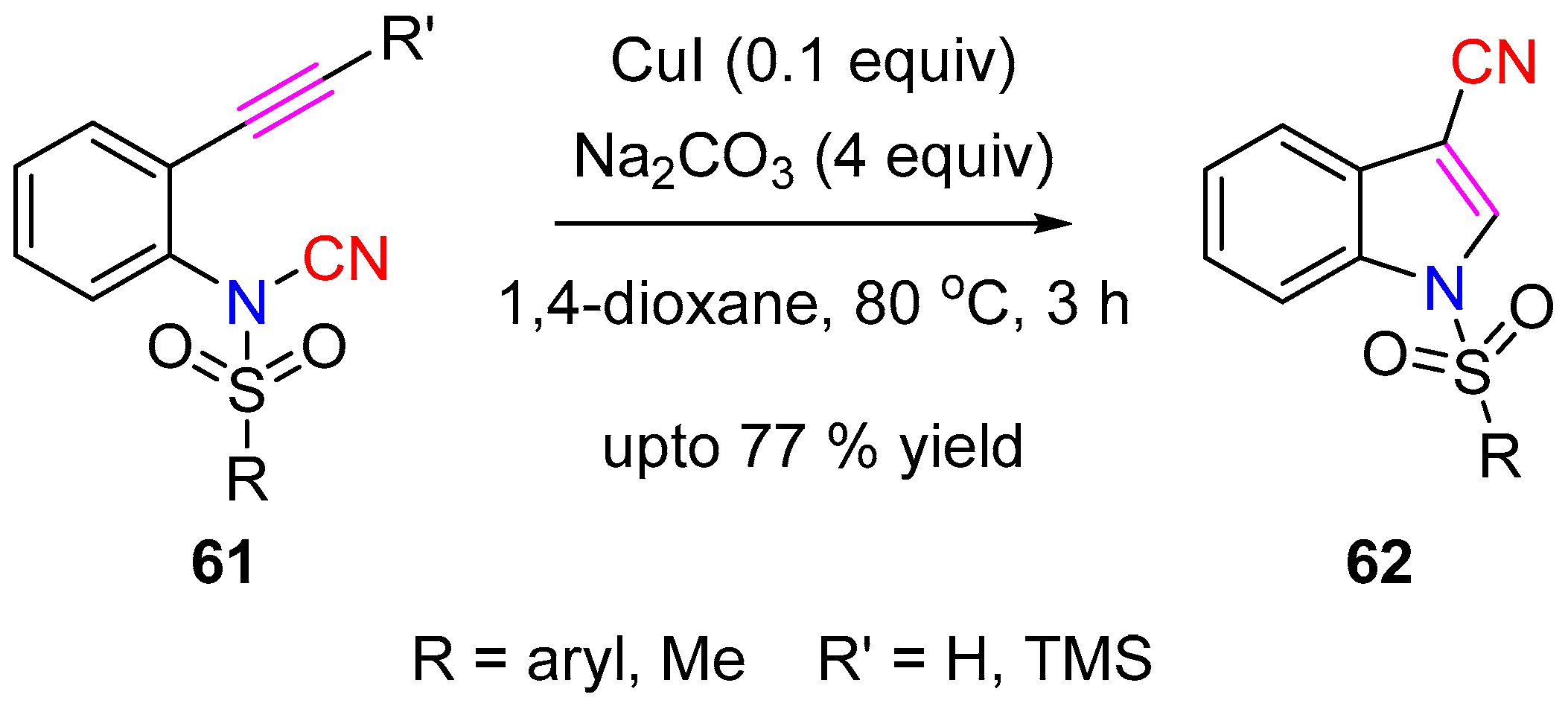{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Synthesis of Mono and Disubstituted Cyanamides
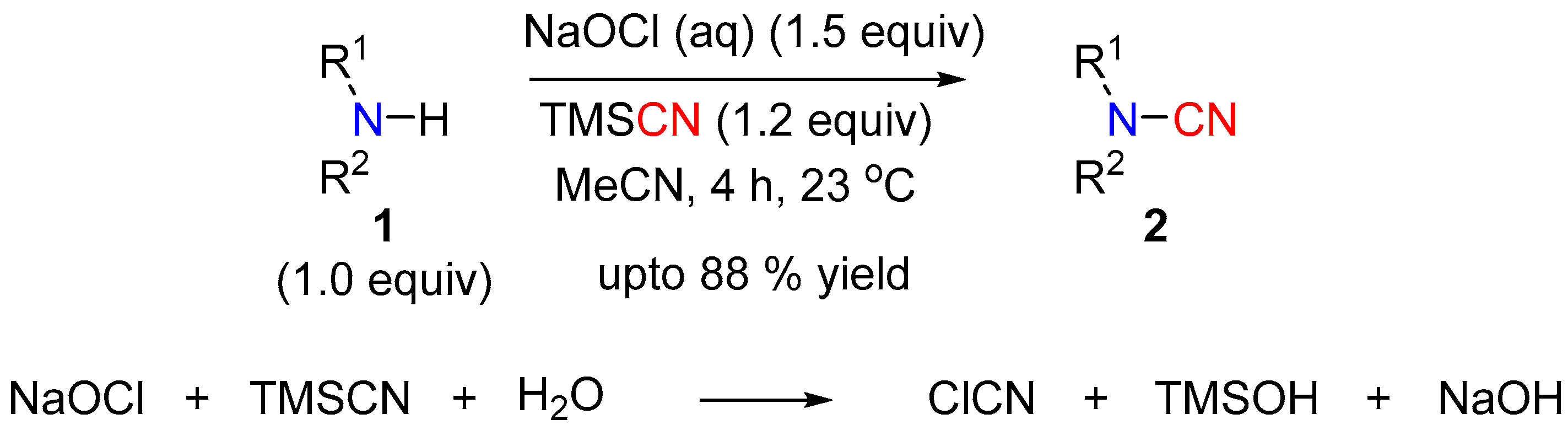
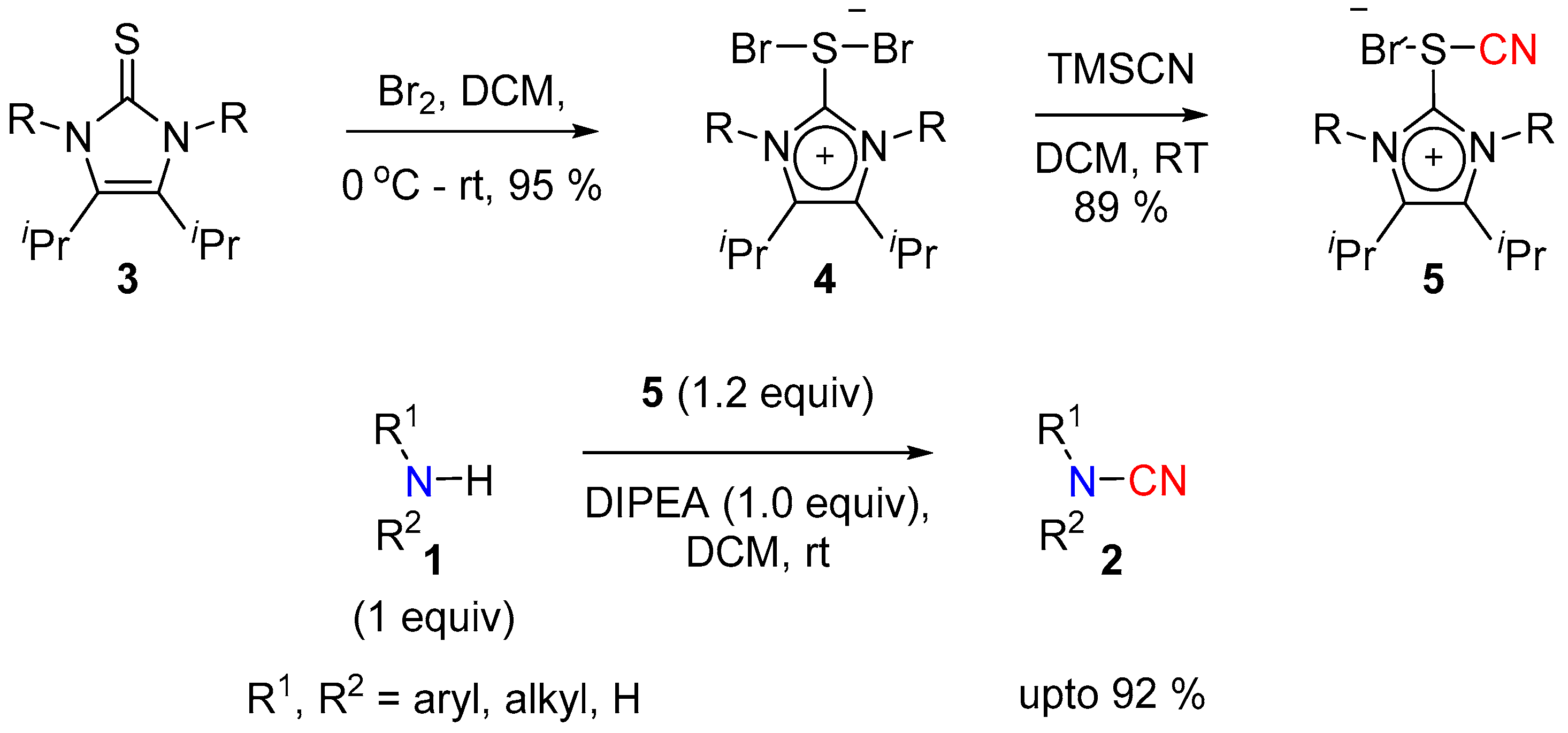
2.1. N-Cyanation of Secondary Amine
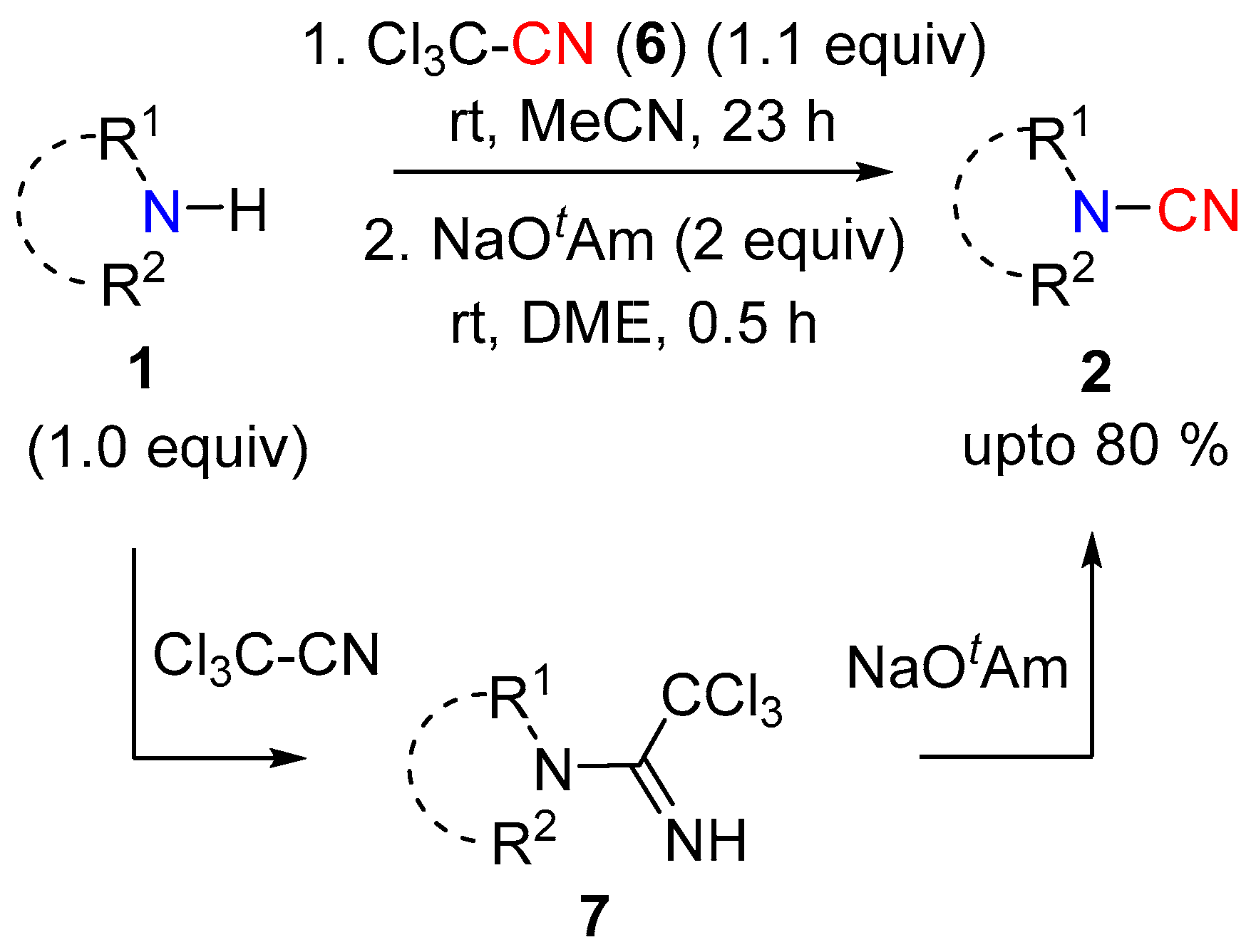
2.1.1. Using Electrophilic Nitrile [CN]+ Reagents
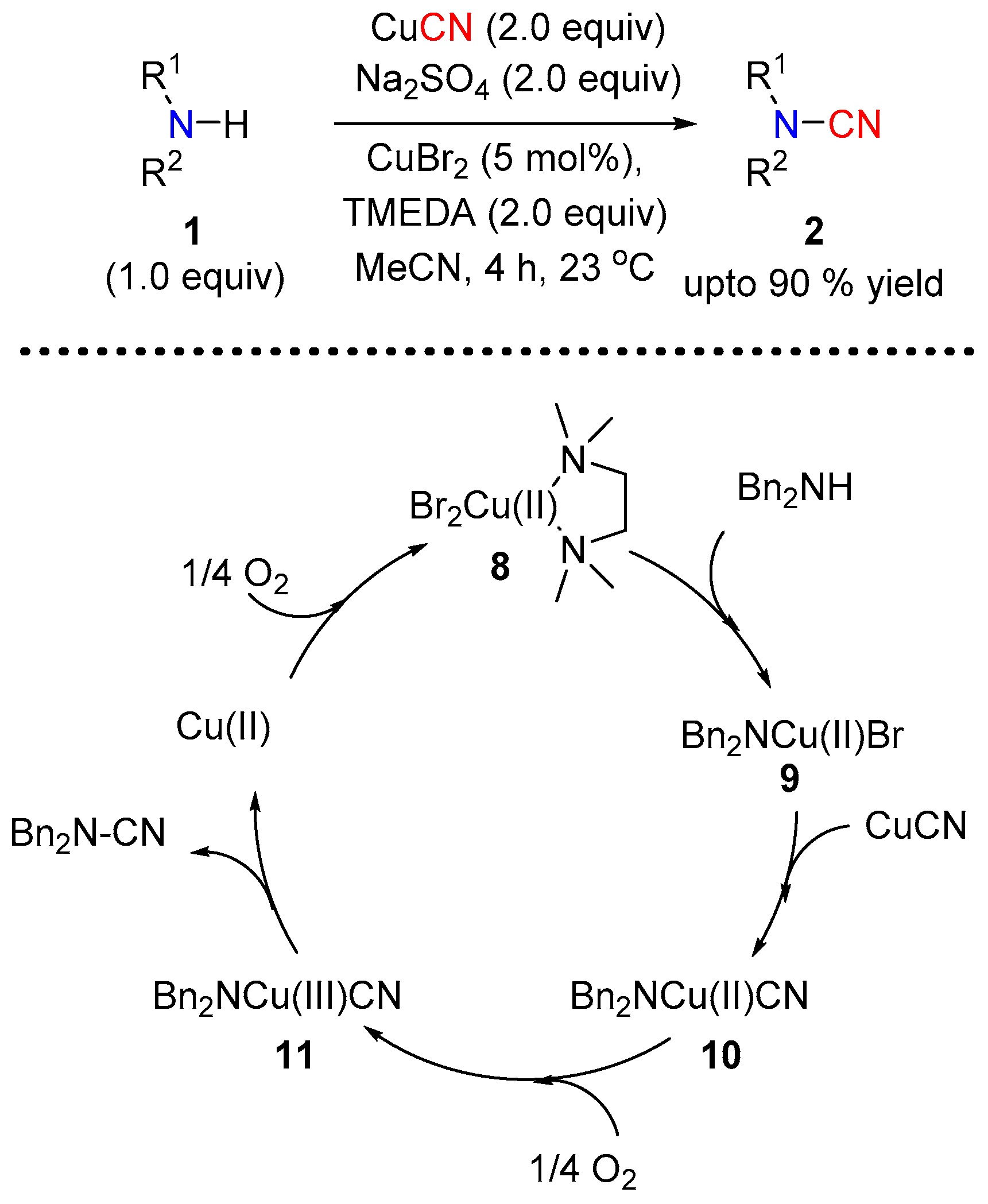
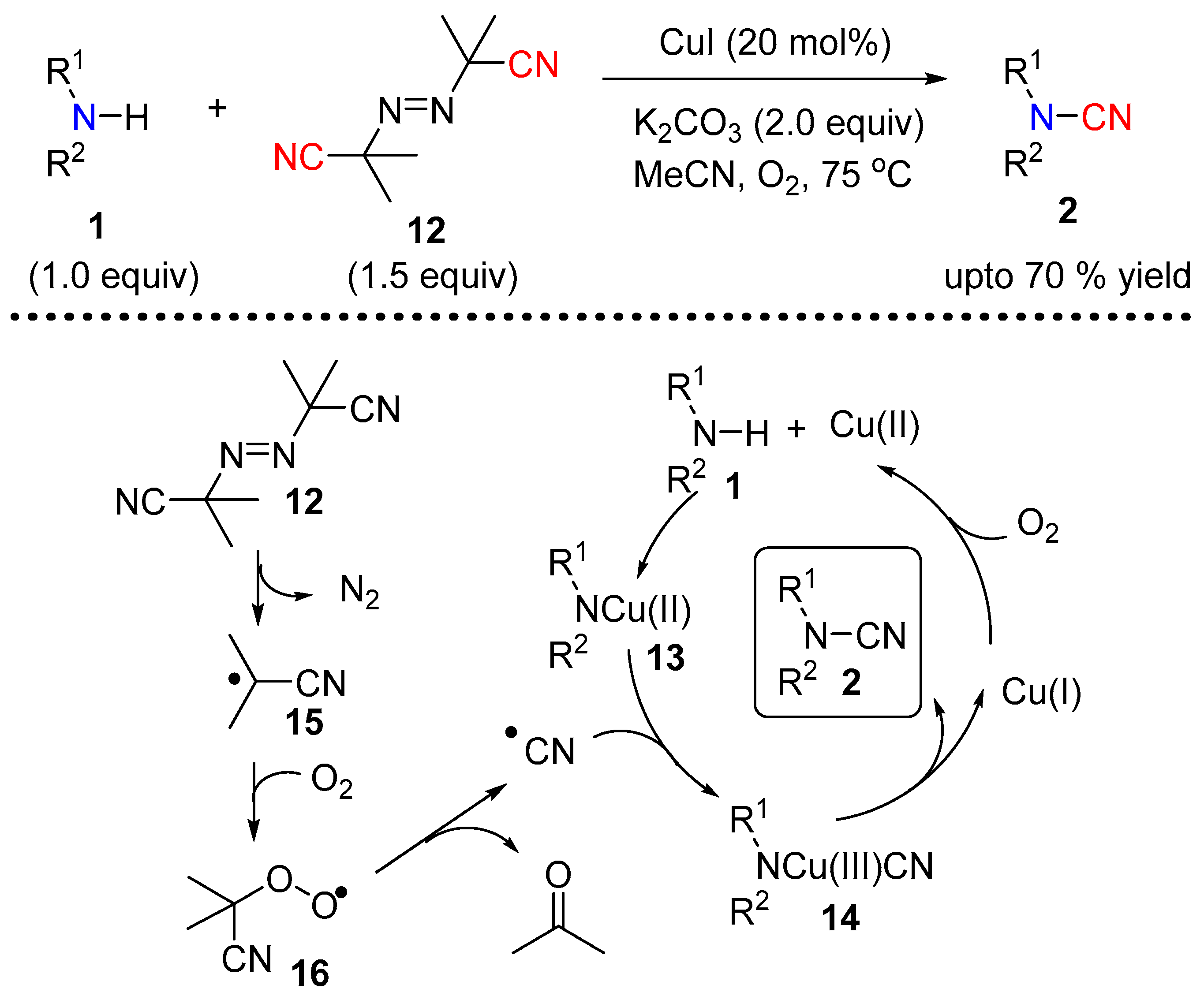
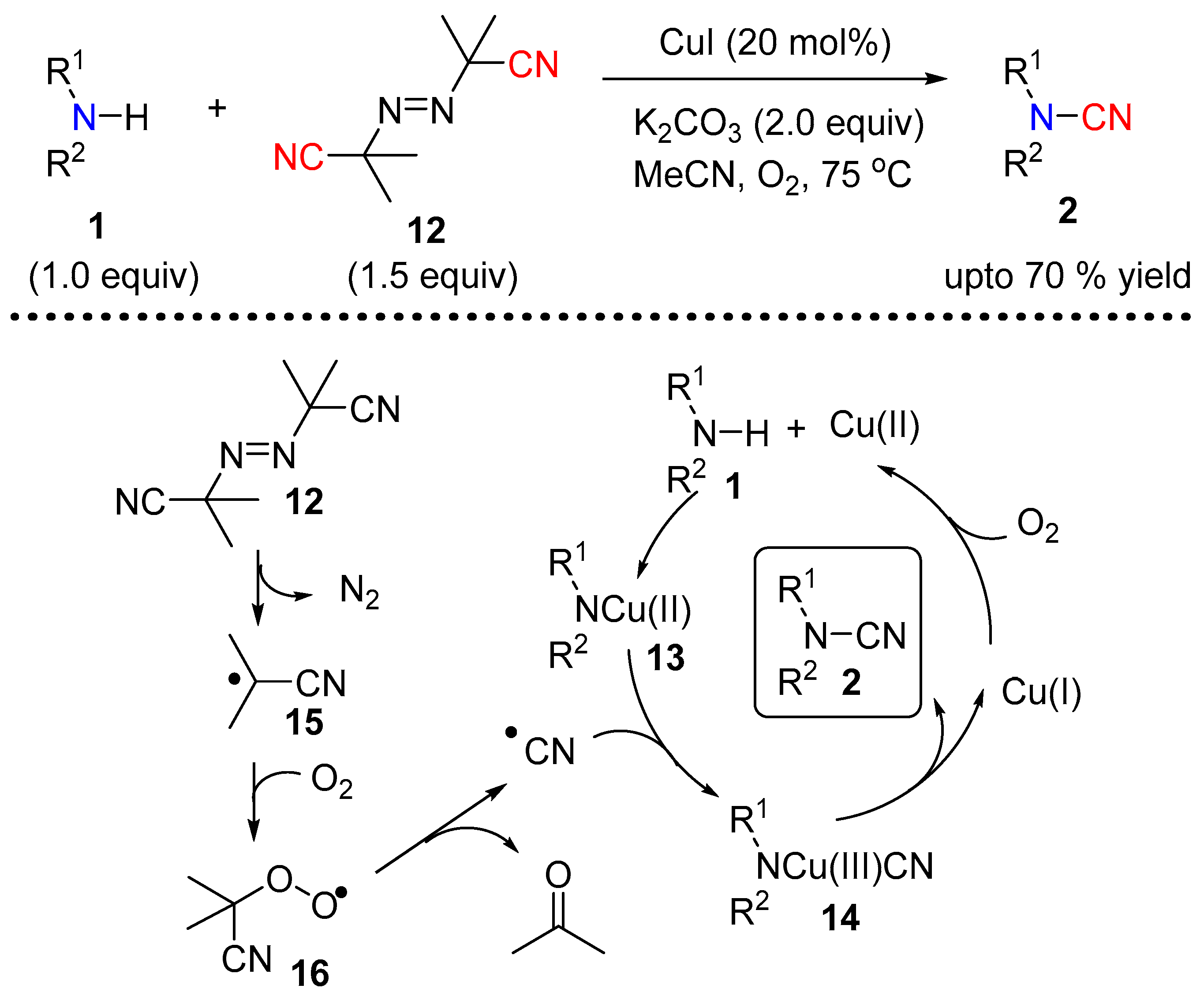
2.1.2. Under Copper Catalysis
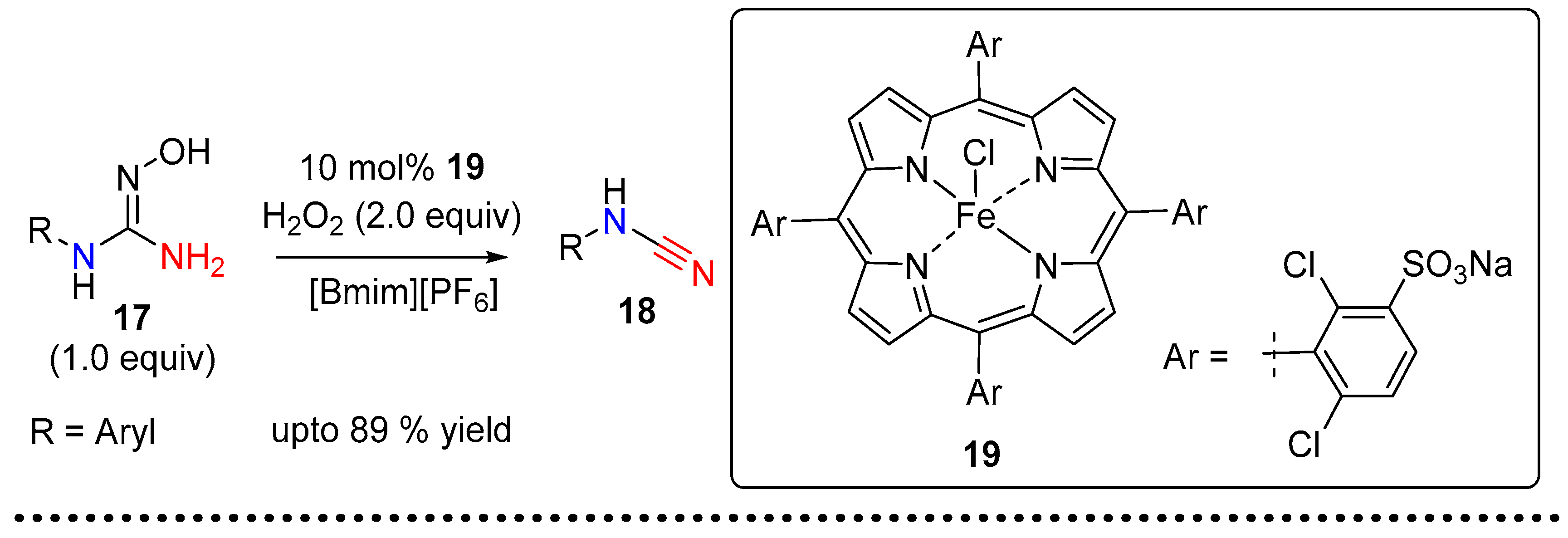
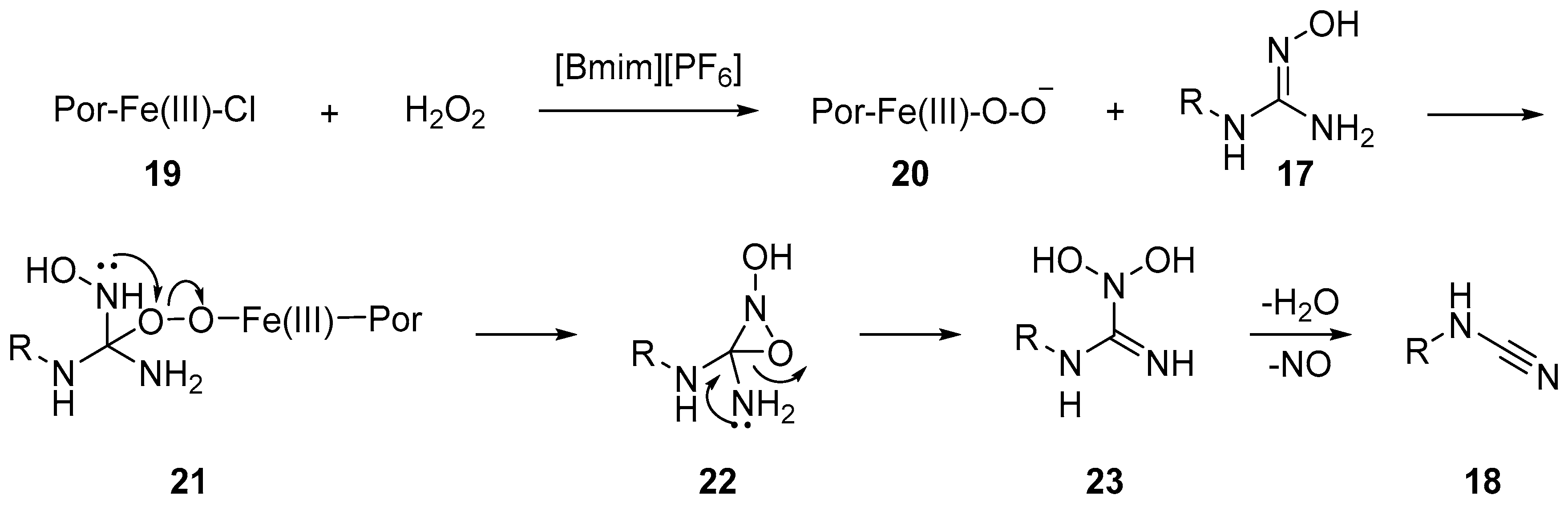
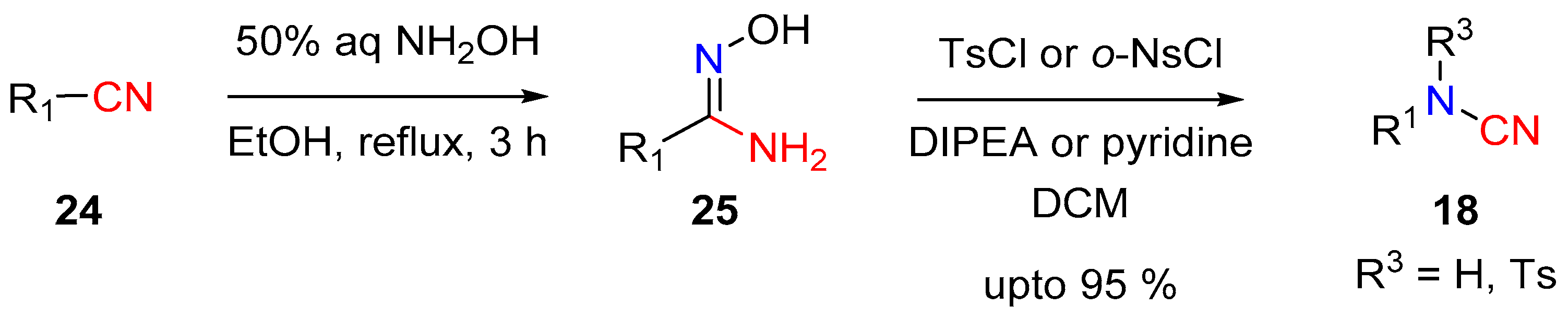
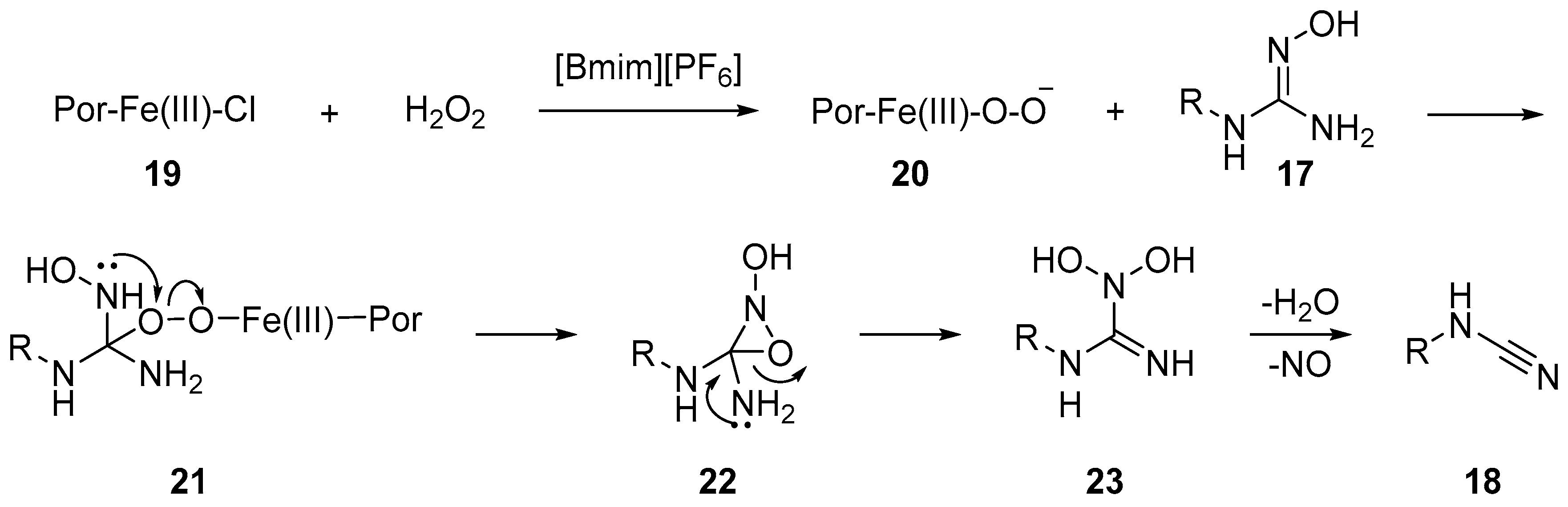
2.2. Cyanamides from Amidoximes and Guanidoximes
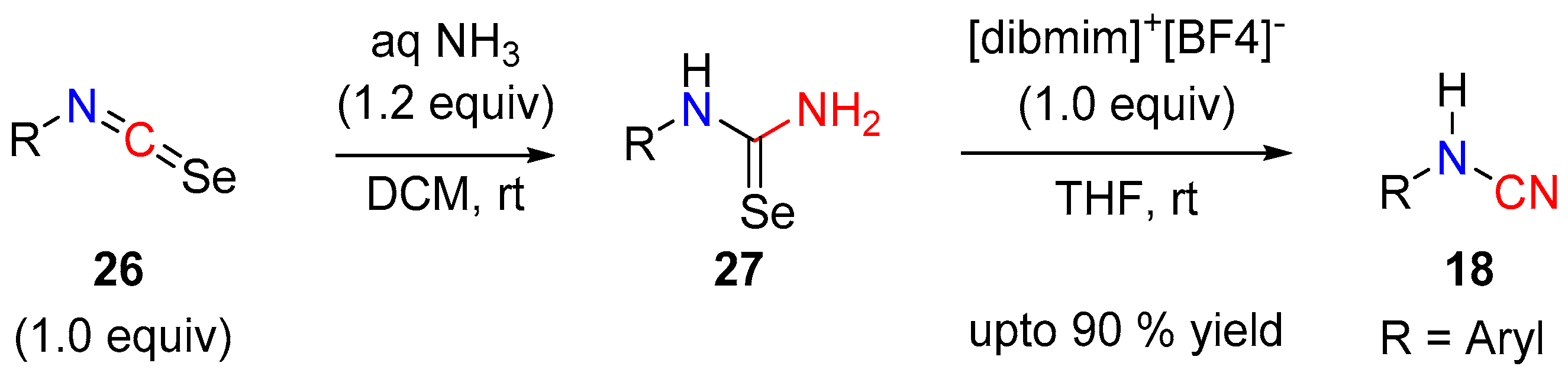
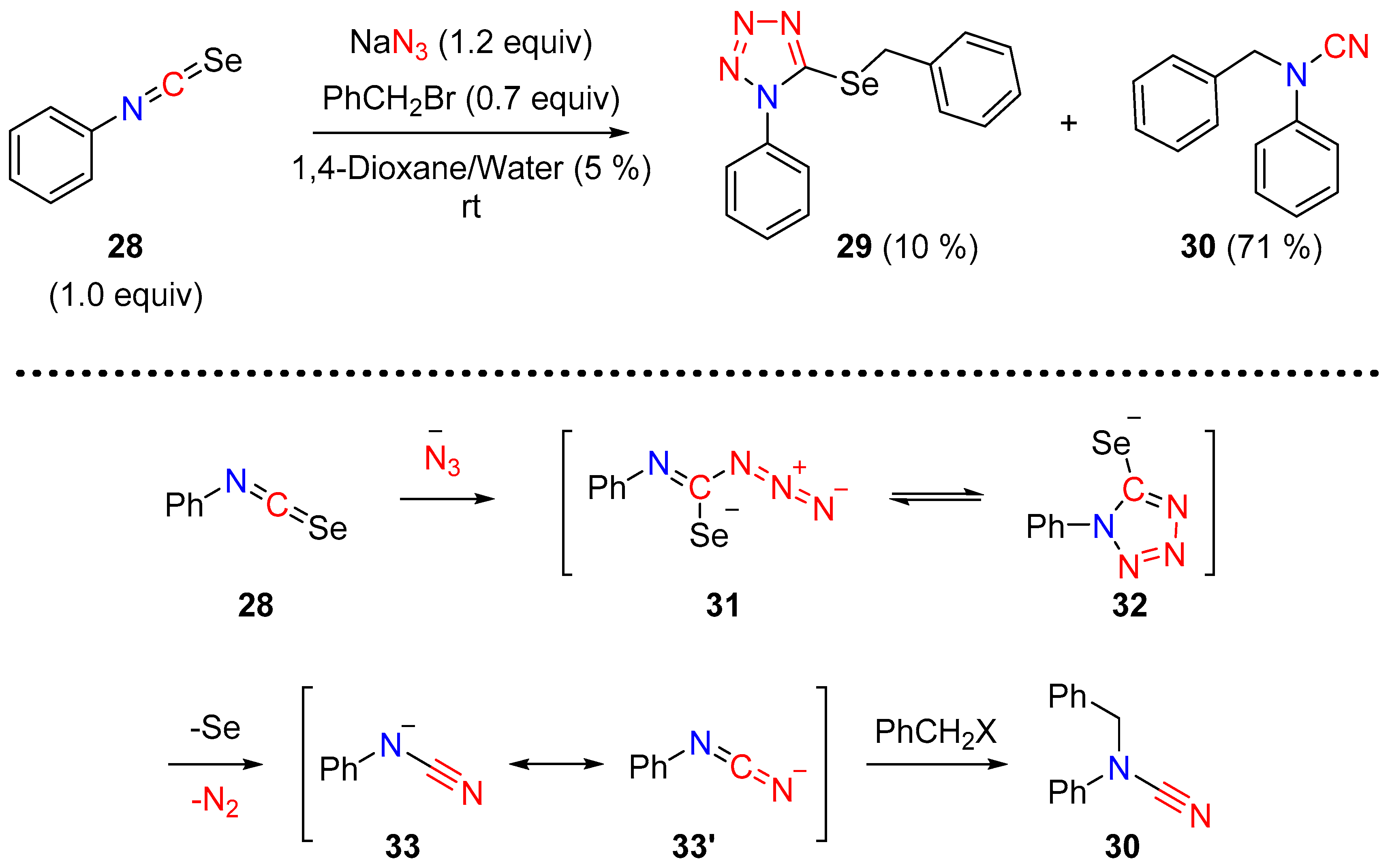
2.3. Cyanamides from Isoselenocyanates
3. Synthetic Applications of Substituted Cyanamides
3.1. Cycloaddition Reactions
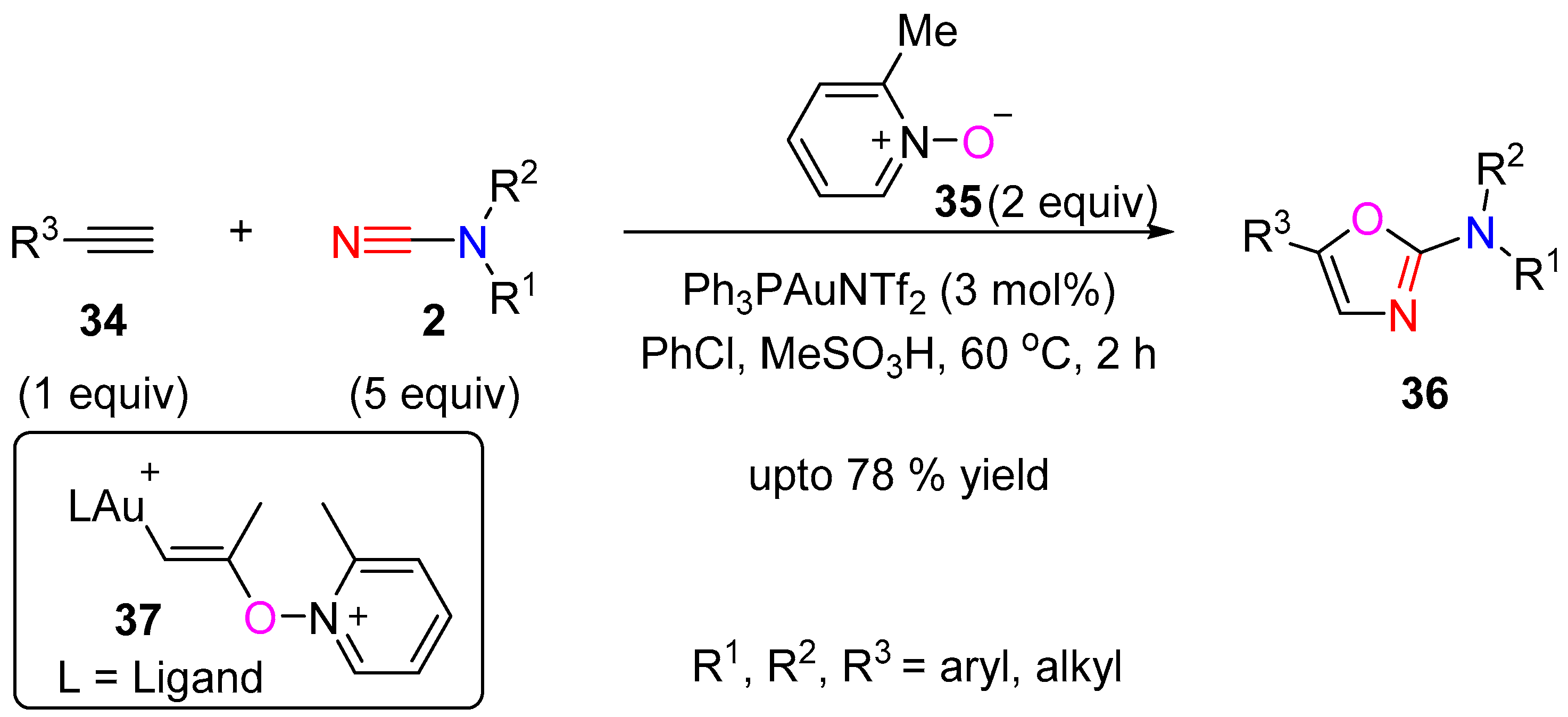
3.1.1. [3 + 2] Cycloaddition
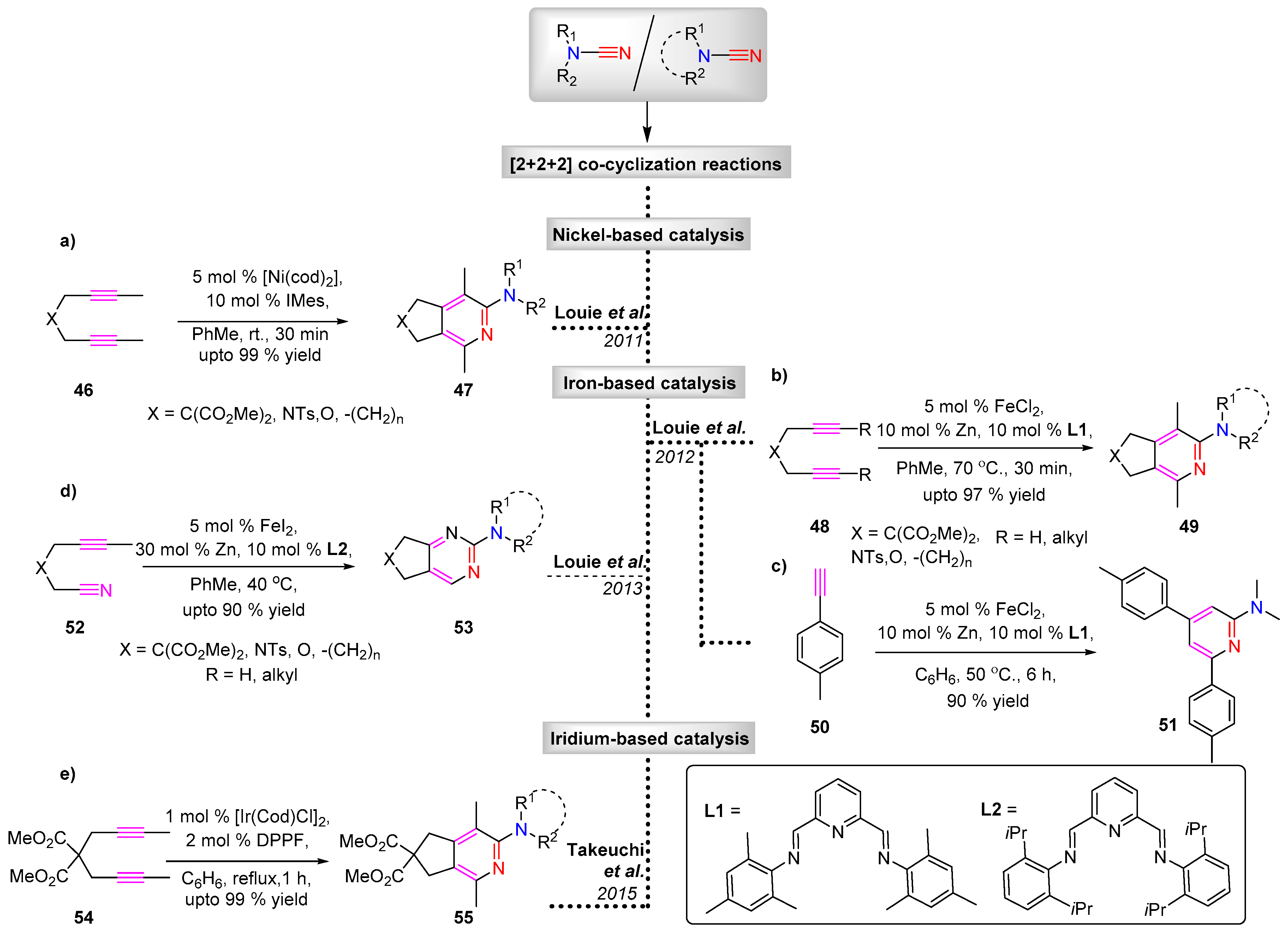
3.1.2. [2 + 2 + 2] Cycloaddition
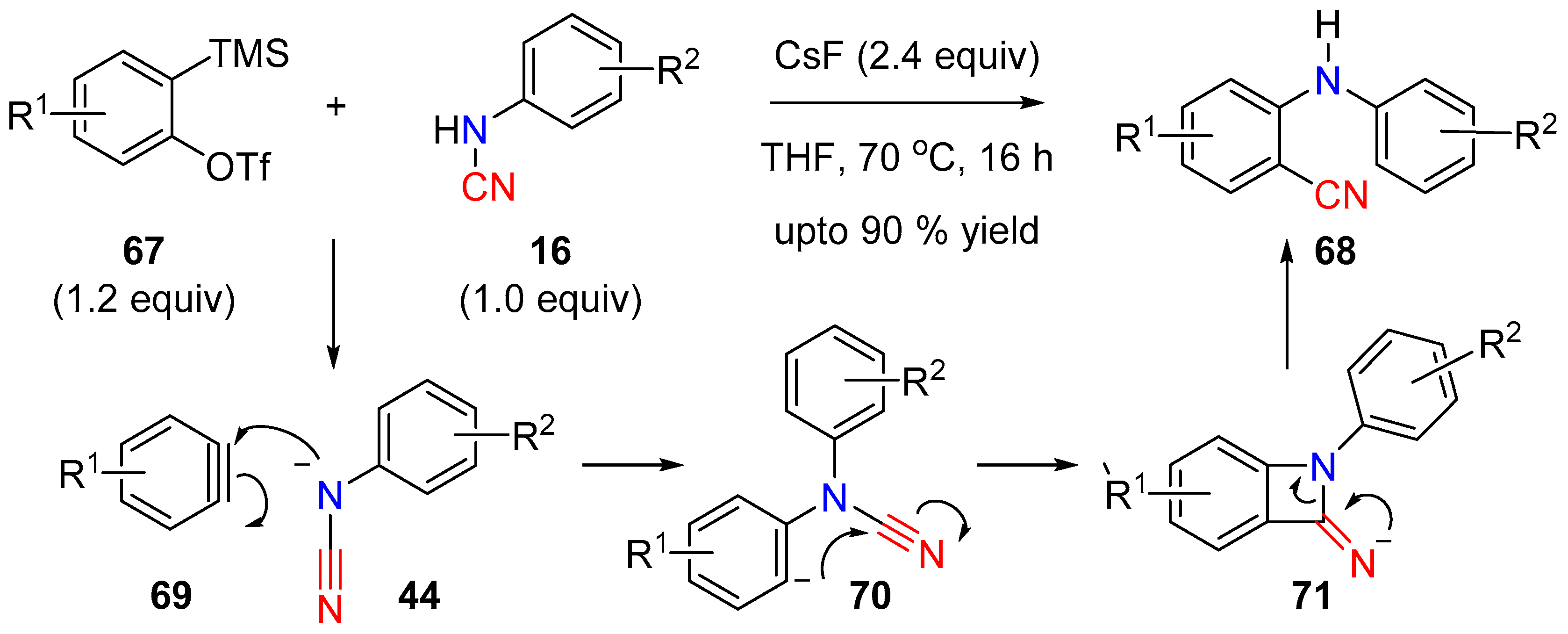
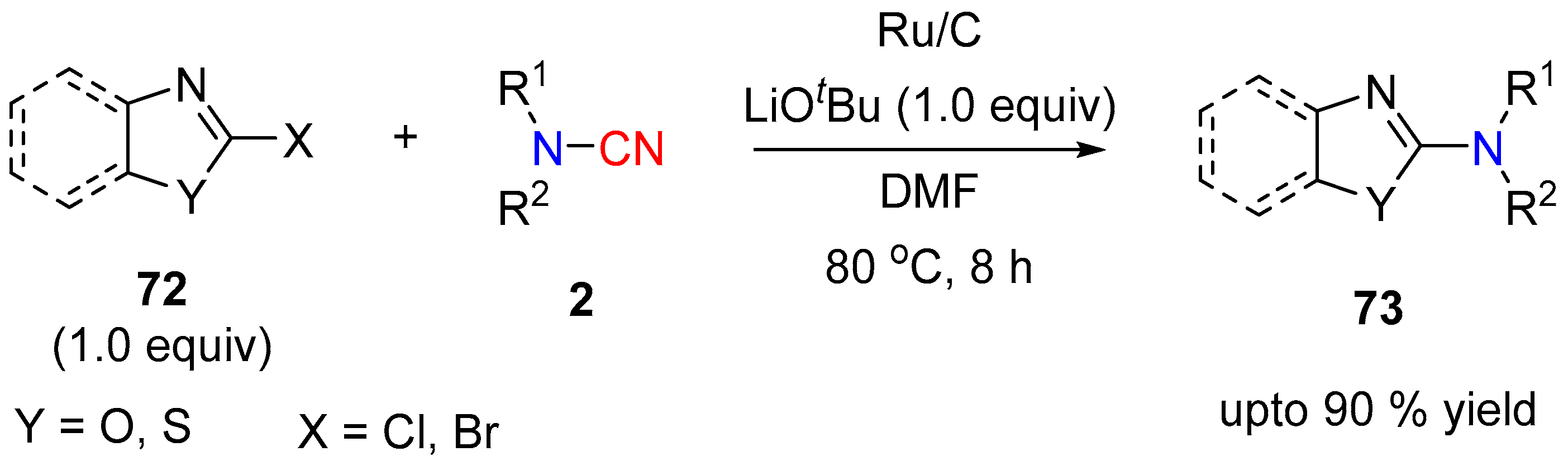
3.2. N-CN Bond Cleavage
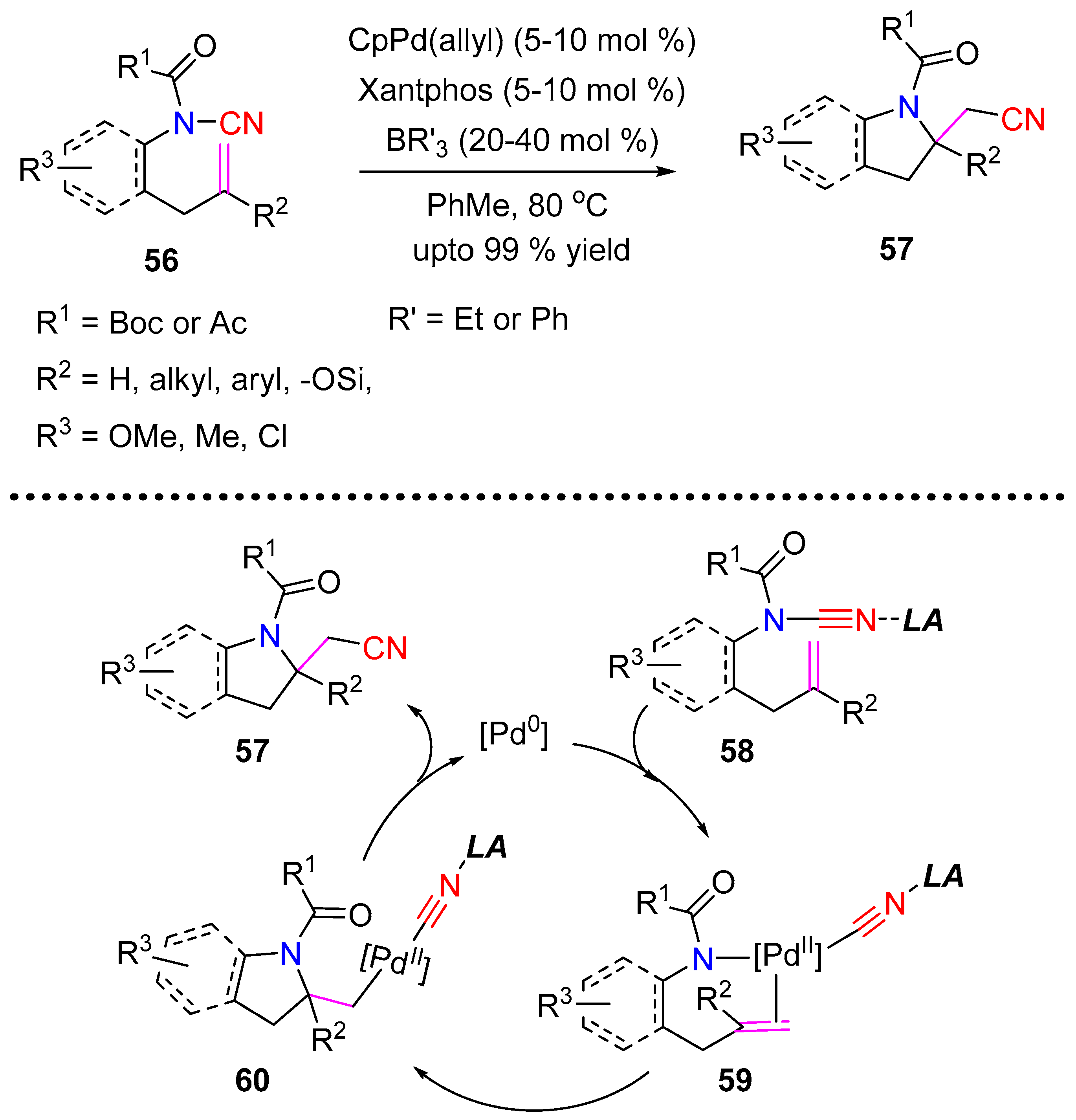
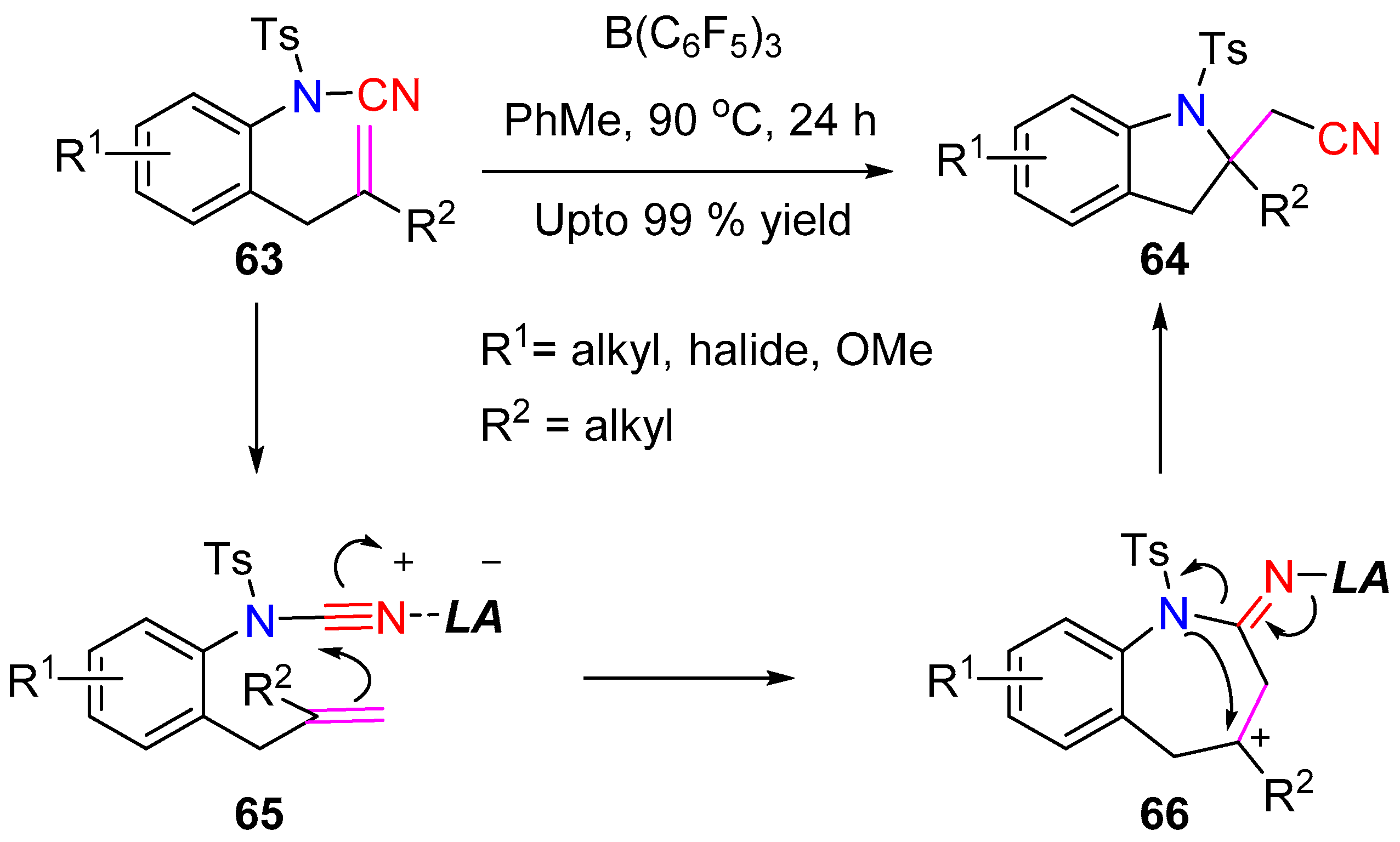
3.2.1. Aminocyanation
3.2.2. Aminating Agent
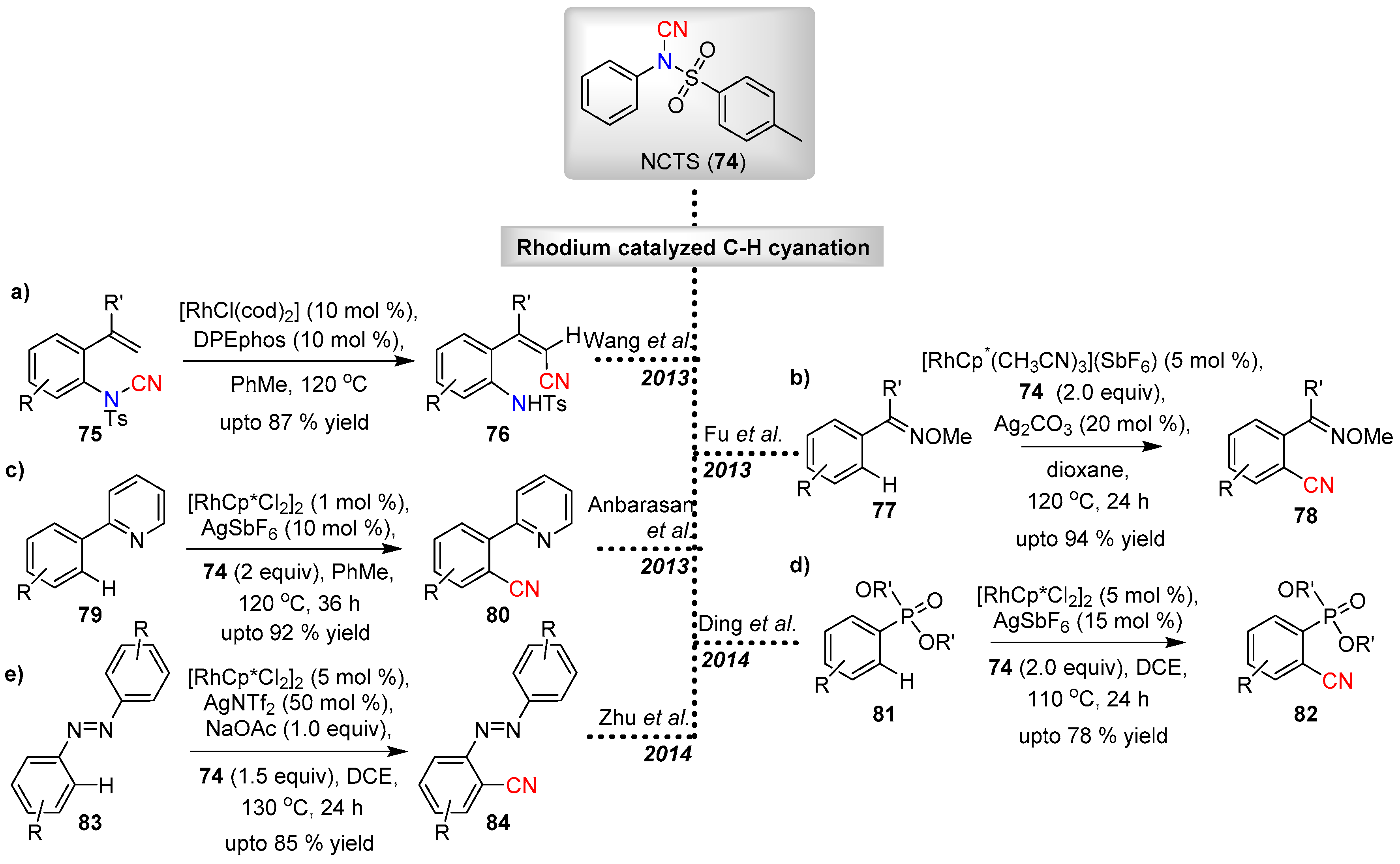
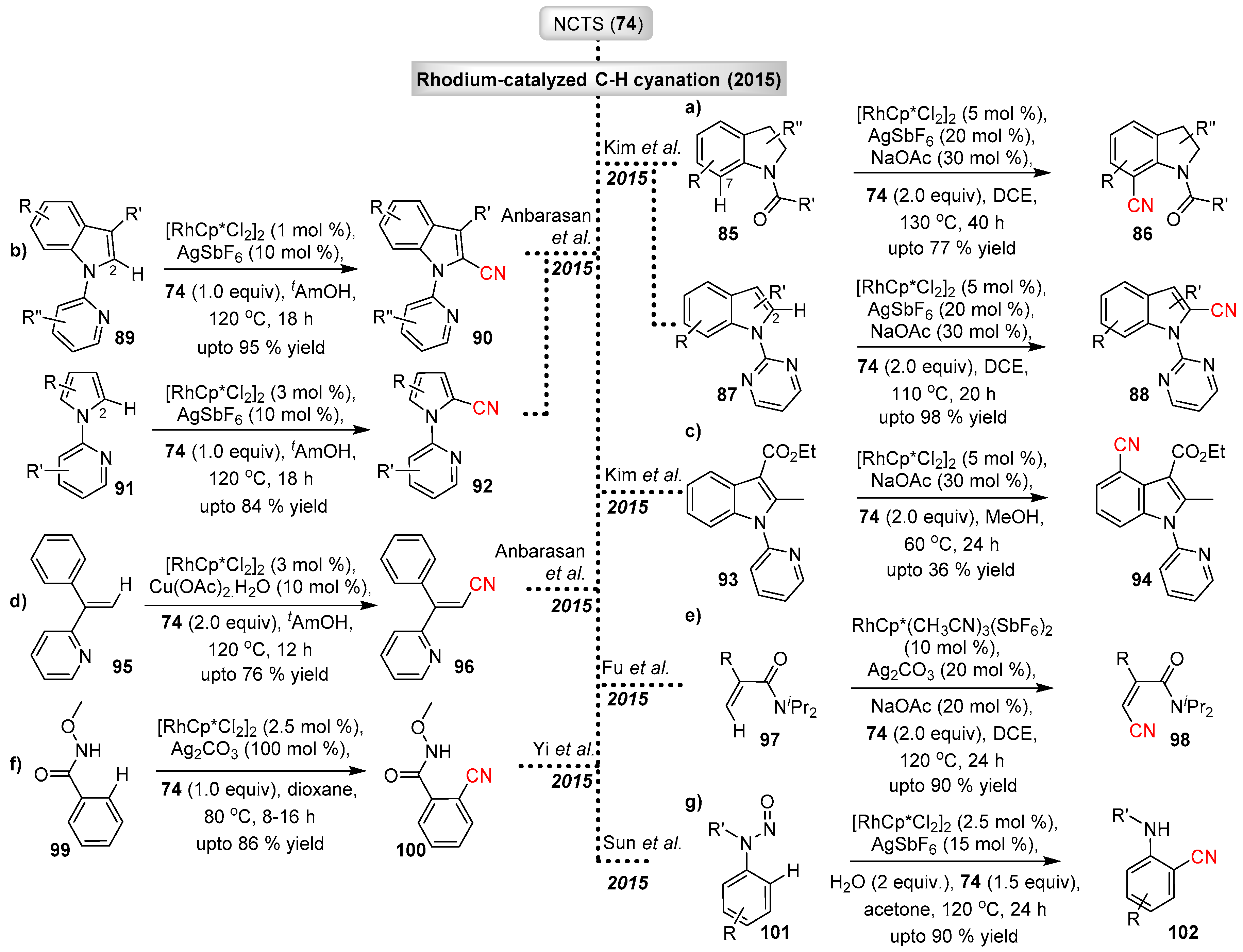
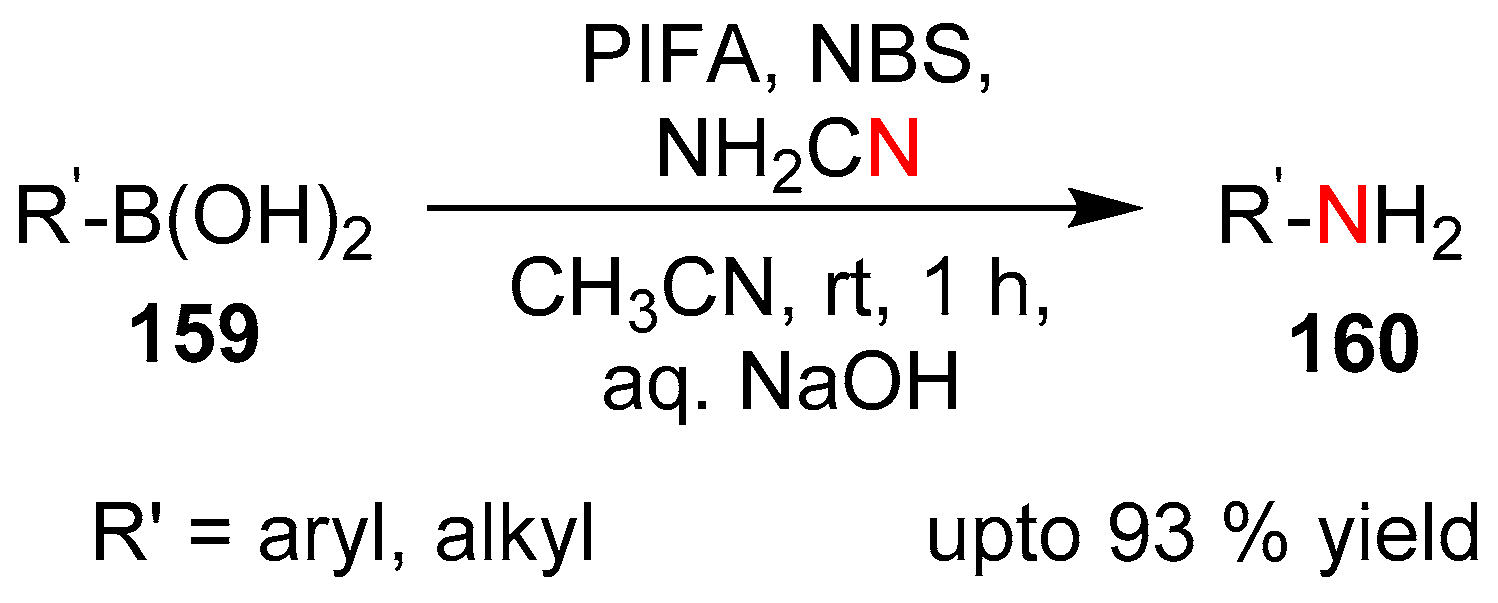
3.2.3. Electrophilic Cyanation
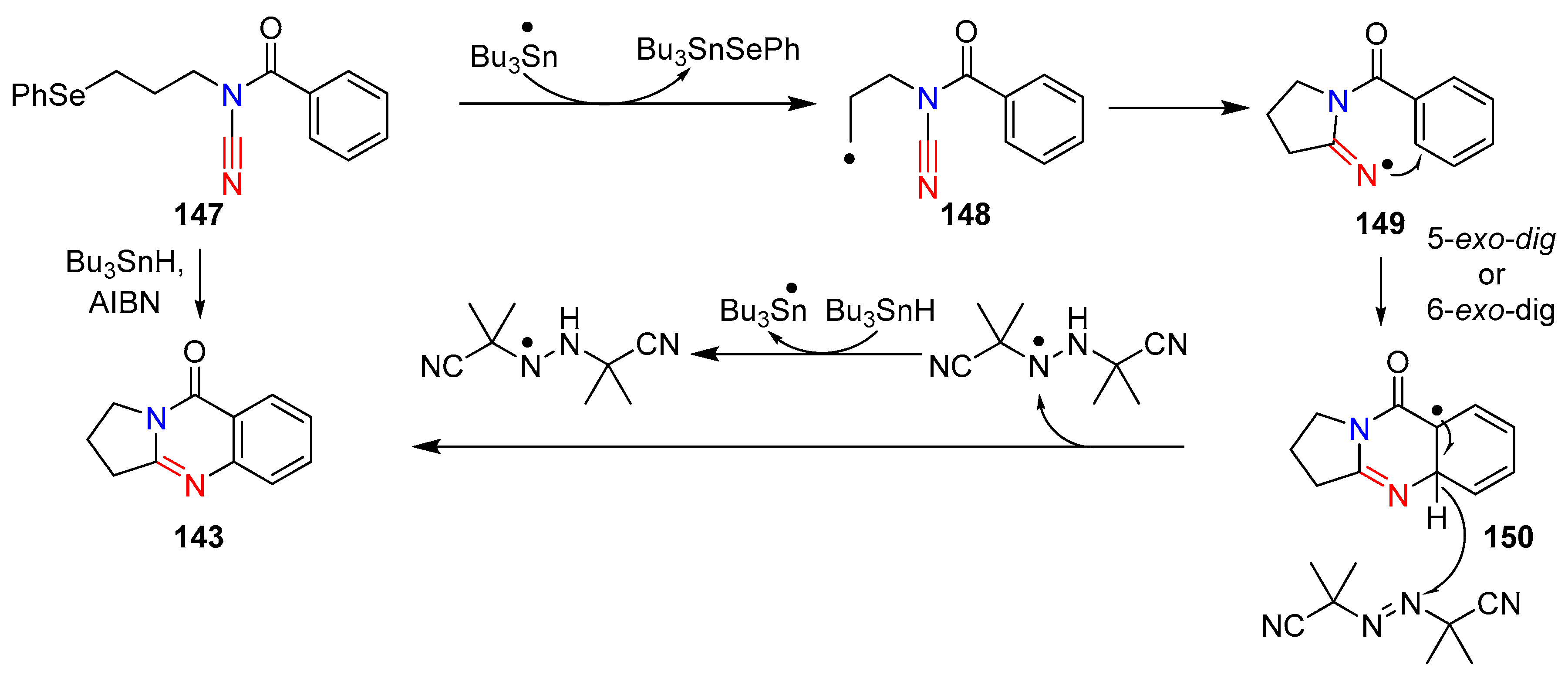
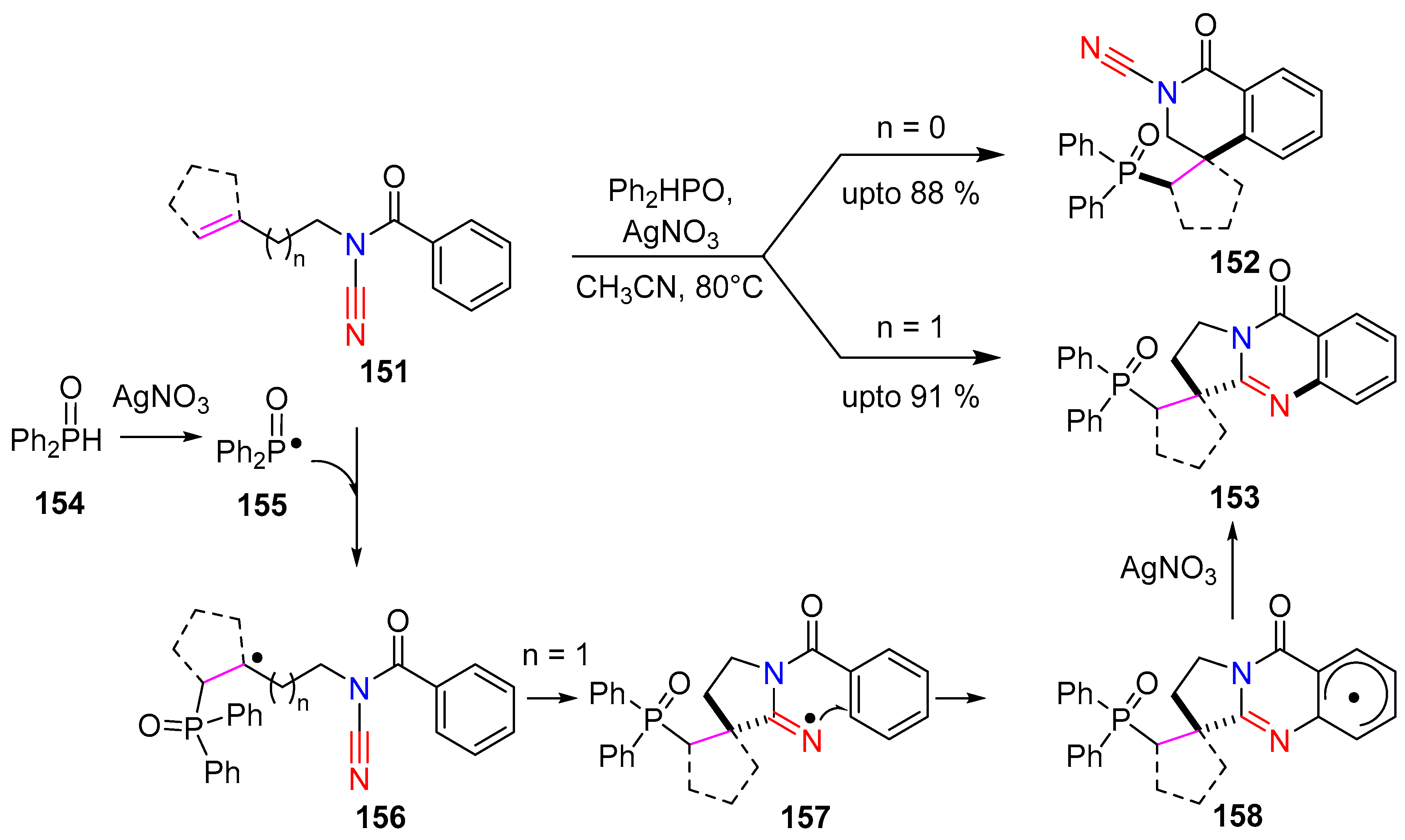
3.3. Cyanamides in Radical Reactions
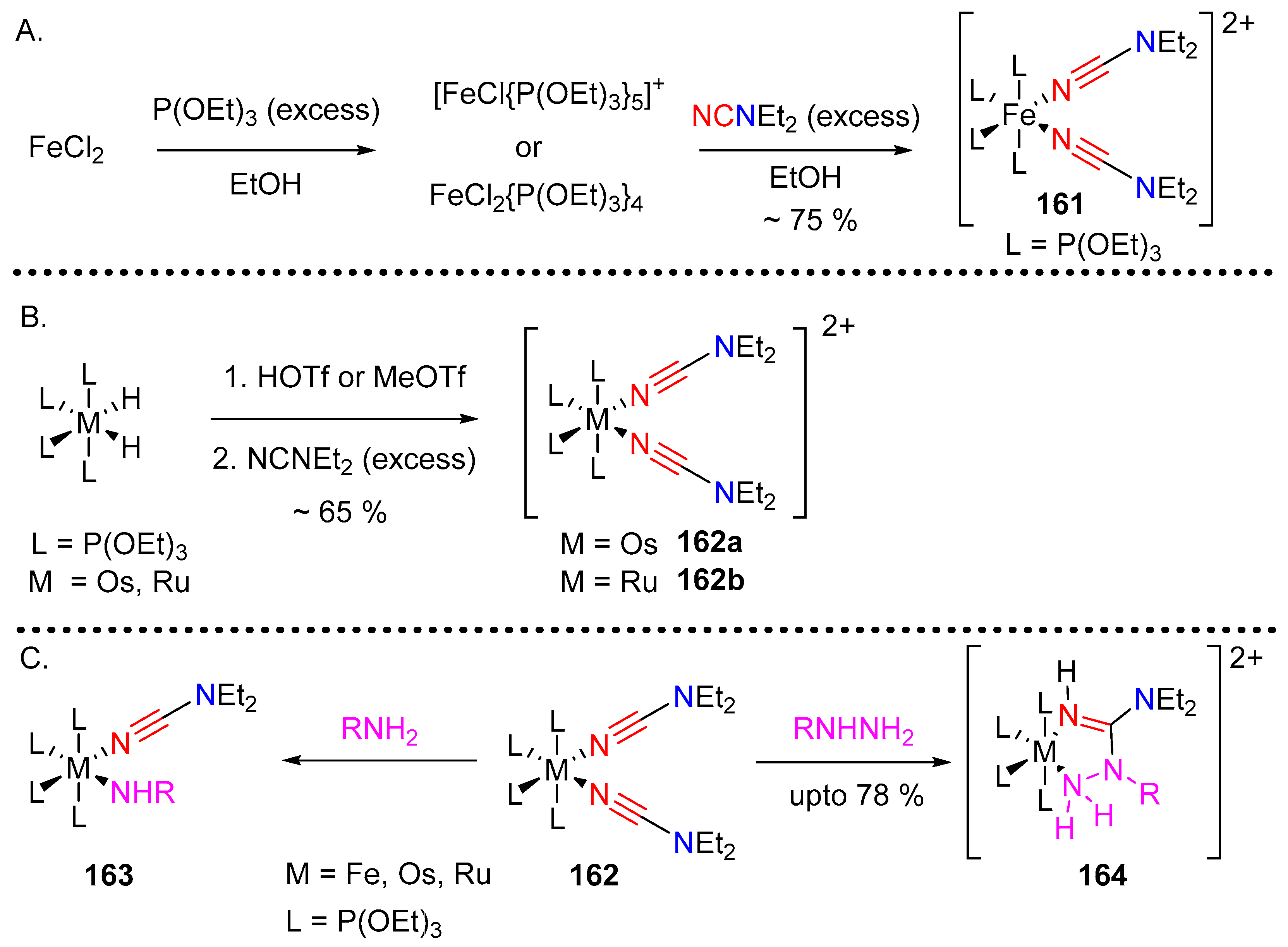
3.4. Coordination Chemistry of Cyanamides
3.4.1. Dialkyl-Cyanamide Complexes
3.4.2. Aryl Cyanamide Complexes
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Conflicts of Interest
References
- Nekrasov, D.D. Synthesis and Chemical Transformations of Mono- and Disubstituted Cyanamides. Russ. J. Org. Chem. 2004, 40, 1387–1402. [Google Scholar] [CrossRef]
- Larraufie, M.H.; Maestri, G.; Malacria, M.; Ollivier, C.; Fensterbank, L.; Lacôte, E. The Cyanamide Moiety, Synthesis and Reactivity. Synthesis 2012, 44, 1279–1292. [Google Scholar]
- Yu, J.-T.; Teng, F.; Cheng, J. The construction of X-CN (x = C, N, O) Bonds. Adv. Synth. Catal. 2017, 359, 26–38. [Google Scholar] [CrossRef]
- Kim, J.-J.; Kweon, D.-H.; Cho, S.-D.; Kim, H.-K.; Jung, E.-Y.; Lee, S.-G.; Falck, J.R.; Yoon, Y.-J. 2-Cyanopyridazin-3(2H)-ones: Effective and chemoselective electrophilic cyanating agents. Tetrahedron 2005, 61, 5889–5894. [Google Scholar] [CrossRef]
- Anbarasan, P.; Neumann, H.; Beller, M. A Convenient Synthesis of Benzonitriles via Electrophilic Cyanation with N-Cyanobenzimidazole. Chem. Eur. J. 2010, 16, 4725–4728. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-Q.; Limburg, D.C.; Wilkinson, D.E.; Hamilton, G.S. 1-Cyanoimidazole as a Mild and Efficient Electrophilic Cyanating Agent. Org. Lett. 2000, 2, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Xia, J.-B.; Chen, C. A Simple Method for the Electrophilic Cyanation of Secondary Amines. Org. Lett. 2014, 16, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Peña, J.; Alcarazo, M. Dihalo(imidazolium)sulfuranes: A Versatile Platform for the Synthesis of New Electrophilic Group-Transfer Reagents. J. Am. Chem. Soc. 2015, 137, 8704–8707. [Google Scholar] [CrossRef] [PubMed]
- Ayres, J.N.; Ling, K.B.; Morrill, L.C. N-Cyanation of Secondary Amines Using Trichloroacetonitrile. Org. Lett. 2016, 18, 5528–5531. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Yu, J.-T.; Jiang, Y.; Yang, H.; Cheng, J. A copper-mediated oxidative N-cyanation reaction. Chem. Commun. 2014, 50, 8412–8415. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; Yu, J.-T.; Zhou, Z.; Chu, H.; Cheng, J. Copper-Catalyzed N‑Cyanation of Sulfoximines by AIBN. J. Org. Chem. 2015, 80, 2822–2826. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Nagpal, R.; Chauhan, P.; Yatindranath, V.; Chauhan, S.M.S. Efficient iron(III) porphyrins-catalyzed oxidation of guanidoximes to cyanamides in ionic liquids. J. Chem. Sci. 2015, 127, 13–18. [Google Scholar] [CrossRef]
- Tiemann, F. Ueber die Einwirkung von Benzolsulfonsäurechlorid auf Amidoxime. Ber. Dtsch. Chem. Ges. 1891, 24, 4162–4167. [Google Scholar] [CrossRef]
- Lin, C.-C.; Hsieh, T.-H.; Liao, P.-Y.; Liao, Z.-Y.; Chang, C.-W.; Shih, Y.-C.; Yeh, W.-H.; Chien, T.-C. Practical Synthesis of N-Substituted Cyanamides via Tiemann Rearrangement of Amidoximes. Org. Lett. 2014, 16, 892–895. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Huang, Y.; Gan, B.; Mi, Z.; Xie, Y. Synthesis of cyanamides from isoselenocyanates promoted by recyclable ionic liquid-supported (diacetoxyiodo)benzene. J. Chem. Res. 2015, 39, 631–634. [Google Scholar] [CrossRef]
- Karabanovich, G.; Roh, J.; Padělková, Z.; Novák, Z.; Vávrová, K.; Hrabálek, A. One-pot synthesis of 1-substituted-5-alkylselanyl-1H-tetrazoles from isoselenocyanates: Unexpected formation of N-alkyl-N-arylcyanamides and (Z)-Se-alkyl-N-cyano-N,N′-diarylisoselenoureas. Tetrahedron 2013, 69, 8798–8808. [Google Scholar] [CrossRef]
- Rassadin, V.A.; Boyarskiy, V.P.; Kukushkin, V.Y. Facile Gold-Catalyzed Heterocyclization of Terminal Alkynes and Cyanamides Leading to Substituted 2-Amino-1,3-Oxazoles. Org. Lett. 2015, 17, 3502–3505. [Google Scholar] [CrossRef] [PubMed]
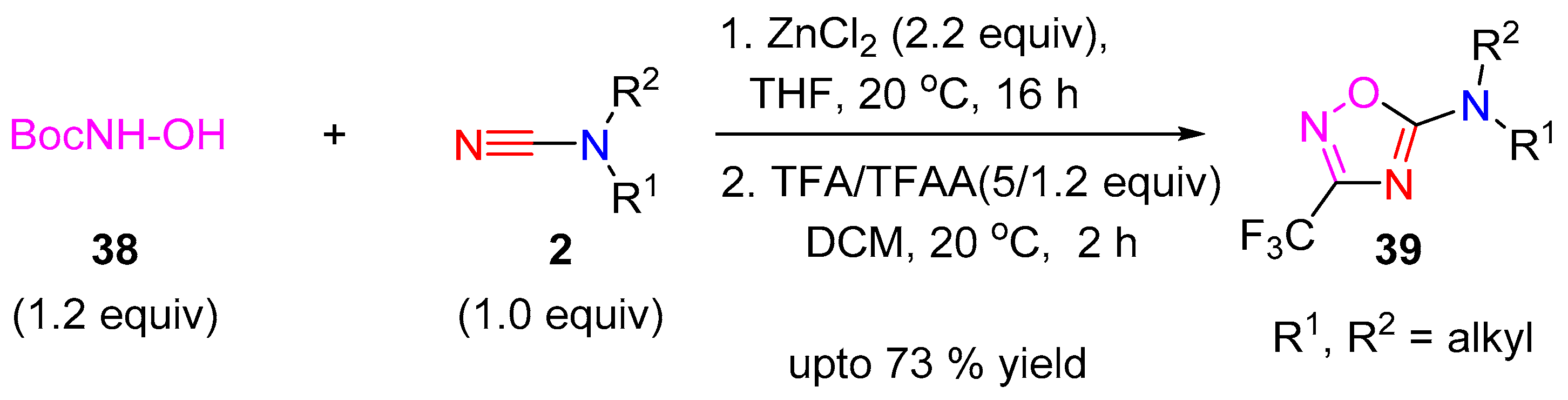
- Goldberg, K.; Clarke, D.S.; Scott, J.S. A facile synthesis of 3-trifluoromethyl-1,2,4-oxadiazoles from cyanamides. Tetrahedron Lett. 2014, 55, 4433–4436. [Google Scholar] [CrossRef]
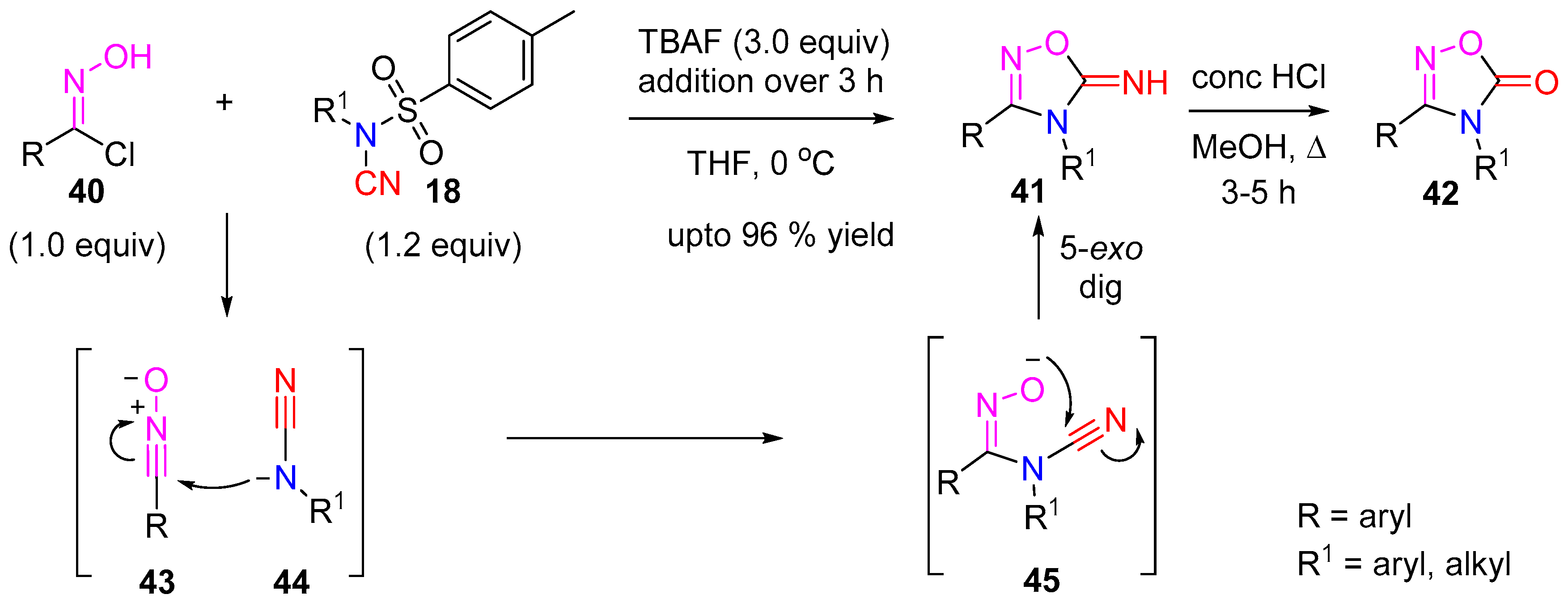
- Bhat, S.V.; Robinson, D.; Moses, J.E.; Sharma, P. Synthesis of Oxadiazol-5-imines via the Cyclizative Capture of in situ Generated Cyanamide Ions and Nitrile Oxides. Org. Lett. 2016, 18, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Heller, B.; Sundermann, B.; Buschmann, H.; Drexler, H.-J.; You, J.; Holzgrabe, U.; Heller, E.; Oehme, G. Photocatalyzed [2 + 2 + 2]-Cycloaddition of Nitriles with Acetylene: An Effective Method for the Synthesis of 2-Pyridines under Mild Conditions. J. Org. Chem. 2002, 67, 4414–4422. [Google Scholar] [CrossRef] [PubMed]
- Boñaga, L.V.R.; Zhang, H.C.; Moretto, A.F.; Ye, H.; Gauthier, D.A.; Li, J.; Leo, G.C.; Maryanoff, B.E. Synthesis of Macrocycles via Cobalt-Mediated [2 + 2 + 2] Cycloadditions. J. Am. Chem. Soc. 2005, 127, 3473–3485. [Google Scholar] [CrossRef] [PubMed]
- Stolley, R.M.; Maczka, M.T.; Louie, J. Nickel-Catalyzed [2 + 2 + 2] Cycloaddition of Diynes and Cyanamides. Eur. J. Org. Chem. 2011, 2011, 3815–3824. [Google Scholar] [CrossRef] [PubMed]
- Lane, T.K.; D’Souza, B.R.; Louie, J. Iron-Catalyzed Formation of 2-Aminopyridines from Diynes and Cyanamides. J. Org. Chem. 2012, 77, 7555–7563. [Google Scholar] [CrossRef] [PubMed]
- Spahn, N.A.; Nguyen, M.H.; Renner, J.; Lane, T.K.; Louie, J. Regioselective Iron-Catalyzed [2 + 2 + 2] Cycloaddition Reaction Forming 4,6-Disubstituted 2-Aminopyridines from Terminal Alkynes and Cyanamides. J. Org. Chem. 2017, 82, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, D.; Xu, F.; Pan, B.; Wan, B. Iron-Catalyzed Cycloaddition Reaction of Diynes and Cyanamides at Room Temperature. J. Org. Chem. 2013, 78, 3065–3072. [Google Scholar] [CrossRef] [PubMed]
- Lane, T.K.; Nguyen, M.H.; D’Souza, B.R.; Spahn, N.A.; Louie, J. The iron-catalyzed construction of 2-aminopyrimidines from alkynenitriles and cyanamides. Chem. Commun. 2013, 49, 7735–7737. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Ishii, S.; Yano, R.; Miura, H.; Sakata, K.; Takeuchi, R. Iridium-Catalyzed [2 + 2 + 2] Cycloaddition of α,ω-Diynes with Cyanamides. Adv. Synth. Catal. 2015, 357, 3901–3916. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Ohta, N.; Semba, K.; Nakao, Y. Intramolecular Aminocyanation of Alkenes by Cooperative Palladium/Boron Catalysis. J. Am. Chem. Soc. 2014, 136, 3732–3735. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.-Y.; Liao, P.-Y.; Chien, T.-C. CuI-Catalyzed intramolecular aminocyanation of terminal alkynes in N-(2-ethynylphenyl)-N-sulfonylcyanamides via Cu-vinylidene intermediates. Chem. Commun. 2016, 52, 14404–14407. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Pound, S.M.; Rondla, N.R.; Douglas, C.J. Intramolecular Aminocyanation of Alkenes by N-CN Bond Cleavage. Angew. Chem. Int. Ed. 2014, 53, 5170–5174. [Google Scholar]
- Rao, B.; Zeng, X. Aminocyanation by the Addition of N–CN Bonds to Arynes: Chemoselective Synthesis of 1,2-Bifunctional Aminobenzonitriles. Org. Lett. 2014, 16, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.H.V.; Kumar, B.S.P.A.; Reddy, V.P.; Kumar, R.U.; Nageswar, Y.V.D. Ru/C: A simple heterogeneous catalyst for the amination of azoles under ligand free conditions. RSC Adv. 2014, 4, 45579–45584. [Google Scholar] [CrossRef]
- Kurzer, F. Cyanamides. Part 1. The Synthesis of Substituted Arylsulfonylcyanamides. J. Chem. Soc. 1949, 1034–1038. [Google Scholar] [CrossRef]
- Anbarasan, P.; Neumann, H.; Beller, M. A Novel and Convenient Synthesis of Benzonitriles: Electrophilic Cyanation of Aryl and Heteroaryl Bromides. Chem. Eur. J. 2011, 17, 4217–4222. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.N.; Nikumbh, S.P.; Kumar, S.P.; Rao, L.V.; Raghunadh, A. An efficient one pot synthesis of 2-amino quinazolin-4(3H)-one derivative via MCR strategy. Tetrahedron Lett. 2015, 56, 5767–5770. [Google Scholar] [CrossRef]
- Kasthuri, M.; Babu, H.S.; Kumar, K.S.; Sudhakar, C.; Kumar, P.V.N. A Facile Synthesis of 2-Aminobenzoxazoles and 2-Aminobenzimidazoles Using N-Cyano-N-phenyl-p-toluenesulfonamide (NCTS) as an Efficient Electrophilic Cyanating Agent. Synlett 2015, 26, 897–900. [Google Scholar] [CrossRef]
- Anbarasan, P.; Neumann, H.; Beller, M. A General Rhodium-Catalyzed Cyanation of Aryl and Alkenyl Boronic Acids. Angew. Chem. Int. Ed. 2011, 50, 519–522. [Google Scholar] [CrossRef] [PubMed]
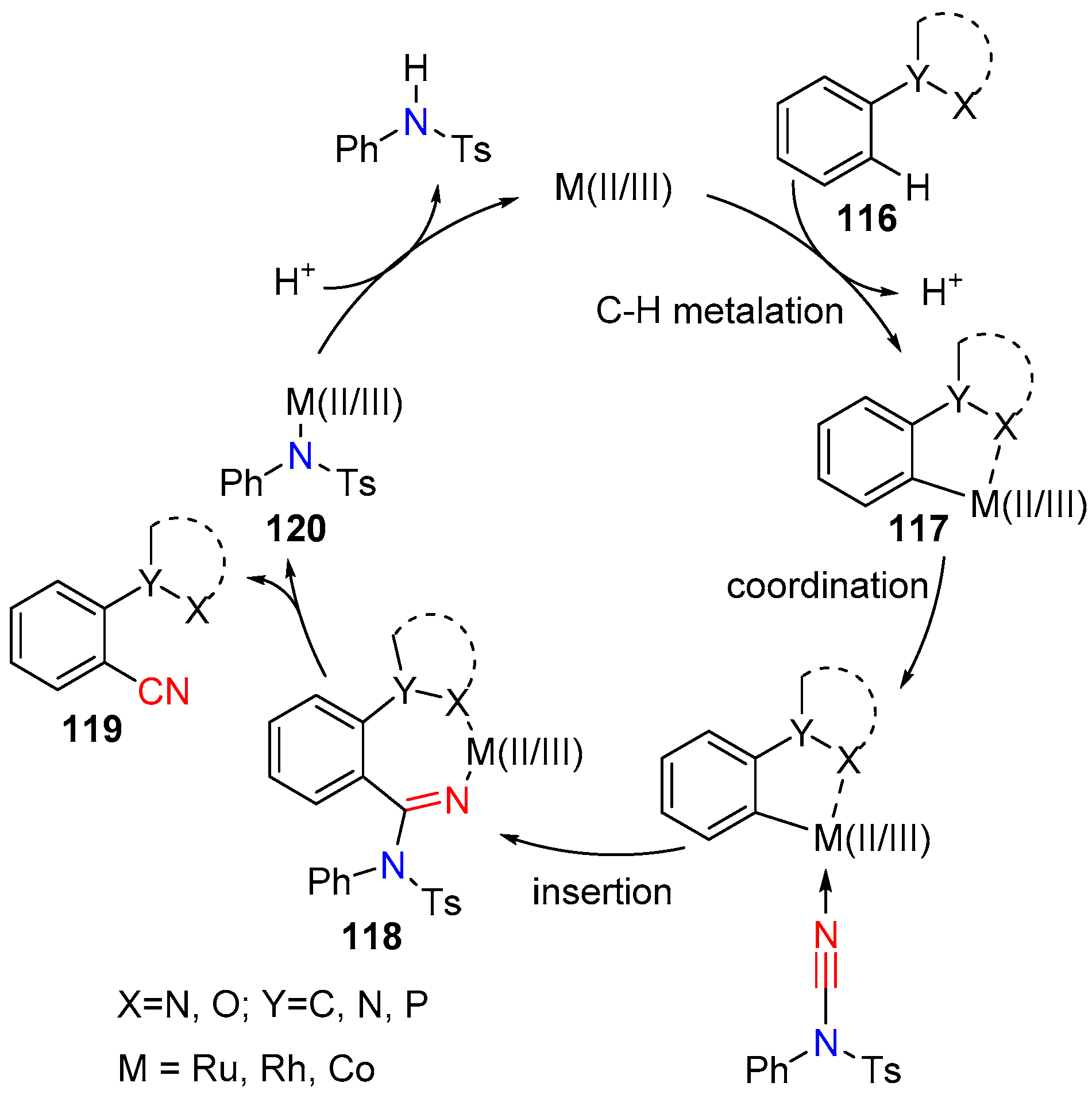
- Ping, Y.; Ding, Q.; Peng, Y. Advances in C−CN Bond Formation via C−H Bond Activation. ACS Catal. 2016, 6, 5989–6005. [Google Scholar] [CrossRef]
- Jiang, H.; Gao, S.; Xu, J.; Wu, X.; Lin, A.; Yao, H. Multiple Roles of the Pyrimidyl Group in the Rhodium-Catalyzed Regioselective Synthesis and Functionalization of Indole-3-carboxylic Acid Esters. Adv. Synth. Catal. 2016, 358, 188–194. [Google Scholar] [CrossRef]
- Wang, R.; Falck, J.R. Rhodium(i)-catalyzed N-CN bond cleavage: Intramolecular β-cyanation of styrenes. Chem. Commun. 2013, 49, 6516–6518. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.-J.; Xiao, B.; Cheng, W.-M.; Su, W.; Xu, J.; Liu, Z.-J.; Liu, L.; Fu, Y. Rhodium-Catalyzed Directed C–H Cyanation of Arenes with N-Cyano-N-phenyl-p-toluenesulfonamide. J. Am. Chem. Soc. 2013, 135, 10630–10633. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, M.; Yadagiri, D.; Anbarasan, P. Rhodium Catalyzed Cyanation of Chelation Assisted C–H Bonds. Org. Lett. 2013, 15, 4960–4963. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.-J.; Jin, C.; Wang, R.; Ding, H.-Y. Rhodium Catalyzed ortho-Cyanation of Arylphosphates with N-cyano-N-phenyl-p-toluenesulfonamide. ChemCatChem 2014, 6, 1225–1228. [Google Scholar]
- Han, J.; Pan, C.; Jia, X.; Zhu, C. Rhodium-catalyzed ortho-cyanation of symmetrical azobenzenes with N-cyano-N-phenyl-p-toluenesulfonamide. Org. Biomol. Chem. 2014, 12, 8603–8606. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.K.; Jeong, T.; Sharma, S.; Shin, Y.; Han, S.; Park, J.; Oh, J.S.; Kwak, J.H.; Jung, Y.H.; Kim, I.S. Rhodium(III)-Catalyzed Selective C–H Cyanation of Indolines and Indoles with an Easily Accessible Cyano Source. Adv. Synth. Catal. 2015, 357, 1293–1298. [Google Scholar] [CrossRef]
- Mishra, N.K.; Choi, M.; Jo, H.; Oh, Y.; Sharma, S.; Han, S.H.; Jeong, T.; Han, S.; Lee, S.-Y.; Kim, I.S. Direct C–H alkylation and indole formation of anilines with diazo compounds under rhodium catalysis. Chem. Commun. 2015, 51, 17229–17232. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, M.; Anbarasan, P. Rhodium Catalyzed C2-Selective Cyanation of Indoles and Pyrroles. J. Org. Chem. 2015, 80, 3695–3700. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, M.; Anbarasan, P. Rhodium-Catalyzed Cyanation of C(sp2)–H Bond of Alkenes. Org. Lett. 2015, 17, 3766–3769. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Gong, T.J.; Xiao, B.; Fu, Y. Rhodium(III)-catalyzed cyanation of vinylic C–H bonds: N-cyano-N-phenyl-p-toluenesulfonamide as a cyanation reagent. Chem. Commun. 2015, 51, 11848–11851. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, J.; Shi, J.; Wang, M.; Xu, H.E.; Yi, W. Rh(III)-catalyzed direct C–H cyanation of N-methoxybenzamides using N-cyano-N-phenyl-p-toluenesulfonamide. Chin. J. Catal. 2015, 36, 1175–1182. [Google Scholar] [CrossRef]
- Dong, J.; Wu, Z.; Liu, Z.; Liu, P.; Sun, P. Rhodium(III)-Catalyzed Direct Cyanation of Aromatic C–H Bond to Form 2-(Alkylamino)benzonitriles Using N-Nitroso As Directing Group. J. Org. Chem. 2015, 80, 12588–12593. [Google Scholar] [CrossRef] [PubMed]
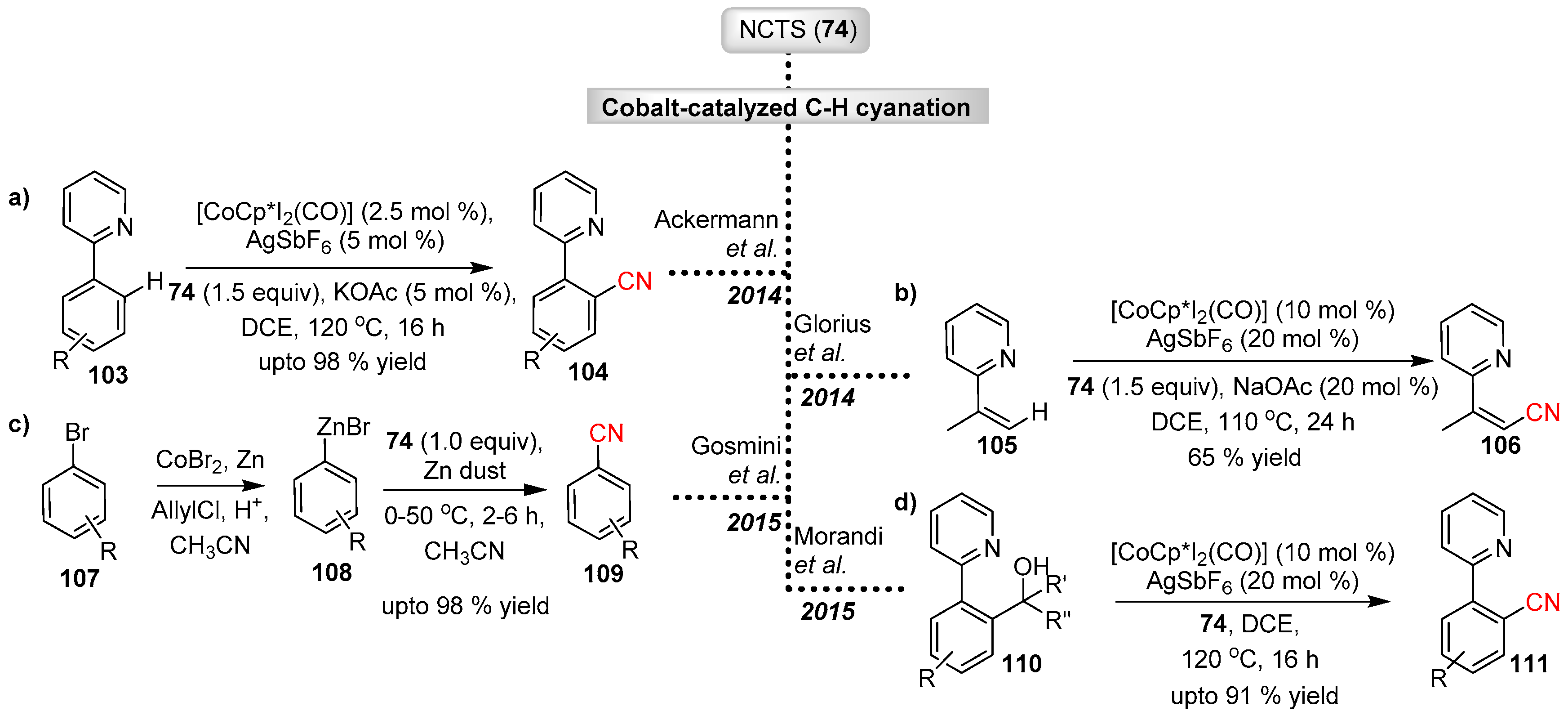
- Li, J.; Ackermann, L. Cobalt-Catalyzed C–H Cyanation of Arenes and Heteroarenes. Angew. Chem. Int. Ed. 2015, 54, 3635–3638. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-G.; Gensch, T.; de Azambuja, F.; Vásquez-Céspedes, S.; Glorius, F. Co(III)-Catalyzed C–H Activation/Formal SN-Type Reactions: Selective and Efficient Cyanation, Halogenation, and Allylation. J. Am. Chem. Soc. 2014, 136, 17722–17725. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Qian, X.; Rérat, A.; Auffrant, A.; Gosmini, C. Cobalt-Catalyzed Electrophilic Cyanation of Arylzinc Halides with N-Cyano-N-phenyl-p-methylbenzenesulfonamide (NCTS). Adv. Synth. Catal. 2015, 357, 3419–3423. [Google Scholar] [CrossRef]
- Ozkal, E.; Cacherat, B.; Morandi, B. Cobalt(III)-Catalyzed Functionalization of Unstrained Carbon–Carbon Bonds through β-Carbon Cleavage of Alcohols. ACS Catal. 2015, 5, 6458–6462. [Google Scholar] [CrossRef]
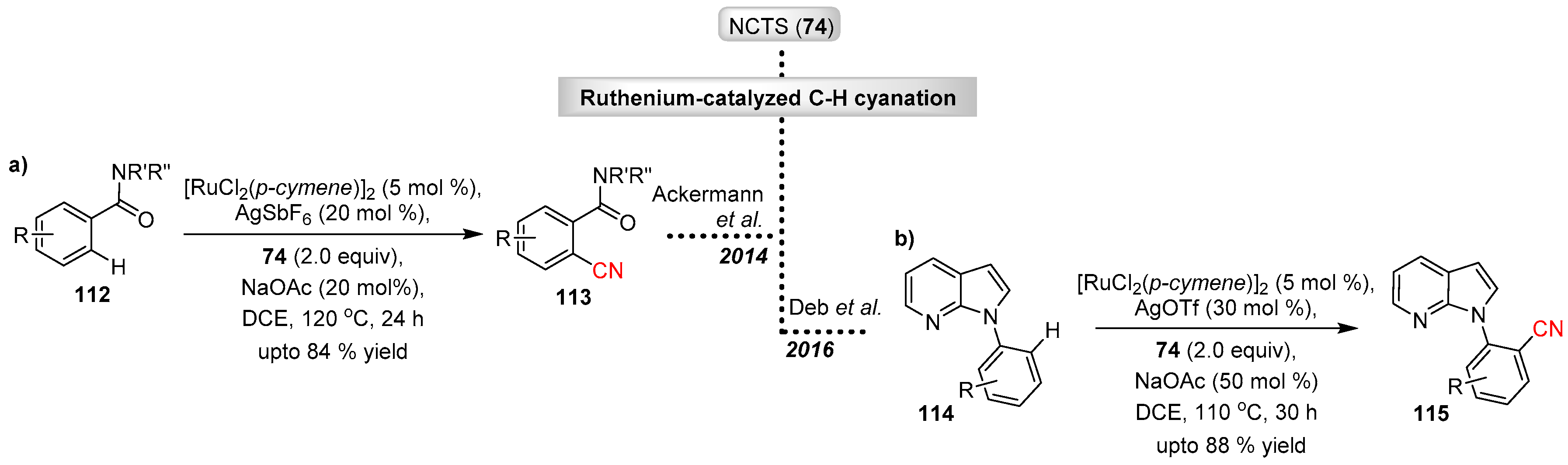
- Liu, W.; Ackermann, L. Versatile ruthenium(II)-catalyzed C–H cyanations of benzamides. Chem. Commun. 2014, 50, 1878–1881. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Vats, T.K.; Deb, I. Ruthenium-Catalyzed Direct and Selective C–H Cyanation of N-(Hetero)aryl-7-azaindoles. J. Org. Chem. 2016, 81, 6525–6534. [Google Scholar] [CrossRef] [PubMed]
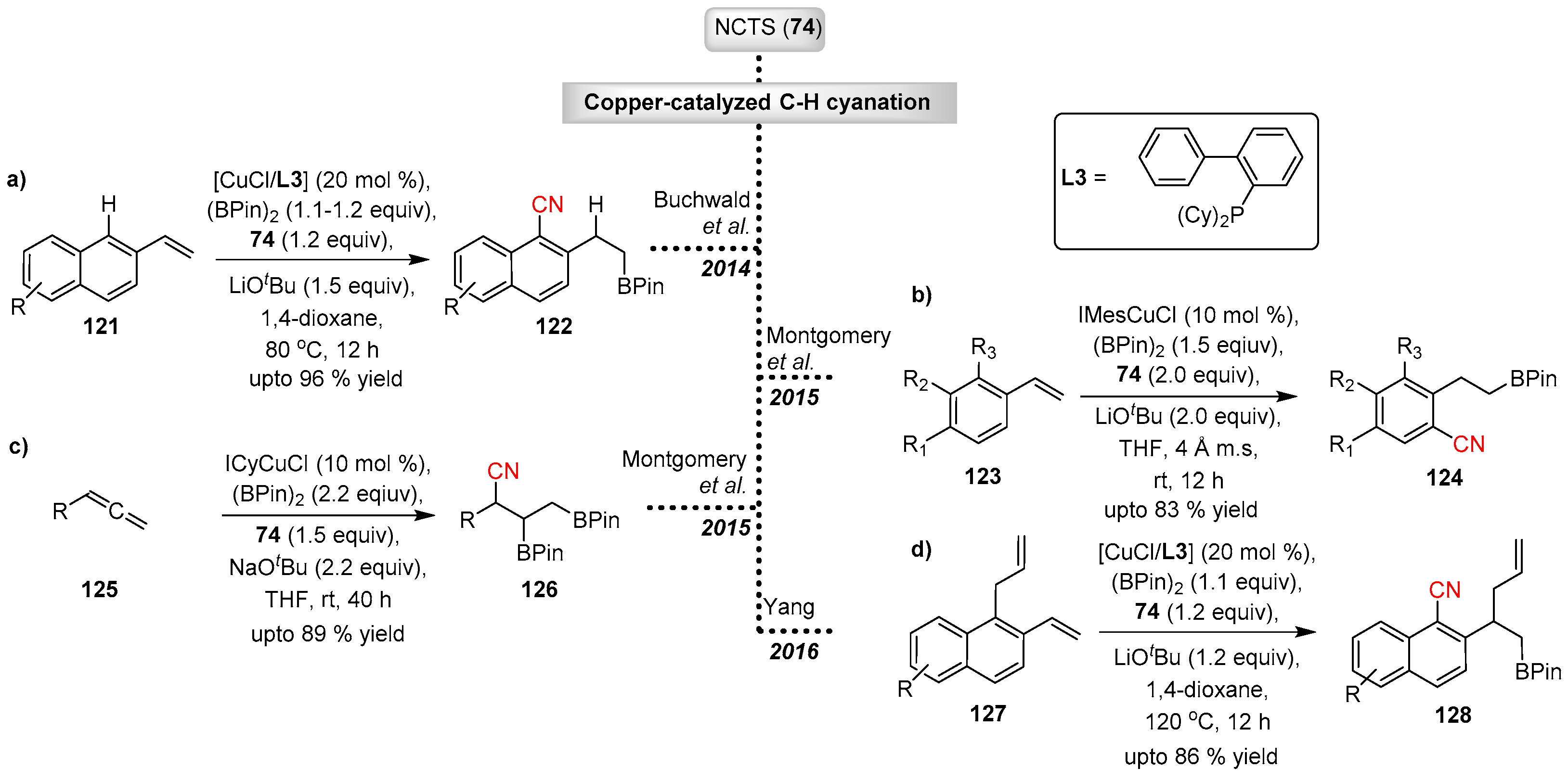
- Yang, Y.; Buchwald, S.L. Copper-Catalyzed Regioselective ortho C–H Cyanation of Vinylarenes. Angew. Chem. Int. Ed. 2014, 53, 8677–8681. [Google Scholar] [CrossRef] [PubMed]
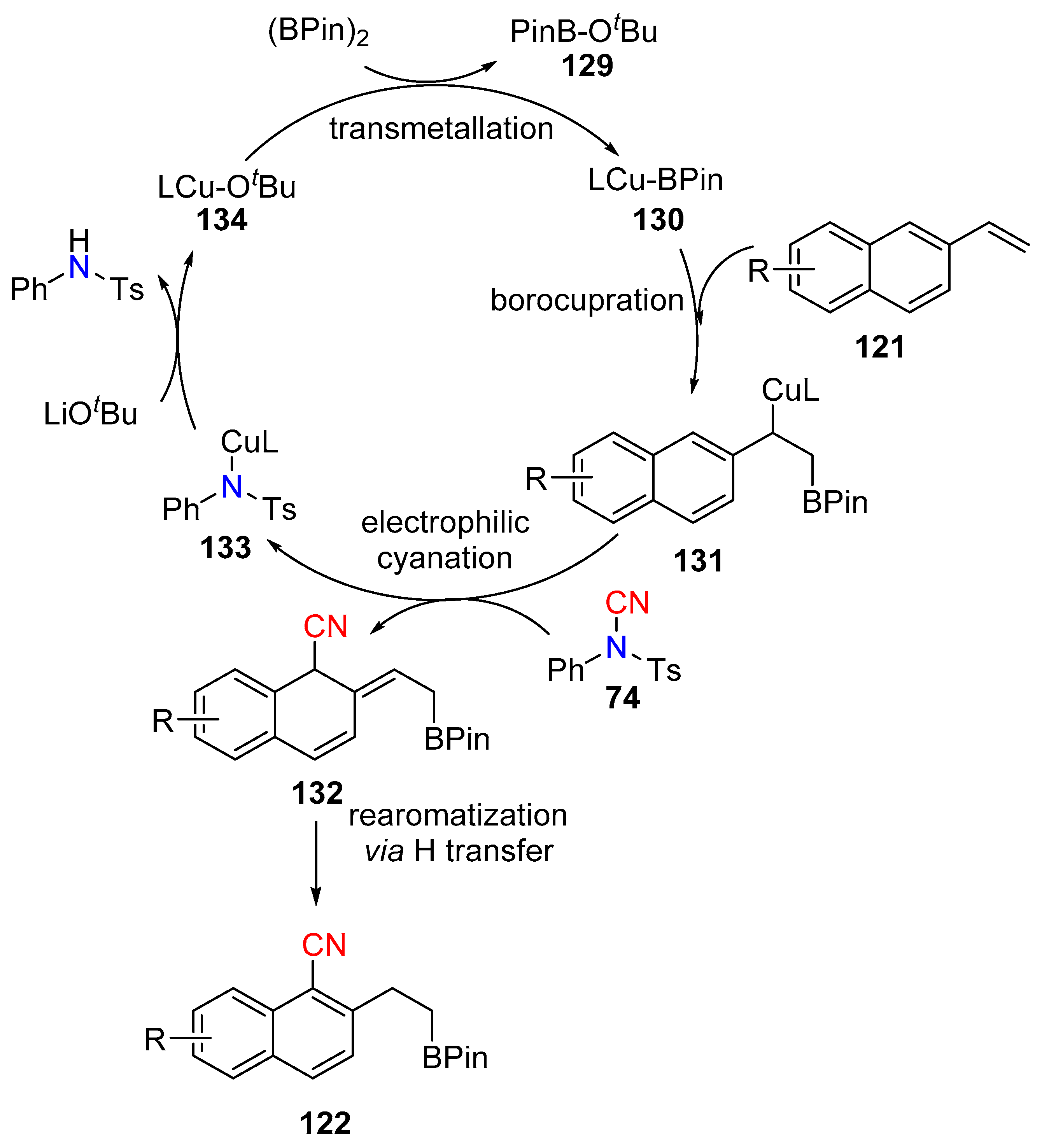
- Zhao, W.; Montgomery, J. Functionalization of Styrenes by Copper-Catalyzed Borylation/ ortho-Cyanation and Silver-Catalyzed Annulation Processes. Angew. Chem. Int. Ed. 2015, 54, 12683–12686. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Montgomery, J. Cascade Copper-Catalyzed 1,2,3-Trifunctionalization of Terminal Allenes. J. Am. Chem. Soc. 2016, 138, 9763–9766. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y. Regio- and Stereospecific 1,3-Allyl Group Transfer Triggered by a Copper-Catalyzed Borylation/ortho-Cyanation Cascade. Angew. Chem. Int. Ed. 2016, 55, 345–349. [Google Scholar] [CrossRef] [PubMed]
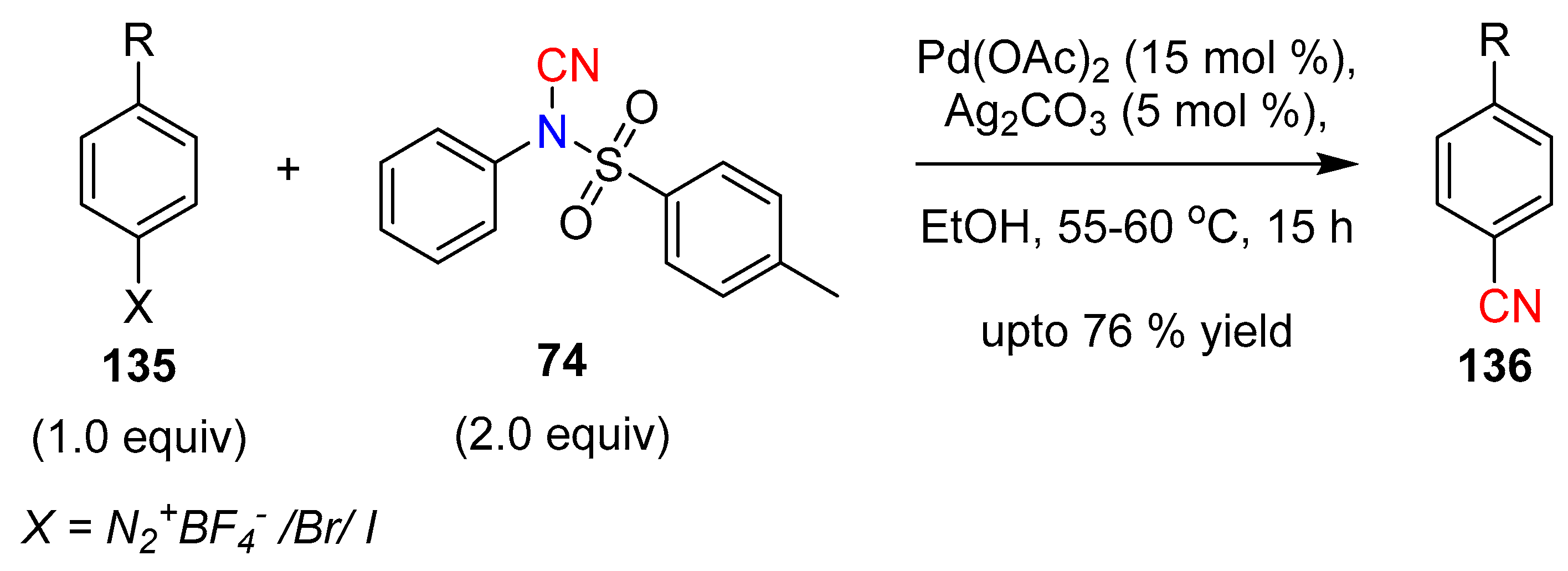
- Li, J.; Xu, W.; Ding, J.; Lee, K.-H. The application of NCTS (N-cyano-N-phenyl-p-toluenesulfonamide) in palladium-catalyzed cyanation of arenediazonium tetrafluoroborates and aryl halides. Tetrahedron Lett. 2016, 57, 1205–1209. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Wang, J. Lewis Acid Catalyzed Direct Cyanation of Indoles and Pyrroles with N-Cyano-N-phenyl-p-toluenesulfonamide (NCTS). Org. Lett. 2011, 13, 5608–5611. [Google Scholar] [CrossRef] [PubMed]
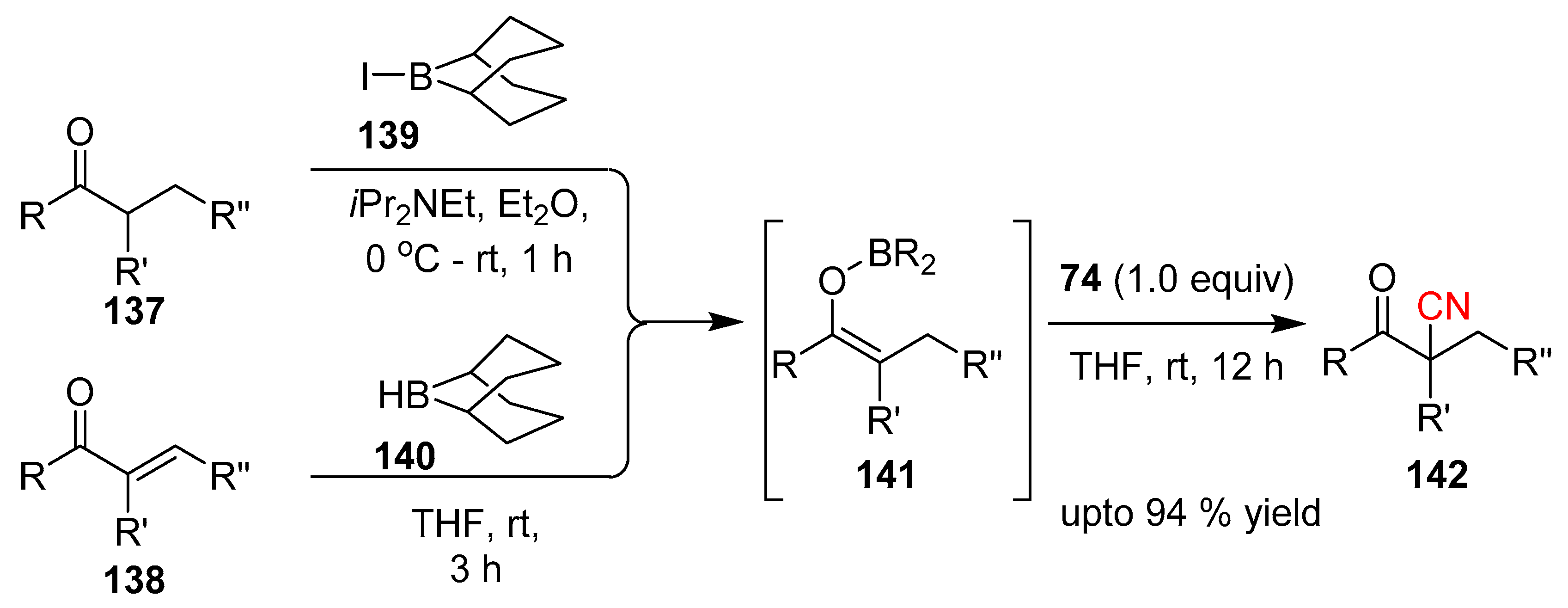
- Kiyokawa, K.; Nagata, T.; Minakata, S. Electrophilic Cyanation of Boron Enolates: Efficient Access to Various β-Ketonitrile Derivatives. Angew. Chem. Int. Ed. 2016, 55, 10458–10462. [Google Scholar] [CrossRef] [PubMed]
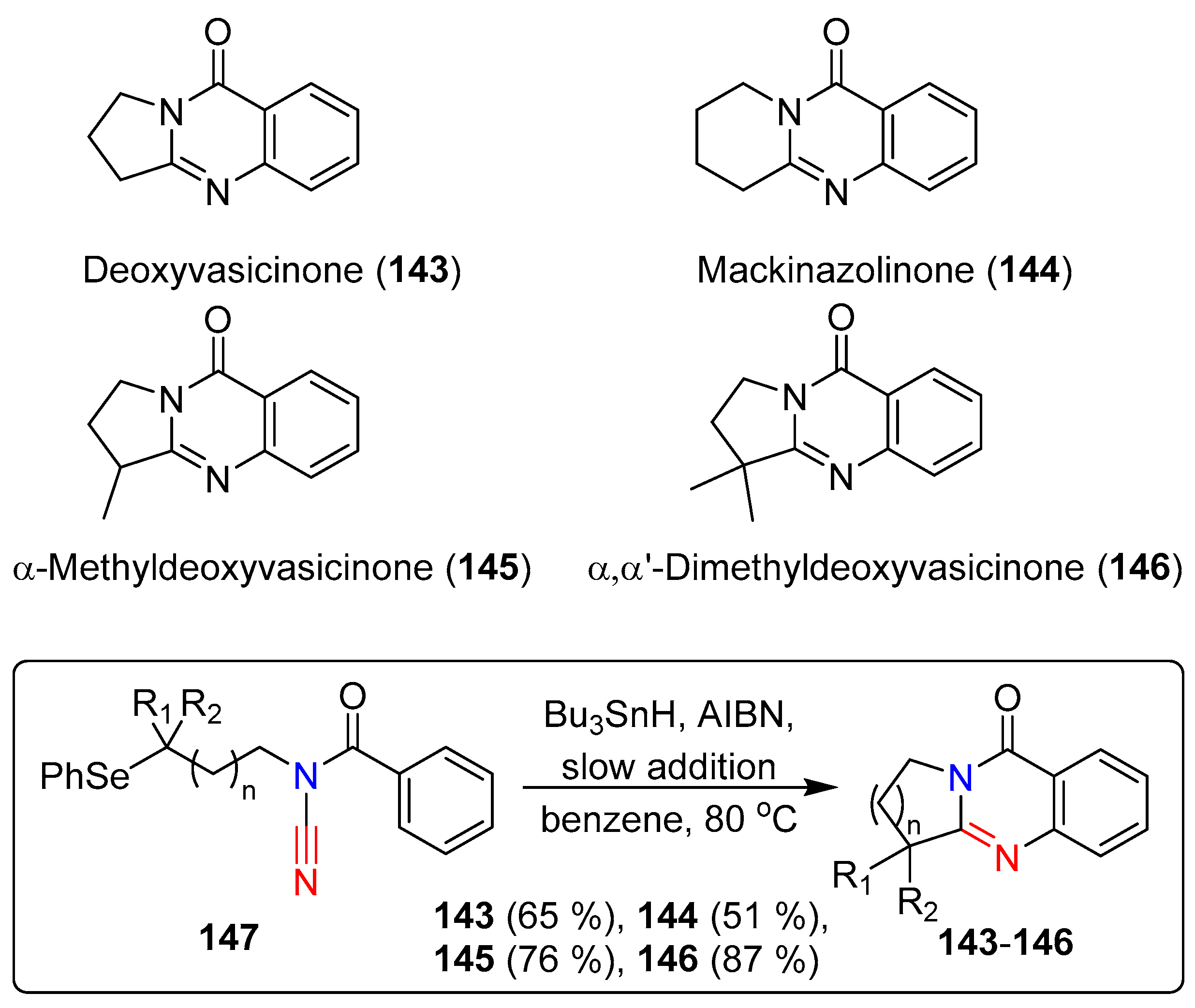
- Larraufie, M.-H.; Ollivier, C.; Fensterbank, L.; Malacria, M.; Lacôte, E. Radical Synthesis of Guanidines from N-Acyl Cyanamides. Angew. Chem. Int. Ed. 2010, 49, 2178–2181. [Google Scholar] [CrossRef] [PubMed]
- Larraufie, M.-H.; Courillon, C.; Ollivier, C.; Lacôte, E.; Malacria, M.; Fensterbank, L. Radical Migration of Substituents of Aryl Groups on Quinazolinones Derived from N-Acyl Cyanamides. J. Am. Chem. Soc. 2010, 132, 4381–4387. [Google Scholar] [CrossRef] [PubMed]
- Larraufie, M.-H.; Malacria, M.; Courillon, C.; Ollivier, C.; Fensterbank, L.; Lacôte, E. Synthesis of natural quinazolinones and some of their analogues through radical cascade reactions involving N-acylcyanamides. Tetrahedron 2013, 69, 7699–7705. [Google Scholar] [CrossRef]
- Zheng, J.; Zhang, Y.; Wang, D.; Cui, S. Silver(I)-Mediated Phosphorylation/Cyclization Cascade of N‑Cyanamide Alkenes for Divergent Access to Quinazolinones and Dihydroisoquinolinones. Org. Lett. 2016, 18, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Arfeen, M.; Bharatam, P.V.; Goswami, A. A Metal and Base-Free Chemoselective Primary Amination of Boronic Acids Using Cyanamidyl/Arylcyanamidyl Radical as Aminating Species: Synthesis and Mechanistic Studies by Density Functional Theory. J. Org. Chem. 2016, 81, 5120–5127. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, L.; Chiniforoshan, H. Sonochemical synthesis of Au nanowires in the III–I oxidation state bridged by 4,4′-dicyanamidobiphenyl and their application as selective CO gas sensors. Dalton Trans. 2015, 44, 2488–2495. [Google Scholar] [CrossRef] [PubMed]
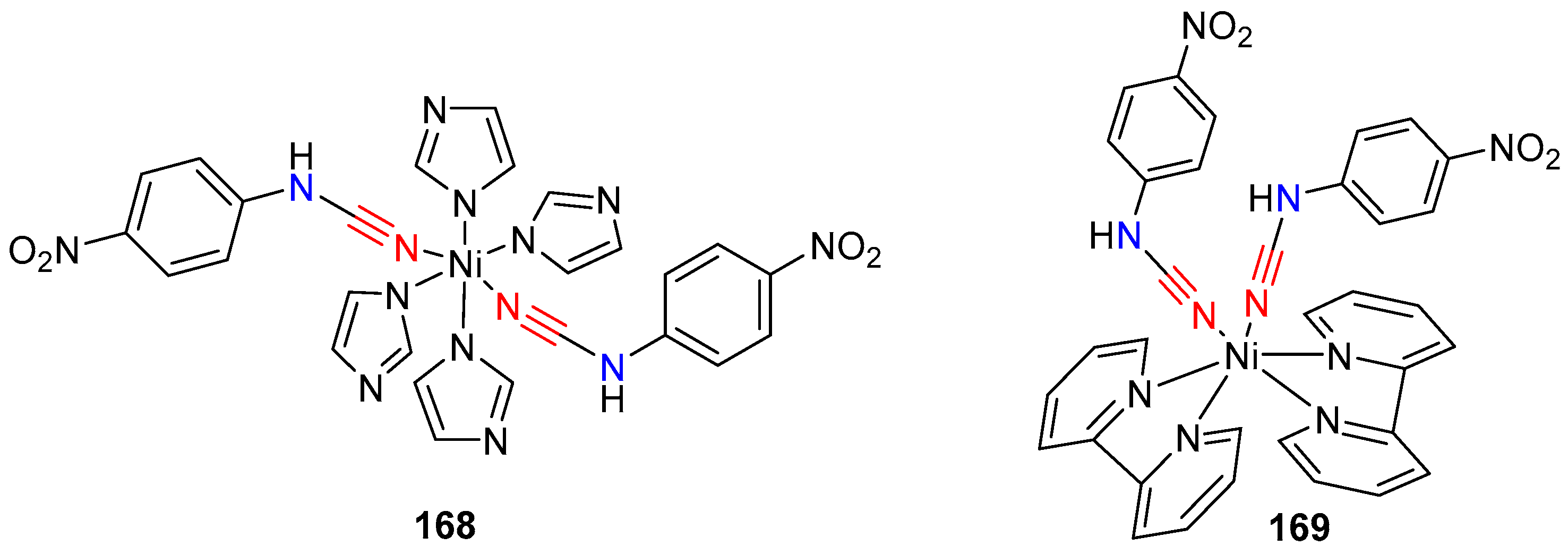
- Jazestani, M.; Chiniforoshan, H.; Tabrizi, L.; McArdle, P.; Notash, B. Synthesis, crystal structure of nickel(II) complexes of 4-nitro phenylcyanamide: Comparative in vitro evaluations of biological perspectives. Inorg. Chim. Acta 2016, 450, 402–410. [Google Scholar] [CrossRef]
- Crutchley, R.J. Phenylcyanamide Ligands. In Comprehensive Coordination Chemistry II; Meyer, T.J., McCleverty, J.A., Eds.; Pergamon: Oxford, UK, 2003; pp. 117–124. [Google Scholar]
- Bokach, N.A.; Kukushkin, V.Y. Coordination chemistry of dialkylcyanamides: Binding properties, synthesis of metal complexes, and ligand reactivity. Coord. Chem. Rev. 2013, 257, 2293–2316. [Google Scholar] [CrossRef]
- Crutchley, R.J. Phenylcyanamide ligands and their metal complexes. Coord. Chem. Rev. 2001, 219–221, 125–155. [Google Scholar] [CrossRef]
- Albertin, G.; Antoniutti, S.; Caia, A.; Castro, J. Reactivity with Amines of Bis(cyanamide) and Bis(cyanoguanidine) Complexes of the Iron Triad. Z. Anorg. Allg. Chem. 2015, 641, 814–819. [Google Scholar] [CrossRef]
- Albertin, G.; Antoniutti, S.; Caia, A.; Castro, J. Preparation and Reactivity Towards Hydrazines of bis(cyanamide) and bis(cyanoguanidine) Complexes of the Iron Triad. Dalton Trans. 2014, 43, 7314–7323. [Google Scholar] [CrossRef] [PubMed]
- Andrusenko, E.V.; Novikov, A.S.; Starova, G.L.; Bokach, N.A. Three-dimensional hydrogen bonding network in the structures of (dimethylcyanamide)cobalt(II) complexes. Inorg. Chim. Acta 2016, 447, 142–149. [Google Scholar] [CrossRef]
- Aquino, M.A.S.; Lee, F.L.; Gabe, E.J.; Bensimon, C.; Greedan, J.E.; Crutchley, R.J. Superexchange metal-metal coupling in dinuclear pentaammineruthenium complexes incorporating a 1,4-dicyanamidobenzene dianion bridging ligand. J. Am. Chem. Soc. 1992, 114, 5130–5140. [Google Scholar] [CrossRef]
- Harb, C.; Kravtsov, P.; Choudhuri, M.; Sirianni, E.R.; Yap, G.P.A.; Lever, A.B.P.; Crutchley, R.J. Phenylcyanamidoruthenium Scorpionate Complexes. Inorg. Chem. 2013, 52, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Chiniforoshan, H.; Jazestani, M.; Notash, B. Cis-Bis[(4-nitrophenyl)cyanamido-κN1]bis(1,10-phenanthroline-κ2N,N')nickel(II) methanol monosolvate. Acta Crystallogr. Sect. E 2012, 68, m417–m418. [Google Scholar] [CrossRef] [PubMed]
- Chiniforoshan, H.; Jazestani, M.; Notash, B. Catena-Poly[[bis(dimethylformamide-κO)cadmium]-bis([μ]-4-nitrophenylcyanamido-κ2N1:N3)]. Acta Crystallogr. Sect. E 2012, 68, m232. [Google Scholar] [CrossRef] [PubMed]
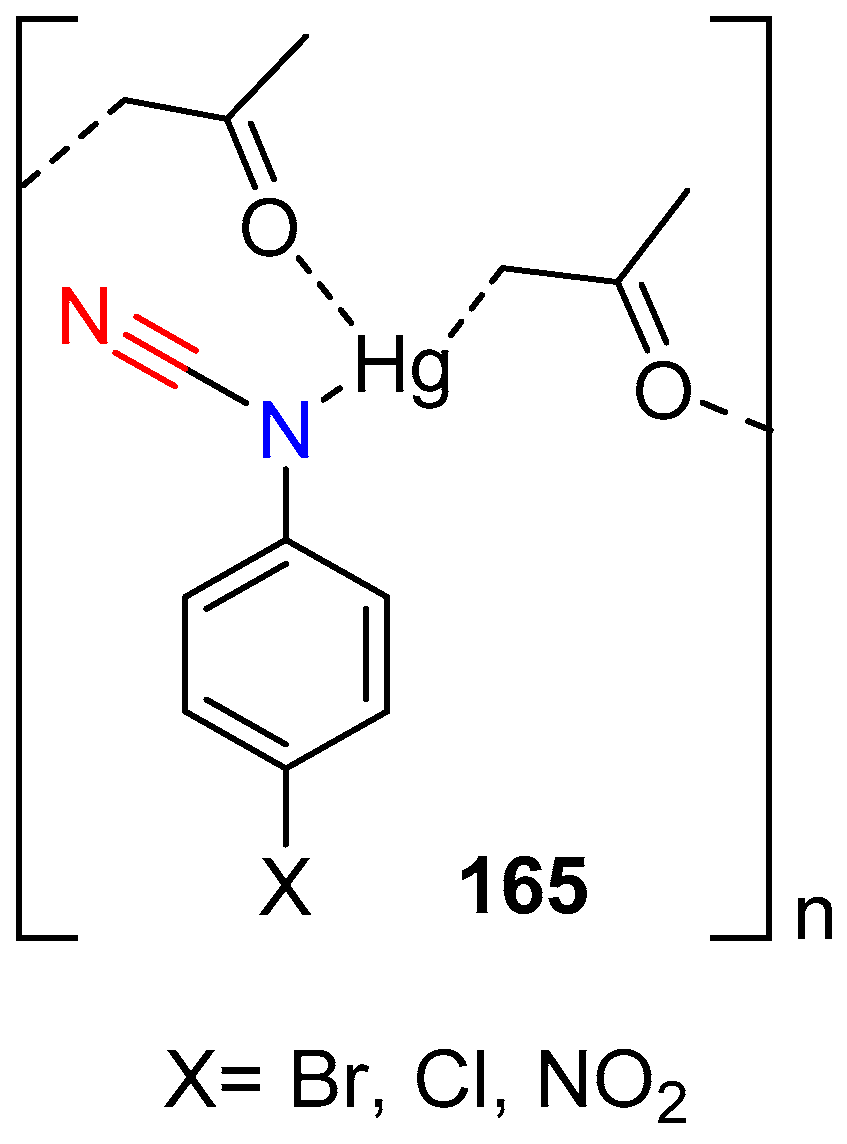
- Chiniforoshan, H.; Pourrahim, N.; Tabrizi, L.; Tavakol, H.; Sabzalian, M.R.; Notash, B. Syntheses, studies and crystal structure of new coordination polymers of mercury(II) with phenylcyanamide derivative ligands. Inorg. Chim. Acta 2014, 416, 85–92. [Google Scholar] [CrossRef]
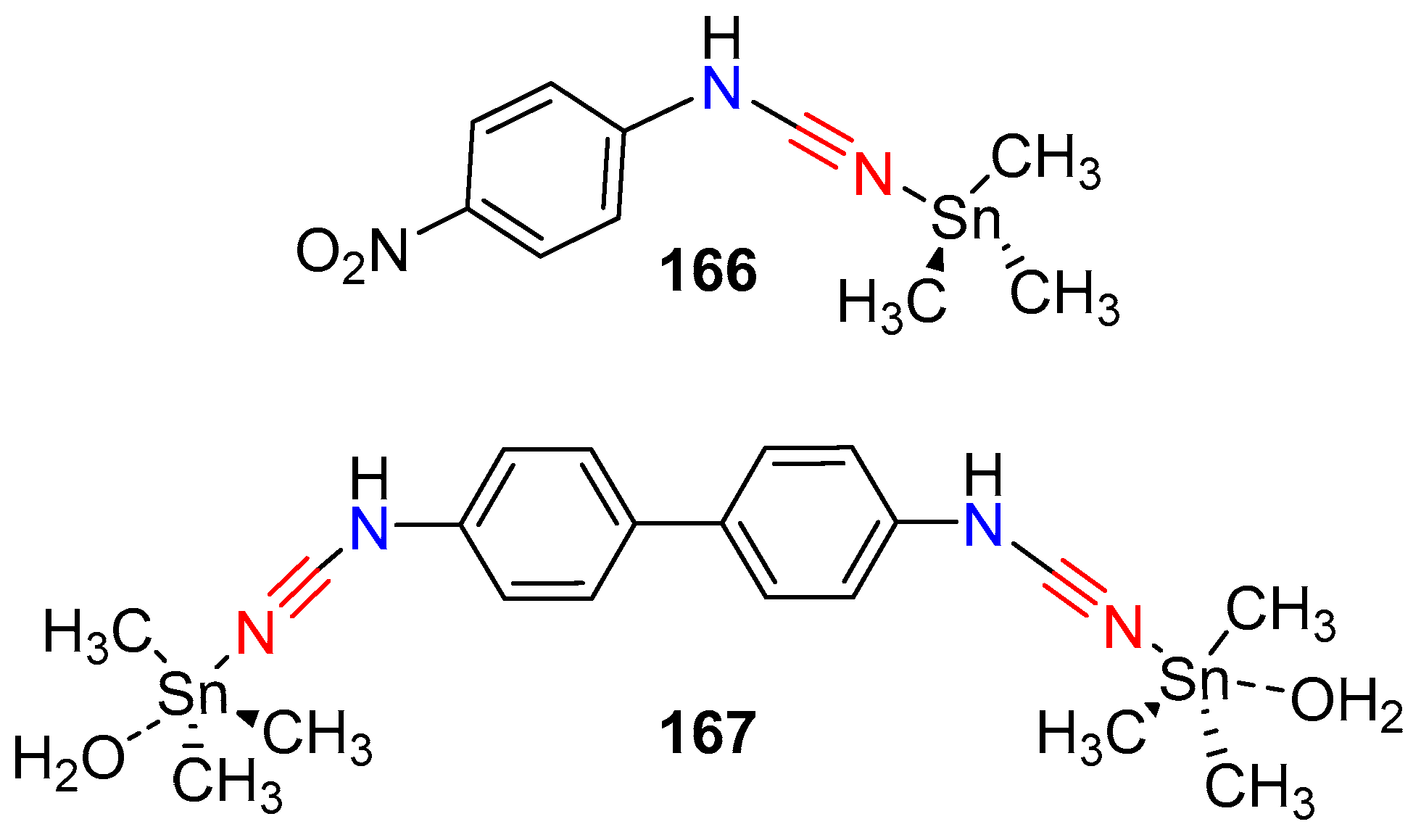
- Tabrizi, L.; McArdle, P.; Erxleben, A.; Chiniforoshan, H. Cytotoxicity and antimicrobial activity of triorganotin(IV) complexes of phenylcyanamide prepared by sonochemical synthesis. Inorg. Chim. Acta 2015, 438, 94–104. [Google Scholar] [CrossRef]


































© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prabhath, M.R.R.; Williams, L.; Bhat, S.V.; Sharma, P. Recent Advances in Cyanamide Chemistry: Synthesis and Applications. Molecules 2017, 22, 615. https://doi.org/10.3390/molecules22040615
Prabhath MRR, Williams L, Bhat SV, Sharma P. Recent Advances in Cyanamide Chemistry: Synthesis and Applications. Molecules. 2017; 22(4):615. https://doi.org/10.3390/molecules22040615
Chicago/Turabian StylePrabhath, M. R. Ranga, Luke Williams, Shreesha V. Bhat, and Pallavi Sharma. 2017. "Recent Advances in Cyanamide Chemistry: Synthesis and Applications" Molecules 22, no. 4: 615. https://doi.org/10.3390/molecules22040615
APA StylePrabhath, M. R. R., Williams, L., Bhat, S. V., & Sharma, P. (2017). Recent Advances in Cyanamide Chemistry: Synthesis and Applications. Molecules, 22(4), 615. https://doi.org/10.3390/molecules22040615