
Heparin and Heparin-Derivatives in Post-Subarachnoid Hemorrhage Brain Injury: A Multimodal Therapy for a Multimodal Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Early Brain Injury, Inflammation, and Blood-Brain Barrier Failure in Aneurysmal Subarachnoid Hemorrhage
3. Heparin—Physiologic and Pharmacologic Roles
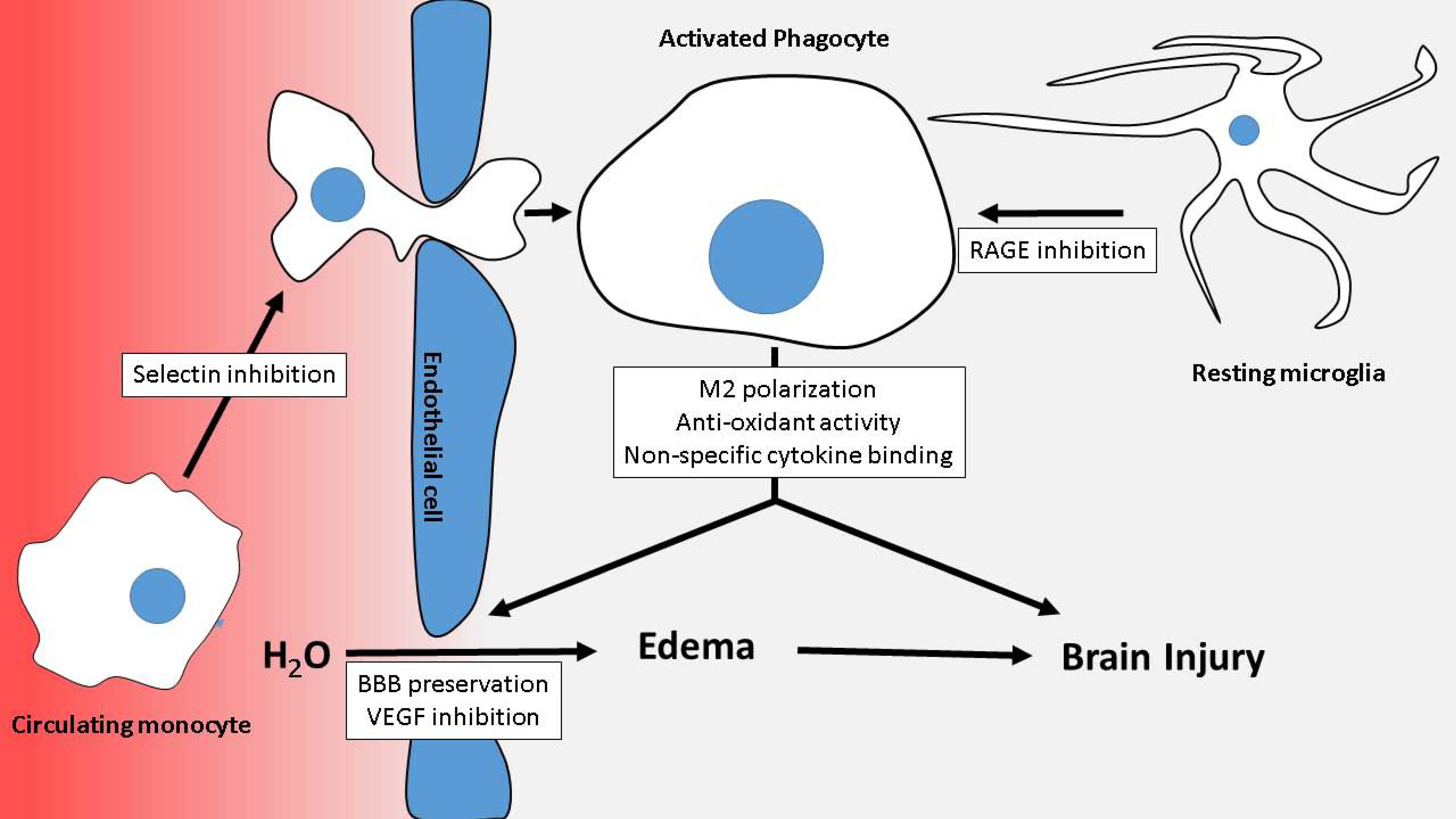
4. Heparin in Post-SAH Brain Injury
4.1. Ischemia-Reperfusion Injury
4.2. Leukocyte Extravasation
4.3. Inflammatory Activation
4.4. Oxidative Stress
4.5. Blood-Brain Barrier Dysfunction and Vasogenic Edema
5. Complications of Heparin Therapy in aSAH
5.1. Heparin Induced Thrombocytopenia (HIT)
5.2. Hemorrhagic Complications
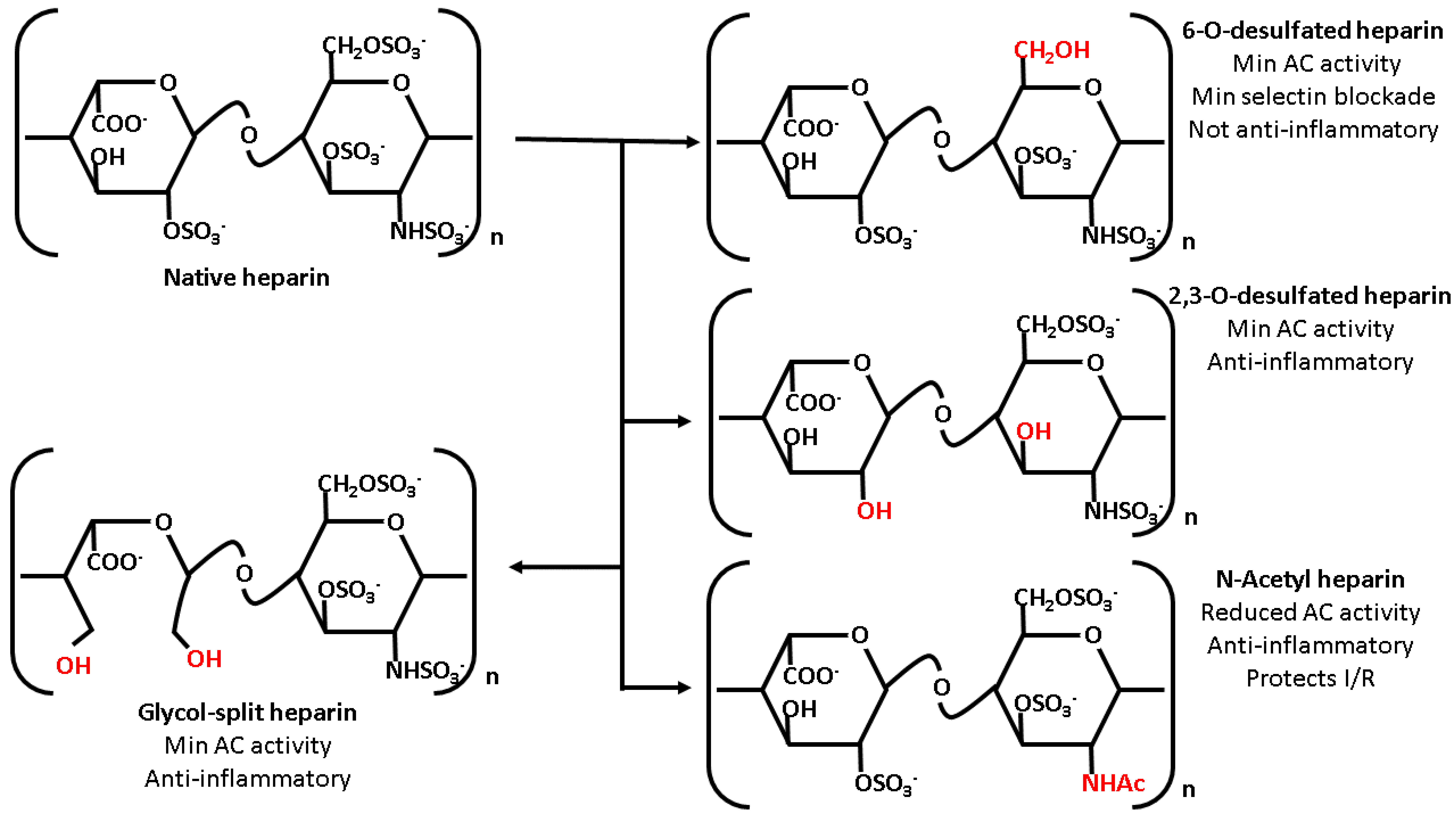
6. Heparin Derivatives
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ogden, J.A.; Utley, T.; Mee, E.W. Neurological and psychosocial outcome 4 to 7 years after subarachnoid hemorrhage. Neurosurgery 1997, 41, 25–34. [Google Scholar] [PubMed]
- Hackett, M.L.; Anderson, C.S. Health outcomes 1 year after subarachnoid hemorrhage: An international population-based study. The australian cooperative research on subarachnoid hemorrhage study group. Neurology 2000, 55, 658–662. [Google Scholar] [PubMed]
- Kreiter, K.T.; Copeland, D.; Bernardini, G.L.; Bates, J.E.; Peery, S.; Claassen, J.; Du, Y.E.; Stern, Y.; Connolly, E.S.; Mayer, S.A. Predictors of cognitive dysfunction after subarachnoid hemorrhage. Stroke 2002, 33, 200–208. [Google Scholar] [PubMed]
- Al-Khindi, T.; Macdonald, R.L.; Schweizer, T.A. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 2010, 41, e519–e536. [Google Scholar] [PubMed]
- Haley, E.C., Jr. Measuring cognitive outcome after subarachnoid hemorrhage. Ann. Neurol. 2006, 60, 502–504. [Google Scholar] [PubMed]
- Kivisaari, R.P.; Salonen, O.; Servo, A.; Autti, T.; Hernesniemi, J.; Ohman, J. Mr imaging after aneurysmal subarachnoid hemorrhage and surgery: A long-term follow-up study. AJNR Am. J. Neuroradiol. 2001, 22, 1143–1148. [Google Scholar] [PubMed]
- Tam, A.K.; Kapadia, A.; Ilodigwe, D.; Li, Z.; Schweizer, T.A.; Macdonald, R.L. Impact of global cerebral atrophy on clinical outcome after subarachnoid hemorrhage. J. Neurosurg. 2013, 119, 198–206. [Google Scholar] [PubMed]
- Macdonald, R.L.; Higashida, R.T.; Keller, E.; Mayer, S.A.; Molyneux, A.; Raabe, A.; Vajkoczy, P.; Wanke, I.; Bach, D.; Frey, A.; et al. Randomised trial of clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid hemorrhage undergoing surgical clipping (conscious-2). Acta Neurochir. Suppl. 2013, 115, 27–31. [Google Scholar] [PubMed]
- Cossu, G.; Messerer, M.; Oddo, M.; Daniel, R.T. To look beyond vasospasm in aneurysmal subarachnoid haemorrhage. Biomed. Res. Int. 2014, 2014, 628597. [Google Scholar] [PubMed]
- Rowland, M.J.; Hadjipavlou, G.; Kelly, M.; Westbrook, J.; Pattinson, K.T. Delayed cerebral ischaemia after subarachnoid haemorrhage: Looking beyond vasospasm. Br. J. Anaesth. 2012, 109, 315–329. [Google Scholar] [PubMed]
- Lucke-Wold, B.P.; Logsdon, A.F.; Manoranjan, B.; Turner, R.C.; McConnell, E.; Vates, G.E.; Huber, J.D.; Rosen, C.L.; Simard, J.M. Aneurysmal subarachnoid hemorrhage and neuroinflammation: A comprehensive review. Int. J. Mol. Sci. 2016, 17, 497. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yin, Q.; Zhong, Q.; Lv, F.L.; Zhou, Y.; Li, J.Q.; Wang, J.Z.; Su, B.Y.; Yang, Q.W. Heme activates tlr4-mediated inflammatory injury via myd88/trif signaling pathway in intracerebral hemorrhage. J. Neuroinflamm. 2012, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.S.; Woo, S.K.; Kurland, D.B.; Yoon, S.H.; Palmer, A.F.; Banerjee, U.; Iqbal, S.; Ivanova, S.; Gerzanich, V.; Simard, J.M. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int. J. Mol. Sci. 2015, 16, 5028–5046. [Google Scholar] [CrossRef] [PubMed]
- Gallia, G.L.; Tamargo, R.J. Leukocyte-endothelial cell interactions in chronic vasospasm after subarachnoid hemorrhage. Neurol. Res. 2006, 28, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Ayer, R.E.; Zhang, J.H. Oxidative stress in subarachnoid haemorrhage: Significance in acute brain injury and vasospasm. Acta Neurochir. Suppl. 2008, 104, 33–41. [Google Scholar] [PubMed]
- Chou, S.H.; Feske, S.K.; Atherton, J.; Konigsberg, R.G.; De Jager, P.L.; Du, R.; Ogilvy, C.S.; Lo, E.H.; Ning, M. Early elevation of serum tumor necrosis factor-alpha is associated with poor outcome in subarachnoid hemorrhage. J. Investig. Med. 2012, 60, 1054–1058. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, K.A.; Morgan Stuart, R.; Fernandez, L.; Schmidt, J.M.; Claassen, J.; Lee, K.; Sander Connolly, E.; Mayer, S.A.; Badjatia, N. Cerebral inflammatory response and predictors of admission clinical grade after aneurysmal subarachnoid hemorrhage. J. Clin. Neurosci. 2010, 17, 22–25. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.J.; Hopkins, S.; Vail, A.; King, A.T.; Smith, D.; Illingworth, K.J.; Clark, S.; Rothwell, N.J.; Tyrrell, P.J. Inflammation as a predictor for delayed cerebral ischemia after aneurysmal subarachnoid haemorrhage. J. Neurointerv. Surg. 2013, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.K.; Ilodigwe, D.; Mocco, J.; Mayer, S.; Kassell, N.; Ruefenacht, D.; Schmiedek, P.; Weidauer, S.; Pasqualin, A.; Macdonald, R.L. Impact of systemic inflammatory response syndrome on vasospasm, cerebral infarction, and outcome after subarachnoid hemorrhage: Exploratory analysis of conscious-1 database. Neurocrit. Care 2010, 13, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Jedrzejowska-Szypulka, H.; Larysz-Brysz, M.; Kukla, M.; Snietura, M.; Lewin-Kowalik, J. Neutralization of interleukin-1beta reduces vasospasm and alters cerebral blood vessel density following experimental subarachnoid hemorrhage in rats. Curr. Neurovasc. Res. 2009, 6, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Aldrich, E.F.; Schreibman, D.; James, R.F.; Polifka, A.; Beaty, N. Low-dose intravenous heparin infusion in patients with aneurysmal subarachnoid hemorrhage: A preliminary assessment. J. Neurosurg. 2013, 119, 1611–1619. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Tosun, C.; Ivanova, S.; Kurland, D.B.; Hong, C.; Radecki, L.; Gisriel, C.; Mehta, R.; Schreibman, D.; Gerzanich, V. Heparin reduces neuroinflammation and transsynaptic neuronal apoptosis in a model of subarachnoid hemorrhage. Transl. Stroke Res. 2012, 3, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Altay, O.; Suzuki, H.; Hasegawa, Y.; Sorar, M.; Chen, H.; Tang, J.; Zhang, J.H. Effects of low-dose unfractionated heparin pretreatment on early brain injury after subarachnoid hemorrhage in mice. Acta Neurochir. Suppl. 2016, 121, 127–130. [Google Scholar] [PubMed]
- Bruder, M.; Won, S.Y.; Kashefiolasl, S.; Wagner, M.; Brawanski, N.; Dinc, N.; Seifert, V.; Konczalla, J. Effect of heparin on secondary brain injury in patients with subarachnoid hemorrhage: An additional “h“ therapy in vasospasm treatment. J. Neurointerv. Surg. 2017. neurintsurg–2016–012925. [Google Scholar] [CrossRef] [PubMed]
- Grote, E.; Hassler, W. The critical first minutes after subarachnoid hemorrhage. Neurosurgery 1988, 22, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, K.A. The role of microglia and the tlr4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Jackowski, A.; Crockard, A.; Burnstock, G.; Russell, R.R.; Kristek, F. The time course of intracranial pathophysiological changes following experimental subarachnoid haemorrhage in the rat. J. Cereb. Blood Flow Metab. 1990, 10, 835–849. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.; Grille, S.; Morelli, P.; Mila, R.; Trias, N.; Brugnini, A.; Luberas, L.N.; Biestro, A.; Lens, D. Immune cells subpopulations in cerebrospinal fluid and peripheral blood of patients with aneurysmal subarachnoid hemorrhage. Springerplus 2015, 4, 195. [Google Scholar] [CrossRef] [PubMed]
- Provencio, J.J.; Swank, V.; Lu, H.; Brunet, S.; Baltan, S.; Khapre, R.V.; Seerapu, H.; Kokiko-Cochran, O.N.; Lamb, B.T.; Ransohoff, R.M. Neutrophil depletion after subarachnoid hemorrhage improves memory via nmda receptors. Brain Behav. Immun. 2016, 54, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Provencio, J.J.; Fu, X.; Siu, A.; Rasmussen, P.A.; Hazen, S.L.; Ransohoff, R.M. Csf neutrophils are implicated in the development of vasospasm in subarachnoid hemorrhage. Neurocrit. Care 2010, 12, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Testai, F.D.; Valyi-Nagy, T.; M, N.P.; Zhai, F.; Nanegrungsunk, D.; Paisansathan, C.; Pelligrino, D.A. vap-1 blockade prevents subarachnoid hemorrhage-associated cerebrovascular dilating dysfunction via repression of a neutrophil recruitment-related mechanism. Brain Res. 2015, 1603, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Satoh, S.; Yamamoto, Y.; Toshima, Y.; Ikegaki, I.I.; Asano, T.; Suzuki, Y.; Shibuya, M. Fasudil, a protein kinase inhibitor, prevents the development of endothelial injury and neutrophil infiltration in a two-haemorrhage canine subarachnoid model. J. Clin. Neurosci. 1999, 6, 394–399. [Google Scholar] [CrossRef]
- Pradilla, G.; Wang, P.P.; Legnani, F.G.; Ogata, L.; Dietsch, G.N.; Tamargo, R.J. Prevention of vasospasm by anti-cd11/cd18 monoclonal antibody therapy following subarachnoid hemorrhage in rabbits. J. Neurosurg. 2004, 101, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Clatterbuck, R.E.; Gailloud, P.; Ogata, L.; Gebremariam, A.; Dietsch, G.N.; Murphy, K.J.; Tamargo, R.J. Prevention of cerebral vasospasm by a humanized anti-cd11/cd18 monoclonal antibody administered after experimental subarachnoid hemorrhage in nonhuman primates. J. Neurosurg. 2003, 99, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Hayman, E.G.; Hong, C.; Stokum, J.A.; Kurland, D.B.; Gerzanich, V.; Simard, J.M. Inducible nitric oxide synthase (nos-2) in subarachnoid hemorrhage: Regulatory mechanisms and therapeutic implications. Brain Circ. 2016, 2, 8–19. [Google Scholar] [PubMed]
- Kooijman, E.; Nijboer, C.H.; van Velthoven, C.T.; Mol, W.; Dijkhuizen, R.M.; Kesecioglu, J.; Heijnen, C.J. Long-term functional consequences and ongoing cerebral inflammation after subarachnoid hemorrhage in the rat. PLoS ONE 2014, 9, e90584. [Google Scholar] [CrossRef] [PubMed]
- Schallner, N.; Pandit, R.; LeBlanc, R.; Thomas, A.J.; Ogilvy, C.S.; Zuckerbraun, B.S.; Gallo, D.; Otterbein, L.E.; Hanafy, K.A. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Investig. 2015, 125, 2609–2625. [Google Scholar] [CrossRef] [PubMed]
- Matz, P.; Turner, C.; Weinstein, P.R.; Massa, S.M.; Panter, S.S.; Sharp, F.R. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res. 1996, 713, 211–222. [Google Scholar] [CrossRef]
- Bernardino, L.; Agasse, F.; Silva, B.; Ferreira, R.; Grade, S.; Malva, J.O. Tumor necrosis factor-alpha modulates survival, proliferation, and neuronal differentiation in neonatal subventricular zone cell cultures. Stem Cells 2008, 26, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Shigemoto-Mogami, Y.; Hoshikawa, K.; Goldman, J.E.; Sekino, Y.; Sato, K. Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J. Neurosci. 2014, 34, 2231–2243. [Google Scholar] [CrossRef] [PubMed]
- Sgubin, D.; Aztiria, E.; Perin, A.; Longatti, P.; Leanza, G. Activation of endogenous neural stem cells in the adult human brain following subarachnoid hemorrhage. J. Neurosci. Res. 2007, 85, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Hayman, E.G.; Wessell, A.; Gerzanich, V.; Sheth, K.N.; Simard, J.M. Mechanisms of global cerebral edema formation in aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2017, 26, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Geng, Z.; Woo, S.K.; Ivanova, S.; Tosun, C.; Melnichenko, L.; Gerzanich, V. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 2009, 29, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Germano, A.; Avella, D.; Imperatore, C.; Caruso, G.; Tomasello, F. Time-course of blood-brain barrier permeability changes after experimental subarachnoid haemorrhage. Acta Neurochir. (Wien) 2000, 142, 575–580; discussion 580–581. [Google Scholar] [CrossRef] [PubMed]
- Claassen, J.; Carhuapoma, J.R.; Kreiter, K.T.; Du, E.Y.; Connolly, E.S.; Mayer, S.A. Global cerebral edema after subarachnoid hemorrhage: Frequency, predictors, and impact on outcome. Stroke 2002, 33, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Ivanidze, J.; Kesavabhotla, K.; Kallas, O.N.; Mir, D.; Baradaran, H.; Gupta, A.; Segal, A.Z.; Claassen, J.; Sanelli, P.C. Evaluating blood-brain barrier permeability in delayed cerebral infarction after aneurysmal subarachnoid hemorrhage. AJNR Am. J. Neuroradiol. 2015, 36, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Egashira, Y.; Zhao, H.; Hua, Y.; Keep, R.F.; Xi, G. White matter injury after subarachnoid hemorrhage: Role of blood-brain barrier disruption and matrix metalloproteinase-9. Stroke 2015, 46, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Carvey, P.M.; Hendey, B.; Monahan, A.J. The blood-brain barrier in neurodegenerative disease: A rhetorical perspective. J. Neurochem. 2009, 111, 291–314. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, D.; Keeling, D. The story of the discovery of heparin and warfarin. Br. J. Haematol. 2008, 141, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Capila, I.; Linhardt, R.J. Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 2002, 41, 391–412. [Google Scholar] [CrossRef]
- Gray, E.; Mulloy, B.; Barrowcliffe, T.W. Heparin and low-molecular-weight heparin. Thromb. Haemost. 2008, 99, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Gao, Y.; Tian, M.; Li, N.; Hao, S.; Zeng, X. Selectively desulfated heparin inhibits p-selectin-mediated adhesion of human melanoma cells. Cancer Lett. 2005, 229, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, G.F.; Mendes, A.; Castro, R.A.; Bau, E.C.; Nader, H.B.; Dietrich, C.P. Distribution of sulfated glycosaminoglycans in the animal kingdom: Widespread occurrence of heparin-like compounds in invertebrates. Biochim. Biophys. Acta 2000, 1475, 287–294. [Google Scholar] [CrossRef]
- Ronnberg, E.; Melo, F.R.; Pejler, G. Mast cell proteoglycans. J. Histochem. Cytochem. 2012, 60, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Artis, D. New paradigms in type 2 immunity. Science 2012, 337, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H.; Mulloy, B. Current structural biology of the heparin interactome. Curr. Opin. Struct. Biol. 2015, 34, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Lever, R.; Page, C.P. Non-anticoagulant effects of heparin: An overview. Handb. Exp. Pharmacol. 2012, 281–305. [Google Scholar]
- Li, X.; Li, Z.; Zheng, Z.; Liu, Y.; Ma, X. Unfractionated heparin ameliorates lipopolysaccharide-induced lung inflammation by downregulating nuclear factor-kappab signaling pathway. Inflammation 2013, 36, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zheng, Z.; Li, X.; Ma, X. Unfractionated heparin inhibits lipopolysaccharide-induced inflammatory response through blocking p38 mapk and nf-kappab activation on endothelial cell. Cytokine 2012, 60, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Norgard-Sumnicht, K.; Linhardt, R.; Varki, A. Differential interactions of heparin and heparan sulfate glycosaminoglycans with the selectins. Implications for the use of unfractionated and low molecular weight heparins as therapeutic agents. J. Clin. Investig. 1998, 101, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.L.; Choi, S.H.; Varki, A. Differential metastasis inhibition by clinically relevant levels of heparins—Correlation with selectin inhibition, not antithrombotic activity. Clin. Cancer Res. 2005, 11, 7003–7011. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Najjam, S.; Gordon, M.Y.; Gibbs, R.V.; Rider, C.C. Il-12 is a heparin-binding cytokine. J. Immunol. 1999, 162, 1064–1070. [Google Scholar] [PubMed]
- Najjam, S.; Gibbs, R.V.; Gordon, M.Y.; Rider, C.C. Characterization of human recombinant interleukin 2 binding to heparin and heparan sulfate using an elisa approach. Cytokine 1997, 9, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Mazor, M.; Mazor, R.; Kristal, B.; Kistler, E.B.; Ziv, I.; Chezar, J.; Sela, S. Heparin interaction with the primed polymorphonuclear leukocyte cd11b induces apoptosis and prevents cell activation. J. Immunol. Res. 2015, 2015, 751014. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ling, Y.; Huang, M.; Yin, T.; Gou, S.M.; Zhan, N.Y.; Xiong, J.X.; Wu, H.S.; Yang, Z.Y.; Wang, C.Y. Heparin inhibits the inflammatory response induced by lps and hmgb1 by blocking the binding of hmgb1 to the surface of macrophages. Cytokine 2015, 72, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.L.; Stone, P.J.; Nugent, M.A. New insights into the inhibition of human neutrophil elastase by heparin. Biochemistry 2006, 45, 9104–9120. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.J.; Myszka, D.G.; Katsamba, P.S.; Ohnuki, L.E.; Gleich, G.J.; Acharya, K.R. Eosinophil-granule major basic protein, a c-type lectin, binds heparin. Biochemistry 2005, 44, 14152–14158. [Google Scholar] [CrossRef] [PubMed]
- Shastri, M.D.; Stewart, N.; Horne, J.; Zaidi, S.T.; Sohal, S.S.; Peterson, G.M.; Korner, H.; Gueven, N.; Patel, R.P. Non-anticoagulant fractions of enoxaparin suppress inflammatory cytokine release from peripheral blood mononuclear cells of allergic asthmatic individuals. PLoS ONE 2015, 10, e0128803. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chi, C.; Guo, L.; Wang, X.; Guo, L.; Sun, J.; Sun, B.; Liu, S.; Chang, X.; Li, E. Heparin therapy reduces 28-day mortality in adult severe sepsis patients: A systematic review and meta-analysis. Crit. Care 2014, 18, 563. [Google Scholar] [CrossRef] [PubMed]
- Chande, N.; MacDonald, J.K.; Wang, J.J.; McDonald, J.W. Unfractionated or low molecular weight heparin for induction of remission in ulcerative colitis: A cochrane inflammatory bowel disease and functional bowel disorders systematic review of randomized trials. Inflamm. Bowel Dis. 2011, 17, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Duong, M.; Cockcroft, D.; Boulet, L.P.; Ahmed, T.; Iverson, H.; Atkinson, D.C.; Stahl, E.G.; Watson, R.; Davis, B.; Milot, J.; et al. The effect of ivx-0142, a heparin-derived hypersulfated disaccharide, on the allergic airway responses in asthma. Allergy 2008, 63, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Wurm, G.; Tomancok, B.; Nussbaumer, K.; Adelwohrer, C.; Holl, K. Reduction of ischemic sequelae following spontaneous subarachnoid hemorrhage: A double-blind, randomized comparison of enoxaparin versus placebo. Clin. Neurol. Neurosurg. 2004, 106, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Siironen, J.; Juvela, S.; Varis, J.; Porras, M.; Poussa, K.; Ilveskero, S.; Hernesniemi, J.; Lassila, R. No effect of enoxaparin on outcome of aneurysmal subarachnoid hemorrhage: A randomized, double-blind, placebo-controlled clinical trial. J. Neurosurg. 2003, 99, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.M.; Schreibman, D.; Aldrich, E.F.; Stallmeyer, B.; Le, B.; James, R.F.; Beaty, N. Unfractionated heparin: Multitargeted therapy for delayed neurological deficits induced by subarachnoid hemorrhage. Neurocrit. Care 2010, 13, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Alotaibi, N.M.; Akbar, M.A.; Ayling, O.G.; Ibrahim, G.M.; Macdonald, R.L.; Noble, A.; Molyneux, A.; Quinn, A.; Schatlo, B.; et al. Loss of consciousness at onset of aneurysmal subarachnoid hemorrhage is associated with functional outcomes in good-grade patients. World Neurosurg. 2017, 98, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Suwatcharangkoon, S.; Meyers, E.; Falo, C.; Schmidt, J.M.; Agarwal, S.; Claassen, J.; Mayer, S.A. Loss of consciousness at onset of subarachnoid hemorrhage as an important marker of early brain injury. JAMA Neurol. 2016, 73, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Quartermain, D.; Li, Y.S.; Jonas, S. The low molecular weight heparin enoxaparin reduces infarct size in a rat model of temporary focal ischemia. Cerebrovasc. Dis. 2003, 16, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Li, P.A.; He, Q.P.; Siddiqui, M.M.; Shuaib, A. Posttreatment with low molecular weight heparin reduces brain edema and infarct volume in rats subjected to thrombotic middle cerebral artery occlusion. Brain Res. 1998, 801, 220–223. [Google Scholar] [CrossRef]
- Mocco, J.; Shelton, C.E.; Sergot, P.; Ducruet, A.F.; Komotar, R.J.; Otten, M.L.; Sosunov, S.A.; Macarthur, R.B.; Kennedy, T.P.; Connolly, E.S., Jr. O-desulfated heparin improves outcome after rat cerebral ischemia/reperfusion injury. Neurosurgery 2007, 61, 1297–1303; discussion 1303–1304. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Ducker, T.B.; Kempe, L.G. Temporary experimental intracranial vascular occlusion. Effect of massive doses of heparin on brain survival. J. Neurosurg. 1969, 30, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, K.; Spellman, S.R.; McCarthy, J.B.; Oegema, T.R., Jr.; Low, W.C.; Camarata, P.J. Reduction of brain injury using heparin to inhibit leukocyte accumulation in a rat model of transient focal cerebral ischemia. I. Protective mechanism. J. Neurosurg. 1996, 85, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Lu, T.S.; Yuan, H.Y. Neuroprotective effects of ultra-low-molecular-weight heparin in vitro and vivo models of ischemic injury. Fundam. Clin. Pharmacol. 2011, 25, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, Q.Z.; Cheng, Y.N.; Ji, S.L.; Du, G.H. Antagonistic effects of ultra-low-molecular-weight heparin against cerebral ischemia/reperfusion injury in rats. Pharmacol. Res. 2007, 56, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Sun, X.; Zhang, Q.Z.; Yang, H. Neuroprotective effects of ultra-low-molecular-weight heparin on cerebral ischemia/reperfusion injury in rats: Involvement of apoptosis, inflammatory reaction and energy metabolism. Int. J. Mol. Sci. 2013, 14, 1932–1939. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Garcia, M.; Testai, F.; Vetri, F.; Barabanova, A.; Pelligrino, D.A.; Paisansathan, C. Pharmacologic blockade of vascular adhesion protein-1 lessens neurologic dysfunction in rats subjected to subarachnoid hemorrhage. Brain Res. 2014, 1586, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Kumasaka, K.; Browne, K.D.; Li, S.; St-Pierre, J.; Cognetti, J.; Marks, J.; Johnson, V.E.; Smith, D.H.; Pascual, J.L. Unfractionated heparin after tbi reduces in vivo cerebrovascular inflammation, brain edema and accelerates cognitive recovery. J. Trauma Acute Care Surg. 2016, 81, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Marks, J.A.; Eisenstadt, R.; Kumasaka, K.; Samadi, D.; Johnson, V.E.; Holena, D.N.; Allen, S.R.; Browne, K.D.; Smith, D.H.; et al. Enoxaparin ameliorates post-traumatic brain injury edema and neurologic recovery, reducing cerebral leukocyte endothelial interactions and vessel permeability in vivo. J. Trauma Acute Care Surg. 2015, 79, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.R.; Angstwurm, K.; Rosenkranz, T.; Lindauer, U.; Freyer, D.; Burger, W.; Busch, C.; Einhaupl, K.M.; Dirnagl, U. Heparin inhibits leukocyte rolling in pial vessels and attenuates inflammatory changes in a rat model of experimental bacterial meningitis. J. Cereb. Blood Flow Metab. 1997, 17, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.M.; Cecconi, O.; Roberts, W.G.; Aruffo, A.; Linhardt, R.J.; Bevilacqua, M.P. Heparin oligosaccharides bind L- and P-selectin and inhibit acute inflammation. Blood 1993, 82, 3253–3258. [Google Scholar] [PubMed]
- Sudha, T.; Phillips, P.; Kanaan, C.; Linhardt, R.J.; Borsig, L.; Mousa, S.A. Inhibitory effect of non-anticoagulant heparin (s-nach) on pancreatic cancer cell adhesion and metastasis in human umbilical cord vessel segment and in mouse model. Clin. Exp. Metastasis 2012, 29, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, W.; Sun, Q.; Liu, M.; Li, W.; Zhang, X.S.; Zhou, M.L.; Hang, C.H. Expression and cell distribution of receptor for advanced glycation end-products in the rat cortex following experimental subarachnoid hemorrhage. Brain Res. 2014, 1543, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.D.; Mao, H.Y.; Lv, J.; Lu, X.J. Expression of high-mobility group box-1 (hmgb1) in the basilar artery after experimental subarachnoid hemorrhage. J. Clin. Neurosci. 2016, 27, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Z.; Wu, S.C.; Kwan, A.L.; Lin, C.L. 4′-O-beta-d-glucosyl-5-O-methylvisamminol, an active ingredient of saposhnikovia divaricata, attenuates high-mobility group box 1 and subarachnoid hemorrhage-induced vasospasm in a rat model. Behav. Brain Funct. 2015, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Sokol, B.; Wozniak, A.; Jankowski, R.; Jurga, S.; Wasik, N.; Shahid, H.; Grzeskowiak, B. Hmgb1 level in cerebrospinal fluid as a marker of treatment outcome in patients with acute hydrocephalus following aneurysmal subarachnoid hemorrhage. J. Stroke Cerebrovasc. Dis. 2015, 24, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wu, W.; Hu, Y.C.; Li, H.; Zhang, D.; Li, S.; Li, W.; Li, W.D.; Ma, B.; Zhu, J.H.; et al. Early release of high-mobility group box 1 (hmgb1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflamm. 2014, 11, 106. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Tsuruta, R.; Kaneko, T.; Yamashita, S.; Fujita, M.; Kasaoka, S.; Hashiguchi, T.; Suzuki, M.; Maruyama, I.; Maekawa, T. High-mobility group box 1 protein in csf of patients with subarachnoid hemorrhage. Neurocrit. Care 2009, 11, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Koide, M.; Dumont, T.M.; Russell, S.R.; Tranmer, B.I.; Wellman, G.C. Subarachnoid hemorrhage induces gliosis and increased expression of the pro-inflammatory cytokine high mobility group box 1 protein. Transl. Stroke Res. 2011, 2, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kellermann, I.; Kleindienst, A.; Hore, N.; Buchfelder, M.; Brandner, S. Early csf and serum s100b concentrations for outcome prediction in traumatic brain injury and subarachnoid hemorrhage. Clin. Neurol. Neurosurg. 2016, 145, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S.; Xu, Y.; Schmidt, C.; Emtmann, I.; Buchfelder, M.; Kleindienst, A. Shunt-dependent hydrocephalus following subarachnoid hemorrhage correlates with increased S100B levels in cerebrospinal fluid and serum. Acta Neurochir. Suppl. 2012, 114, 217–220. [Google Scholar] [PubMed]
- Stranjalis, G.; Korfias, S.; Psachoulia, C.; Kouyialis, A.; Sakas, D.E.; Mendelow, A.D. The prognostic value of serum S-100B protein in spontaneous subarachnoid haemorrhage. Acta Neurochir. (Wien) 2007, 149, 231–237; discussion 237–238. [Google Scholar] [CrossRef] [PubMed]
- Haruma, J.; Teshigawara, K.; Hishikawa, T.; Wang, D.; Liu, K.; Wake, H.; Mori, S.; Takahashi, H.K.; Sugiu, K.; Date, I.; et al. Anti-high mobility group box-1 (hmgb1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci. Rep. 2016, 6, 37755. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.; Dong, Y.; Huo, R.; Wen, K.; Zhang, Y.; Liang, G. Rutin inhibits neuroinflammation and provides neuroprotection in an experimental rat model of subarachnoid hemorrhage, possibly through suppressing the rage-nf-kappab inflammatory signaling pathway. Neurochem. Res. 2016, 41, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, J.S.; Zhang, D.D.; Yang, Y.Q.; Huang, L.T.; Yu, Z.; Chen, R.D.; Yang, H.K.; Hang, C.H. Inhibition of the receptor for advanced glycation end-products (rage) attenuates neuroinflammation while sensitizing cortical neurons towards death in experimental subarachnoid hemorrhage. Mol. Neurobiol. 2017, 54, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Myint, K.M.; Yamamoto, Y.; Doi, T.; Kato, I.; Harashima, A.; Yonekura, H.; Watanabe, T.; Shinohara, H.; Takeuchi, M.; Tsuneyama, K.; et al. Rage control of diabetic nephropathy in a mouse model: Effects of rage gene disruption and administration of low-molecular weight heparin. Diabetes 2006, 55, 2510–2522. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Mori, S.; Wake, H.; Zhang, J.; Liu, K.; Izushi, Y.; Takahashi, H.K.; Peng, B.; Nishibori, M. Establishment of in vitro binding assay of high mobility group box-1 and s100a12 to receptor for advanced glycation endproducts: Heparin‘s effect on binding. Acta Med. Okayama 2009, 63, 203–211. [Google Scholar] [PubMed]
- Rao, N.V.; Argyle, B.; Xu, X.; Reynolds, P.R.; Walenga, J.M.; Prechel, M.; Prestwich, G.D.; MacArthur, R.B.; Walters, B.B.; Hoidal, J.R.; et al. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of rage with its ligands. Am. J. Physiol. Cell Physiol. 2010, 299, C97–C110. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Yang, Z.Y.; Yin, T.; Li, L.; Yuan, W.W.; Wu, H.S.; Wang, C.Y. Heparin changes the conformation of high-mobility group protein 1 and decreases its affinity toward receptor for advanced glycation endproducts in vitro. Int. Immunopharmacol. 2011, 11, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Yamamoto, Y.; Munesue, S.; Harashima, A.; Watanabe, T.; Yonekura, H.; Yamamoto, H.; Tsuchiya, H. Low molecular weight heparin suppresses receptor for advanced glycation end products-mediated expression of malignant phenotype in human fibrosarcoma cells. Cancer Sci. 2013, 104, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Eisenstadt, R.; Kumasaka, K.; Johnson, V.E.; Marks, J.; Nagata, K.; Browne, K.D.; Smith, D.H.; Pascual, J.L. Does enoxaparin interfere with hmgb1 signaling after tbi? A potential mechanism for reduced cerebral edema and neurologic recovery. J. Trauma Acute Care Surg. 2016, 80, 381–387; discussion 387–389. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Kumar, A. Microglia in the tbi brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275 Pt 3, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Sozen, T.; Tsuchiyama, R.; Hasegawa, Y.; Suzuki, H.; Jadhav, V.; Nishizawa, S.; Zhang, J.H. Role of interleukin-1beta in early brain injury after subarachnoid hemorrhage in mice. Stroke 2009, 40, 2519–2525. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Wang, Z.; Li, H.; Shen, H.; Xu, X.; Jia, G.; Chen, G. Inhibition of mammalian target of rapamycin attenuates early brain injury through modulating microglial polarization after experimental subarachnoid hemorrhage in rats. J. Neurol. Sci. 2016, 367, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, B.L.; Lord, M.S.; Melrose, J.; Whitelock, J.M. Can we produce heparin/heparan sulfate biomimetics using “mother-nature“ as the gold standard? Molecules 2015, 20, 4254–4276. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Chen, L.; Paglia, M.; Gallagher, I.; Allen, J.E.; Vyas, Y.M.; Ray, A.; Ray, P. Simvastatin promotes th2-type responses through the induction of the chitinase family member ym1 in dendritic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 7777–7782. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Kumar, R.K.; Zhou, J.; Foster, P.S.; Webb, D.C. ym1/2 promotes th2 cytokine expression by inhibiting 12/15(s)-lipoxygenase: Identification of a novel pathway for regulating allergic inflammation. J. Immunol. 2009, 182, 5393–5399. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Hung, S.I.; Hwa, K.Y.; Kato, I.; Chen, J.E.; Liu, C.H.; Chang, A.C. A macrophage protein, Ym1, transiently expressed during inflammation is a novel mammalian lectin. J. Biol. Chem. 2001, 276, 17497–17506. [Google Scholar] [CrossRef] [PubMed]
- Lean, Q.Y.; Eri, R.D.; Randall-Demllo, S.; Sohal, S.S.; Stewart, N.; Peterson, G.M.; Gueven, N.; Patel, R.P. Orally administered enoxaparin ameliorates acute colitis by reducing macrophage-associated inflammatory responses. PLoS ONE 2015, 10, e0134259. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhang, D.M.; Chen, H.L.; Lin, Y.X.; Hang, C.H.; Yin, H.X.; Shi, J.X. N-acetylcysteine suppresses oxidative stress in experimental rats with subarachnoid hemorrhage. J. Clin. Neurosci. 2009, 16, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Nito, C.; Kamada, H.; Yu, F.; Chan, P.H. Reduction in oxidative stress by superoxide dismutase overexpression attenuates acute brain injury after subarachnoid hemorrhage via activation of akt/glycogen synthase kinase-3beta survival signaling. J. Cereb. Blood Flow Metab. 2007, 27, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Zhou, C.; Heistad, D.D.; Watanabe, Y.; Zhang, J.H. Gene transfer of extracellular superoxide dismutase failed to prevent cerebral vasospasm after experimental subarachnoid hemorrhage. Stroke 2004, 35, 2512–2517. [Google Scholar] [CrossRef] [PubMed]
- Froehler, M.T.; Kooshkabadi, A.; Miller-Lotan, R.; Blum, S.; Sher, S.; Levy, A.; Tamargo, R.J. Vasospasm after subarachnoid hemorrhage in haptoglobin 2-2 mice can be prevented with a glutathione peroxidase mimetic. J. Clin. Neurosci. 2010, 17, 1169–1172. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, J.; Carlsson, L.; Marklund, S.L.; Edlund, T. The heparin-binding domain of extracellular superoxide dismutase c and formation of variants with reduced heparin affinity. J. Biol. Chem. 1992, 267, 18205–18209. [Google Scholar] [PubMed]
- Adachi, T.; Yamnamoto, M.; Hara, H. Heparin-affinity of human extracellular-superoxide dismutase in the brain. Biol. Pharm. Bull. 2001, 24, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, J.; Nilsson, P.; Karlsson, K.; Marklund, S.L. 10-fold increase in human plasma extracellular superoxide dismutase content caused by a mutation in heparin-binding domain. J. Biol. Chem. 1994, 269, 19163–19166. [Google Scholar] [PubMed]
- Adachi, T.; Yamada, H.; Futenma, A.; Kato, K.; Hirano, K. Heparin-induced release of extracellular-superoxide dismutase form (V) to plasma. J. Biochem. 1995, 117, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Hara, H.; Yamada, H.; Yamazaki, N.; Yamamoto, M.; Sugiyama, T.; Futenma, A.; Katagiri, Y. Heparin-stimulated expression of extracellular-superoxide dismutase in human fibroblasts. Atherosclerosis 2001, 159, 307–312. [Google Scholar] [CrossRef]
- Nakane, H.; Chu, Y.; Faraci, F.M.; Oberley, L.W.; Heistad, D.D. Gene transfer of extracellular superoxide dismutase increases superoxide dismutase activity in cerebrospinal fluid. Stroke 2001, 32, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Y.; Tsai, K.L.; Lee, T.Y.; Chiueh, C.C.; Lee, W.S.; Hsu, C. Sex-specific role of thioredoxin in neuroprotection against iron-induced brain injury conferred by estradiol. Stroke 2010, 41, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Baratz-Goldstein, R.; Deselms, H.; Heim, L.R.; Khomski, L.; Hoffer, B.J.; Atlas, D.; Pick, C.G. Thioredoxin-mimetic-peptides protect cognitive function after mild traumatic brain injury (mtbi). PLoS ONE 2016, 11, e0157064. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Stadtman, T.C. Heparin-binding properties of selenium-containing thioredoxin reductase from hela cells and human lung adenocarcinoma cells. Proc. Natl. Acad. Sci. USA 1997, 94, 6138–6141. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.A.; Long, W.F.; Williamson, F.B. Inhibition by heparin of fe(II)-catalysed free-radical peroxidation of linolenic acid. Biochem. J. 1992, 286 (Pt 3), 717–720. [Google Scholar] [CrossRef] [PubMed]
- Albertini, R.; Rindi, S.; Passi, A.; Pallavicini, G.; De Luca, G. Heparin protection against Fe2+ -and Cu2+ -mediated oxidation of liposomes. FEBS Lett. 1996, 383, 155–158. [Google Scholar] [CrossRef]
- Wahl, F.; Grosjean-Piot, O.; Bareyre, F.; Uzan, A.; Stutzmann, J.M. Enoxaparin reduces brain edema, cerebral lesions, and improves motor and cognitive impairments induced by a traumatic brain injury in rats. J. Neurotrauma 2000, 17, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Pratt, J.; Boudeau, P.; Uzan, A.; Imperato, A.; Stutzmann, J. Enoxaparin reduces cerebral edemaafter photothrombotic injury in the rat. Haemostasis 1998, 28, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Wagner, K.R.; Keep, R.F.; Hua, Y.; de Courten-Myers, G.M.; Broderick, J.P.; Brott, T.G.; Hoff, J.T. Role of blood clot formation on early edema development after experimental intracerebral hemorrhage. Stroke 1998, 29, 2580–2586. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Xi, G.H.; Keep, R.F.; Hoff, J.T.; Hua, Y. Complement inhibition attenuates brain edema and neurological deficits induced by thrombin. Acta Neurochir. Suppl. 2005, 95, 389–392. [Google Scholar] [PubMed]
- Kim, L.; Schuster, J.; Holena, D.N.; Sims, C.A.; Levine, J.; Pascual, J.L. Early initiation of prophylactic heparin in severe traumatic brain injury is associated with accelerated improvement on brain imaging. J. Emerg. Trauma Shock 2014, 7, 141–148. [Google Scholar] [PubMed]
- Bruce, J.N.; Criscuolo, G.R.; Merrill, M.J.; Moquin, R.R.; Blacklock, J.B.; Oldfield, E.H. Vascular permeability induced by protein product of malignant brain tumors: Inhibition by dexamethasone. J. Neurosurg. 1987, 67, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Tessler, S.; Rockwell, P.; Hicklin, D.; Cohen, T.; Levi, B.Z.; Witte, L.; Lemischka, I.R.; Neufeld, G. Heparin modulates the interaction of vegf165 with soluble and cell associated flk-1 receptors. J. Biol. Chem. 1994, 269, 12456–12461. [Google Scholar] [PubMed]
- Marchetti, M.; Vignoli, A.; Russo, L.; Balducci, D.; Pagnoncelli, M.; Barbui, T.; Falanga, A. Endothelial capillary tube formation and cell proliferation induced by tumor cells are affected by low molecular weight heparins and unfractionated heparin. Thromb. Res. 2008, 121, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Oschatz, C.; Maas, C.; Lecher, B.; Jansen, T.; Bjorkqvist, J.; Tradler, T.; Sedlmeier, R.; Burfeind, P.; Cichon, S.; Hammerschmidt, S.; et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity 2011, 34, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Carr, J. The anti-inflammatory action of heparin: Heparin as an antagonist to histamine, bradykinin and prostaglandin e1. Thromb. Res. 1979, 16, 507–516. [Google Scholar] [CrossRef]
- Thal, S.C.; Sporer, S.; Schmid-Elsaesser, R.; Plesnila, N.; Zausinger, S. Inhibition of bradykinin b2 receptors before, not after onset of experimental subarachnoid hemorrhage prevents brain edema formation and improves functional outcome. Crit. Care Med. 2009, 37, 2228–2234. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Tiruvoipati, R.; Green, C.; Botha, J.; Tran, H. Heparin induced thrombocytopenia in critically ill: Diagnostic dilemmas and management conundrums. World J. Crit. Care Med. 2015, 4, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Hoh, B.L.; Aghi, M.; Pryor, J.C.; Ogilvy, C.S. Heparin-induced thrombocytopenia type II in subarachnoid hemorrhage patients: Incidence and complications. Neurosurgery 2005, 57, 243–248; discussion 243–248. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Hahn, D.K.; Kellner, C.P.; Komotar, R.J.; Starke, R.; Garrett, M.C.; Yao, J.; Cleveland, J.; Mayer, S.A.; Connolly, E.S. The incidence of heparin-induced thrombocytopenia type II in patients with subarachnoid hemorrhage treated with heparin versus enoxaparin. J. Neurosurg. 2009, 110, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Mehta, B.P.; Sims, J.R.; Baccin, C.E.; Leslie-Mazwi, T.M.; Ogilvy, C.S.; Nogueira, R.G. Predictors and outcomes of suspected heparin-induced thrombocytopenia in subarachnoid hemorrhage patients. Interv. Neurol. 2014, 2, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, N.; Naraoka, M.; Matsuda, N.; Ohkuma, H. Safety of preprocedural antiplatelet medication in coil embolization of ruptured cerebral aneurysms at the acute stage. Interv. Neuroradiol. 2014, 20, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Hoh, B.L.; Nogueira, R.G.; Ledezma, C.J.; Pryor, J.C.; Ogilvy, C.S. Safety of heparinization for cerebral aneurysm coiling soon after external ventriculostomy drain placement. Neurosurgery 2005, 57, 845–849; discussion 845–849. [Google Scholar] [CrossRef] [PubMed]
- Egashira, Y.; Yoshimura, S.; Enomoto, Y.; Ishiguro, M.; Asano, T.; Iwama, T. Ultra-early endovascular embolization of ruptured cerebral aneurysm and the increased risk of hematoma growth unrelated to aneurysmal rebleeding. J. Neurosurg. 2013, 118, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, J.; Snyder, K.A.; Graffeo, C.S.; Khanal, D.R.; Lanzino, G.; Wijdicks, E.F.; Rabinstein, A.A. Risk of ventriculostomy-associated hemorrhage in patients with aneurysmal subarachnoid hemorrhage treated with anticoagulant thromboprophylaxis. Neurocrit. Care 2016, 25, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Bruder, M.; Schuss, P.; Konczalla, J.; El-Fiki, A.; Lescher, S.; Vatter, H.; Seifert, V.; Guresir, E. Ventriculostomy-related hemorrhage after treatment of acutely ruptured aneurysms: The influence of anticoagulation and antiplatelet treatment. World Neurosurg. 2015, 84, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.A. Fondaparinux: Basic properties and efficacy and safety in venous thromboembolism prophylaxis. Am. J. Orthop. (Belle Mead NJ) 2002, 31, 4–10. [Google Scholar] [PubMed]
- Zhang, Y.; Zhao, Z.; Guan, L.; Mao, L.; Li, S.; Guan, X.; Chen, M.; Guo, L.; Ding, L.; Cong, C.; et al. N-acetyl-heparin attenuates acute lung injury caused by acid aspiration mainly by antagonizing histones in mice. PLoS ONE 2014, 9, e97074. [Google Scholar] [CrossRef] [PubMed]
- Veraldi, N.; Hughes, A.J.; Rudd, T.R.; Thomas, H.B.; Edwards, S.W.; Hadfield, L.; Skidmore, M.A.; Siligardi, G.; Cosentino, C.; Shute, J.K.; et al. Heparin derivatives for the targeting of multiple activities in the inflammatory response. Carbohydr. Polym. 2015, 117, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, J.R.; Varki, A.; Esko, J.D. Heparin‘s anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of l- and p-selectins. J. Clin. Investig. 2002, 110, 127–136. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayman, E.G.; Patel, A.P.; James, R.F.; Simard, J.M. Heparin and Heparin-Derivatives in Post-Subarachnoid Hemorrhage Brain Injury: A Multimodal Therapy for a Multimodal Disease. Molecules 2017, 22, 724. https://doi.org/10.3390/molecules22050724
Hayman EG, Patel AP, James RF, Simard JM. Heparin and Heparin-Derivatives in Post-Subarachnoid Hemorrhage Brain Injury: A Multimodal Therapy for a Multimodal Disease. Molecules. 2017; 22(5):724. https://doi.org/10.3390/molecules22050724
Chicago/Turabian StyleHayman, Erik G., Akil P. Patel, Robert F. James, and J. Marc Simard. 2017. "Heparin and Heparin-Derivatives in Post-Subarachnoid Hemorrhage Brain Injury: A Multimodal Therapy for a Multimodal Disease" Molecules 22, no. 5: 724. https://doi.org/10.3390/molecules22050724
APA StyleHayman, E. G., Patel, A. P., James, R. F., & Simard, J. M. (2017). Heparin and Heparin-Derivatives in Post-Subarachnoid Hemorrhage Brain Injury: A Multimodal Therapy for a Multimodal Disease. Molecules, 22(5), 724. https://doi.org/10.3390/molecules22050724