
Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Isatin-Thiazole Derivatives as α-Glucosidase Inhibitors

Abstract
:
1. Introduction
2. Results and Discussion
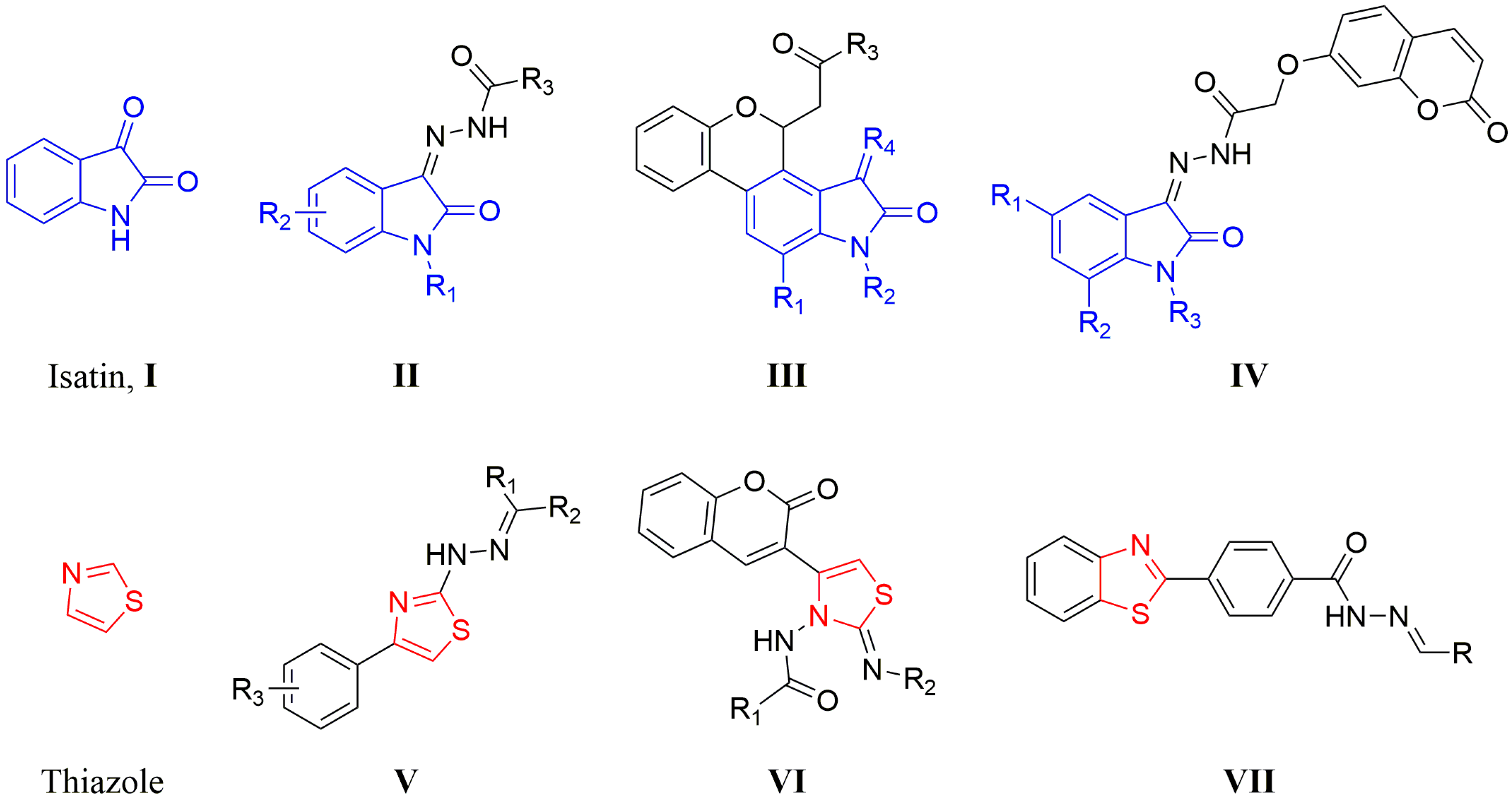
2.1. Chemistry
2.2. α-Glucosidase Inhibition Assay
2.3. Homology Model
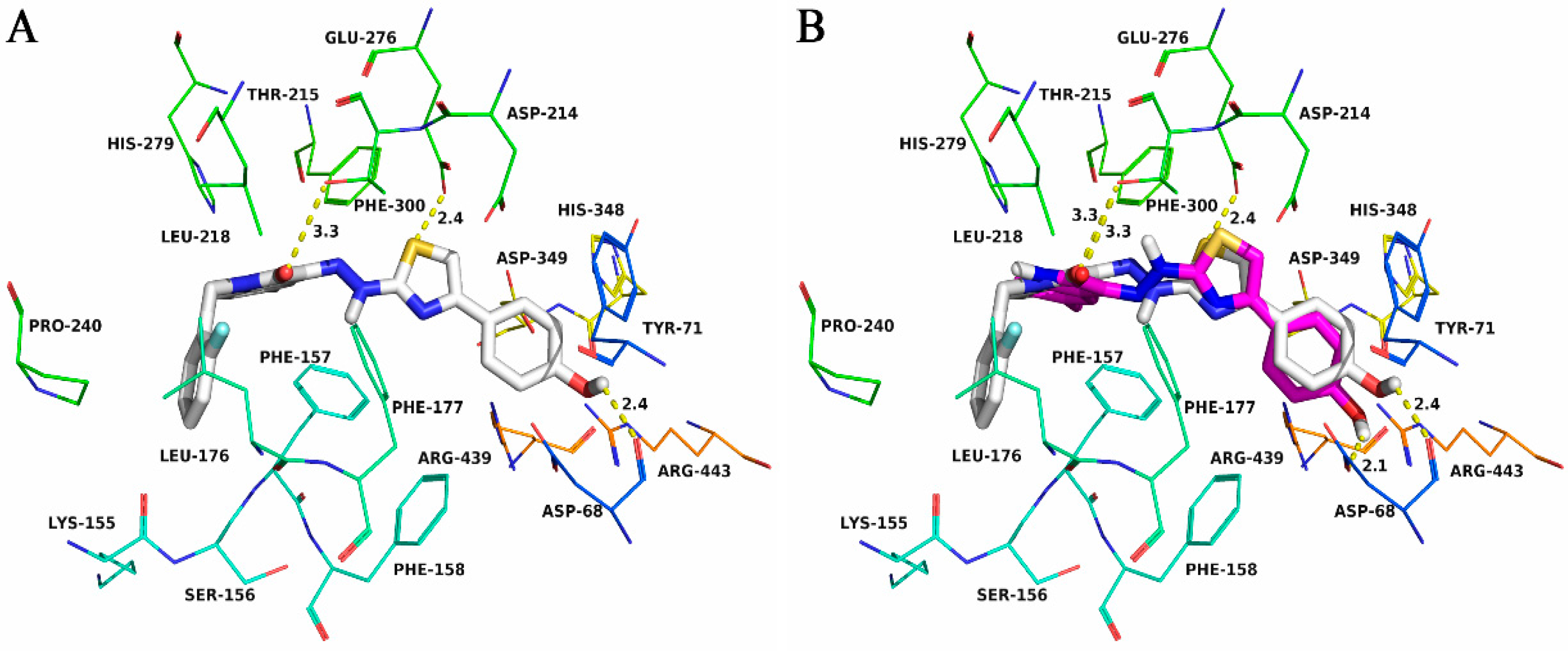
2.4. Molecular Docking
3. Experimental Section
3.1. General
3.2. General Procedures for the Synthesis of 6a–6p
3.3. In Vitro Assay of α-Glucosidase Inhibitory Activity
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Report on Diabetes; WHO: Geneva, Switzerland, 30 August 2016. [Google Scholar]
- Deshpande, A.D.; Harris-Hayes, M.; Schootman, M. Epidemiology of Diabetes and Diabetes-Related Complications. Phys. Ther. 2008, 88, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, F.A. α-glucosidase inhibitors in the early treatment of type 2 diabetes. Vasc. Health Risk Manag. 2008, 4, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.R.; Standl, E.; Tong, N.; Shah, P.; Kalra, S.; Rathod, R. Therapeutic potential of α-glucosidase inhibitors in type 2 diabetes mellitus: An evidence-based review. Expert Opin. Pharmacother. 2015, 16, 1959–1981. [Google Scholar] [CrossRef] [PubMed]
- Pili, R.; Chang, J.; Partis, R.A.; Mueller, R.A.; Chrest, F.J.; Passaniti, A. The α-glucosidase I inhibitor castanospermine alters endothelial cell glycosylation, prevents angiogenesis, and inhibits tumor growth. Cancer Res. 1995, 55, 2920–2926. [Google Scholar] [PubMed]
- Mehta, A.; Zitzmann, N.; Rudd, P.M.; Block, T.M.; Dwek, R.A. α-glucosidase inhibitors as potential broad based anti-viral agents. FEBS Lett. 1998, 430, 17–22. [Google Scholar] [CrossRef]
- Zitzmann, N.; Mehta, A.S.; Carrouée, S.; Butters, T.D.; Platt, F.M.; McCauley, J.; Blumberg, B.S.; Dwek, R.A.; Block, T.M. Imino sugars inhibit the formation and secretion of bovine viral diarrhea virus, a pestivirus model of hepatitis C virus: Implications for the development of broad spectrum anti-hepatitis virus agents. Proc. Natl. Acad. Sci. USA 1999, 96, 11878–11882. [Google Scholar] [CrossRef] [PubMed]
- Hamaue, N.; Minami, M.; Hirafuji, M.; Terado, M.; Machida, M.; Yamazaki, N.; Yoshioka, M.; Ogata, A.; Tashiro, K. Isatin, an endogenous MAO inhibitor, as a new biological modulator. CNS Drug Rev. 1999, 5, 331–346. [Google Scholar] [CrossRef]
- Khan, F.A.; Maalik, A.; Noor, T.; Zaidi, A.; Farooq, U.; Bukhari, S.M. Advances in Pharmacology of Isatin and its Derivatives: A Review. Trop. J. Pharm. Res. 2015, 14, 1937–1942. [Google Scholar] [CrossRef]
- Nguyen-Hai, N.; Tran Lan, H.; Do Thi Mai, D.; Phan Thi Phuong, D.; Dao Thi Kim, O.; Do, Q.; Le Thi, T.; Park, S.H.; Kim, K.R.; Han, B.W.; et al. Novel isatin-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Eur. J. Med. Chem. 2013, 70, 477–486. [Google Scholar]
- Zhang, X.-M.; Guo, H.; Li, Z.-S.; Song, F.-H.; Wang, W.-M.; Dai, H.-Q.; Zhang, L.-X.; Wang, J.-G. Synthesis and evaluation of isatin-beta-thiosemicarbazones as novel agents against antibiotic-resistant Gram-positive bacterial species. Eur. J. Med. Chem. 2015, 101, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Bal, T.R.; Anand, B.; Yogeeswari, P.; Sriram, D. Synthesis and evaluation of anti-HIV activity of isatin beta-thiosemicarbazone derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 4451–4455. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Tang, L.-M.; Li, F.-N.; Guan, L.-P.; Pan, C.-Y.; Wang, S.-H. Structure-based design, synthesis, and anticonvulsant activity of isatin-1-N-phenylacetamide derivatives. Med. Chem. Res. 2014, 23, 2161–2168. [Google Scholar] [CrossRef]
- Sharma, P.K.; Balwani, S.; Mathur, D.; Malhotra, S.; Singh, B.K.; Prasad, A.K.; Len, C.; Van der Eycken, E.V.; Ghosh, B.; Richards, N.G.J.; et al. Synthesis and anti-inflammatory activity evaluation of novel triazolyl-isatin hybrids. J. Enzym. Inhib. Med. Chem. 2016, 31, 1520–1526. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.A.; Raghunathan, R.; SrideviKumari, M.R.; Raman, N. Synthesis, antimicrobial and antifungal activity of a new class of spiro pyrrolidines. Bioorg. Med. Chem. 2003, 11, 407–419. [Google Scholar] [CrossRef]
- Rahim, F.; Malik, F.; Ullah, H.; Wadood, A.; Khan, F.; Javid, M.T.; Taha, M.; Rehman, W.; Rehman, A.U.; Khan, K.M. Isatin based Schiff bases as inhibitors of α-glucosidase: Synthesis, characterization, in vitro evaluation and molecular docking studies. Bioorg. Chem. 2015, 60, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, Y.; Ding, W.; Zhao, X.; Song, X.; Wang, D.; Li, Y.; Han, K.; Yang, Y.; Ma, Y.; et al. Inhibitory activity evaluation and mechanistic studies of tetracyclic oxindole derivatives as α-glucosidase inhibitors. Eur. J. Med. Chem. 2016, 123, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.; He, D.; Li, X.; Li, J.; Peng, Z. Synthesis, in vitro evaluation and molecular docking studies of novel coumarin-isatin derivatives as α-glucosidase inhibitors. Chem. Biol. Drug Des. 2016, 89, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Kumar, S.; Kaushik, P.; Kaushik, D.; Gupta, G.K. Synthesis and pharmacological evaluation of some novel 2-(5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazol-1-yl)-4-(coumarin-3-yl )thiazoles. Eur. J. Med. Chem. 2013, 62, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Azizmohammadi, M.; Khoobi, M.; Ramazani, A.; Emami, S.; Zarrin, A.; Firuzi, O.; Miri, R.; Shafiee, A. 2H-chromene derivatives bearing thiazolidine-2,4-dione, rhodanine or hydantoin moieties as potential anticancer agents. Eur. J. Med. Chem. 2013, 59, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.; Ahsan, W. Triazole incorporated thiazoles as a new class of anticonvulsants: Design, synthesis and in vivo screening. Eur. J. Med. Chem. 2010, 45, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.-J.; Wang, P.-F.; Makawana, J.A.; Wang, Z.-C.; Wang, Z.-N.; Yan, G.; Jiang, A.-Q.; Zhu, H.-L. Design, synthesis and biological evaluation of metronidazole-thiazole derivatives as antibacterial inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 5279–5283. [Google Scholar] [CrossRef] [PubMed]
- Koppireddi, S.; Komsani, J.R.; Tiwari, A.K.; Ali, A.Z.; Yadla, R. Synthesis and biological evaluation of new derivatives of tertiary thiazolamines. Pharm. Chem. 2014, 6, 136–143. [Google Scholar]
- Rahim, F.; Ullah, H.; Javid, M.T.; Wadood, A.; Taha, M.; Ashraf, M.; Shaukat, A.; Junaid, M.; Hussain, S.; Rehman, W.; et al. Synthesis, in vitro evaluation and molecular docking studies of thiazole derivatives as new inhibitors of α-glucosidase. Bioorg. Chem. 2015, 62, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Ismail, N.H.; Lalani, S.; Fatmi, M.Q.; Atia-tul-Wahab; Siddiqui, S.; Khan, K.M.; Imran, S.; Choudhary, M.I. Synthesis of novel inhibitors of α-glucosidase based on the benzothiazole skeleton containing benzohydrazide moiety and their molecular docking studies. Eur. J. Med. Chem. 2015, 92, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Qurban, S.; Salar, U.; Taha, M.; Hussain, S.; Perveen, S.; Hameed, A.; Ismail, N.H.; Riaz, M.; Wadood, A. Synthesis, in vitro α-glucosidase inhibitory activity and molecular docking studies of new thiazole derivatives. Bioorg. Chem. 2016, 68, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Salar, U.; Taha, M.; Khan, K.M.; Ismail, N.H.; Imran, S.; Perveen, S.; Gul, S.; Wadood, A. Syntheses of new 3-thiazolyl coumarin derivatives, in vitro α-glucosidase inhibitory activity, and molecular modeling studies. Eur. J. Med. Chem. 2016, 122, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Viegas-Junior, C.; Danuello, A.; da Silva Bolzani, V.; Barreir, E.J.; Manssour Fraga, C.A. Molecular hybridization: A useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Shaveta; Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar]
- Wang, G.; Wang, J.; He, D.; Li, X.; Li, J.; Peng, Z. Synthesis and biological evaluation of novel 1,2,4-triazine derivatives bearing carbazole moiety as potent α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2806–2809. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, J.; He, D.; Li, X.; Li, J.; Peng, Z. One-pot and three-component synthesis, characterization and biological evaluation of some new 1,2,4-triazine-coumarins. Heterocycles 2016, 92, 1430–1439. [Google Scholar] [CrossRef]
- Wang, G.; Peng, Z.; Wang, J.; Li, J.; Li, X. Synthesis and biological evaluation of novel 2,4,5-triarylimidazole–1,2,3-triazole derivatives via click chemistry as α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 5719–5723. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Peng, Z.; Wang, J.; Li, J.; Li, X. Synthesis, biological evaluation and molecular docking study of N-arylbenzo[d]oxazol-2-amines as potential α-glucosidase inhibitors. Bioorg. Med. Chem. 2016, 24, 5374–5379. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Peng, Z.; Wang, J.; Li, X.; Li, J. Synthesis, in vitro evaluation and molecular docking studies of novel triazine-triazole derivatives as potential α-glucosidase inhibitors. Eur. J. Med. Chem. 2017, 125, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Niaz, H.; Kashtoh, H.; Khan, J.A.J.; Khan, A.; Atiatul, W.; Alam, M.T.; Khan, K.M.; Perveen, S.; Choudhary, M.I. Synthesis of diethyl 4-substituted-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylates as a new series of inhibitors against yeast α-glucosidase. Eur. J. Med. Chem. 2015, 95, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Miyake, H.; Kusunoki, M.; Osaki, S. Crystal structures of isomaltase from Saccharomyces cerevisiae and in complex with its competitive inhibitor maltose. FEBS J. 2010, 277, 4205–4214. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 6a–6p are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R1 | R2 | R3 | IC50 (μm) a |
|---|---|---|---|---|
| 6a | 5-Cl | H | Br | 6.87 ± 0.14 |
| 6b | 5-Me | 2-F-benzyl | Me | 35.76 ± 0.31 |
| 6c | 5-Me | 2-F-benzyl | MeO | 10.34 ± 0.17 |
| 6d | H | Me | Me | 32.17 ± 0.29 |
| 6e | H | Me | F | 33.20 ± 0.24 |
| 6f | H | Me | MeO | 24.17 ± 0.28 |
| 6g | H | H | Cl | 5.98 ± 0.12 |
| 6h | 5.7-Me2 | H | Br | 6.12 ± 0.15 |
| 6i | 5-Me | H | OH | 7.51 ± 0.17 |
| 6j | 5.7-Me2 | H | F | 6.51 ± 0.13 |
| 6k | 5.7-Me2 | H | Me | 15.68 ± 0.24 |
| 6l | 5-F | H | Me | 8.33 ± 0.18 |
| 6m | 5-F | H | F | 7.17 ± 0.15 |
| 6n | H | H | F | 6.36 ± 0.12 |
| 6o | H | H | Me | 11.78 ± 0.21 |
| 6p | 5-Me | 2-F-benzyl | OH | 5.36 ± 0.13 |
| Acarbose | 817.38 ± 6.27 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Z.; Wang, G.; Wang, J.; Chen, M.; Peng, Y.; Li, L.; Deng, B.; Chen, S.; Li, W. Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Isatin-Thiazole Derivatives as α-Glucosidase Inhibitors. Molecules 2017, 22, 659. https://doi.org/10.3390/molecules22040659
Xie Z, Wang G, Wang J, Chen M, Peng Y, Li L, Deng B, Chen S, Li W. Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Isatin-Thiazole Derivatives as α-Glucosidase Inhibitors. Molecules. 2017; 22(4):659. https://doi.org/10.3390/molecules22040659
Chicago/Turabian StyleXie, Zhenzhen, Guangcheng Wang, Jing Wang, Ming Chen, Yaping Peng, Luyao Li, Bing Deng, Shan Chen, and Wenbiao Li. 2017. "Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Isatin-Thiazole Derivatives as α-Glucosidase Inhibitors" Molecules 22, no. 4: 659. https://doi.org/10.3390/molecules22040659
APA StyleXie, Z., Wang, G., Wang, J., Chen, M., Peng, Y., Li, L., Deng, B., Chen, S., & Li, W. (2017). Synthesis, Biological Evaluation, and Molecular Docking Studies of Novel Isatin-Thiazole Derivatives as α-Glucosidase Inhibitors. Molecules, 22(4), 659. https://doi.org/10.3390/molecules22040659