
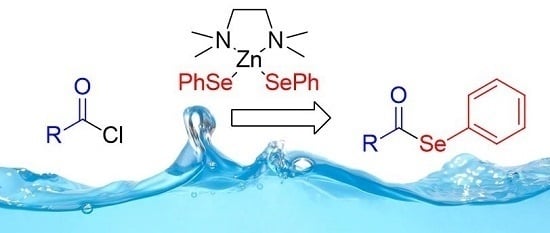
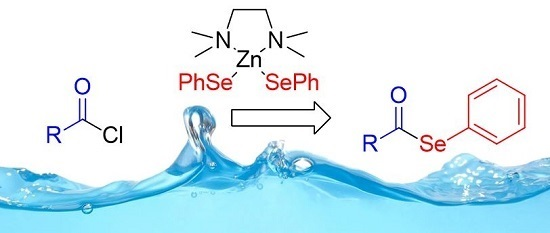
Atom Efficient Preparation of Zinc Selenates for the Synthesis of Selenol Esters under “On Water” Conditions






Abstract
:
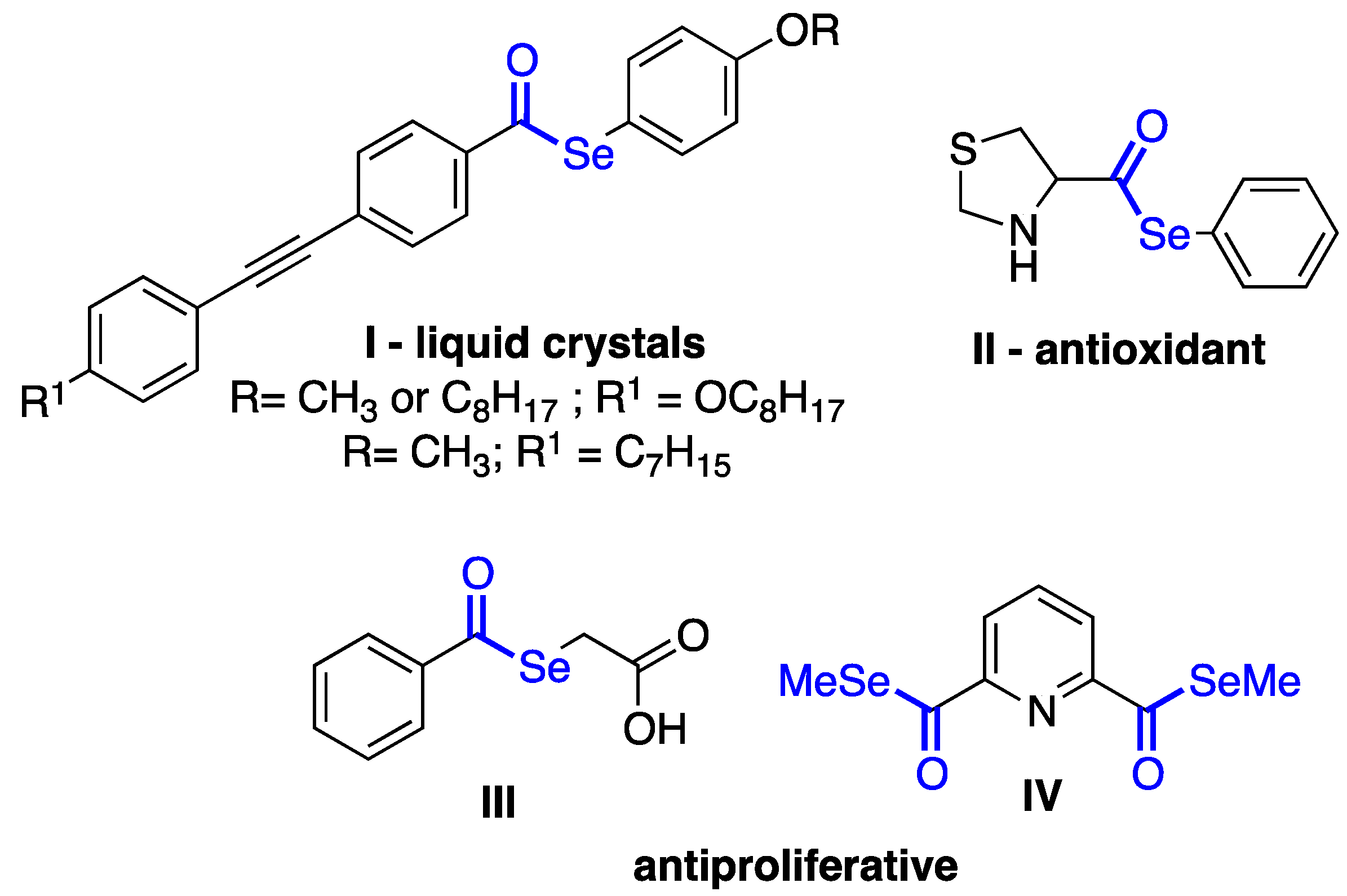
1. Introduction
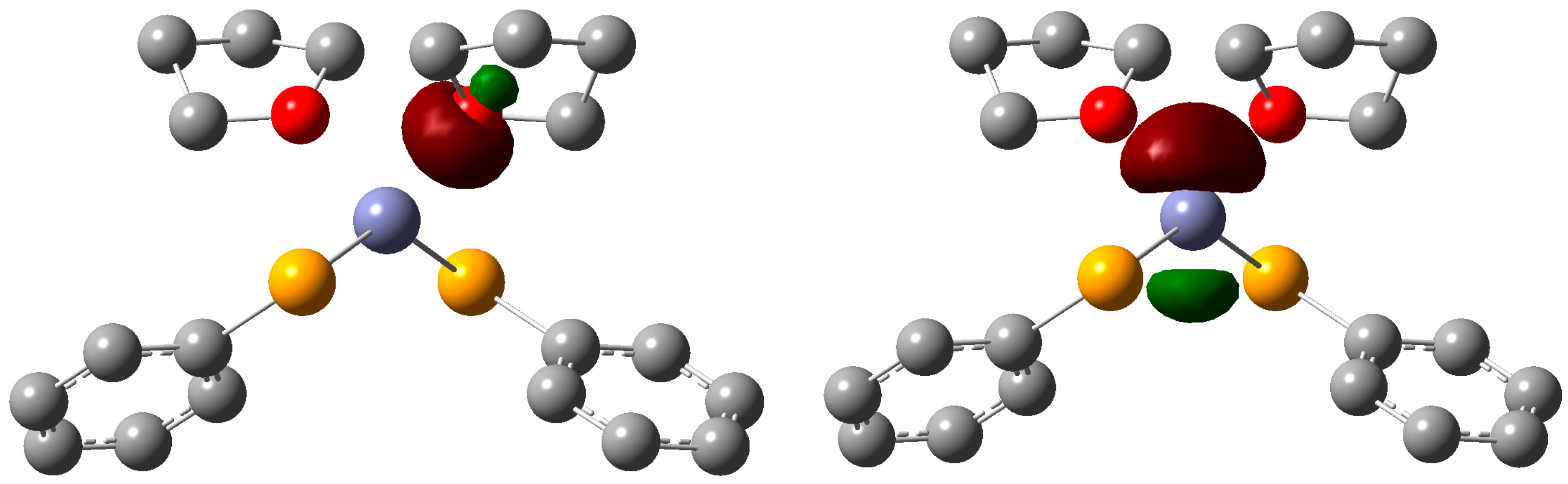
2. Results
3. Materials and Methods
General Procedure for the Synthesis of Selenol Esters 5a–h
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fujiwara, S.; Kambe, N. Thio-, Seleno-, and Telluro-Carboxylic Acid Esters. In Chalcogenocarboxylic Acid Derivatives; Kato, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 87–140. [Google Scholar]
- Pakzad, B.; Praefcke, K.; Simon, H. 5H-[1]Benzoselenino[2,3-b]pyridine—A Novel Heterocyclic Ring System. Angew. Chem. Int. Ed. Eng. 1977, 16, 319–320. [Google Scholar] [CrossRef]
- Rampon, D.S.; Rodembusch, F.S.; Gonçalves, P.F.B.; Lourega, R.V.; Merlo, A.A.; Schneider, P.H. An evaluation of the chalcogen atom effect on the mesomorphic and electronic properties in a new homologous series of chalcogeno esters. J. Braz. Chem. Soc. 2010, 21, 2100–2107. [Google Scholar] [CrossRef]
- Rampon, D.S.; Rodembusch, F.S.; Schneider, J.M.F.M.; Bechtold, I.H.; Gonçalves, P.F.B.; Merlo, A.A.; Schneider, P.H. Novel selenoesters fluorescent liquid crystalline exhibiting a rich phase polymorphism. J. Mater. Chem. 2010, 20, 715–722. [Google Scholar] [CrossRef]
- Victoria, F.N.; Martinez, D.M.; Castro, M.; Casaril, A.M.; Alves, D.; Lenardão, E.J.; Salles, H.D.; Schneider, P.H.; Savegnago, L. Antioxidant properties of (R)-Se-aryl thiazolidine-4-carboselenoate. Chem. Biol. Interact. 2013, 205, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Sanmartín, C.; Plano, D.; Domínguez, E.; Font, M.; Calvo, A.; Prior, C.; Encío, I.; Palop, J.A. Synthesis and Pharmacological Screening of Several Aroyl and Heteroaroyl Selenylacetic Acid Derivatives as Cytotoxic and Antiproliferative Agents. Molecules 2009, 14, 3313–3338. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Álvarez, E.; Plano, D.; Font, M.; Calvo, A.; Prior, C.; Jacob, C.; Palop, J.A.; Sanmartín, C. Synthesis and antiproliferative activity of novel selenoester derivatives. Eur. J. Med. Chem. 2014, 73, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Makriyannis, A.; Guenther, W.H.H.; Mautner, H.G. Selenol esters as specific reagents of the acylation of thiol groups. J. Am. Chem. Soc. 1973, 95, 8403–8406. [Google Scholar] [CrossRef]
- Reinerth, W.A.; Tour, J.M. Protecting Groups for Organoselenium Compounds. J. Org. Chem. 1998, 63, 2397–2400. [Google Scholar] [CrossRef]
- Subramanyam, C.; Noguchi, M.; Weinreb, S.M. An approach to amphimedine and related marine alkaloids utilizing an intramolecular Kondrat’eva pyridine synthesis. J. Org. Chem. 1989, 54, 5580–5585. [Google Scholar] [CrossRef]
- Bennasar, M.-L.; Zulaica, E.; Solé, D.; Roca, T.; García-Díaz, D.; Alonso, S. Total Synthesis of the Bridged Indole Alkaloid Apparicine. J. Org. Chem. 2009, 74, 8359–8368. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.F.; Chen, K.X.; Eary, C.T. An Enantioselective Total Synthesis of (+)-Geissoschizine. Org. Lett. 1999, 1, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Yamashita, S.; Ishihara, Y.; Hirama, M. Two Convergent Routes to the Left-Wing Fragment of Ciguatoxin CTX3C Using O, S-Acetals As Key Intermediates. Org. Lett. 2006, 8, 5805–5808. [Google Scholar] [CrossRef] [PubMed]
- Sviridov, A.F.; Ermolenko, M.S.; Yashunskii, D.V.; Kochetkov, N.K. Selenoesters in organic synthesis. 2. New synthesis of saturated and α,β-unsaturated ketones and the synthesis of the Peach moth pheromone (Carposia niponensis). Bull. Acad. Sci. USSR Div. Chem. Sci. 1985, 34, 1514–1518. [Google Scholar] [CrossRef]
- Durek, T.; Alewood, P.F. Preformed Selenoesters Enable Rapid Native Chemical Ligation at Intractable Sites. Angew. Chem. Int. Ed. Eng. 2011, 50, 12042–12045. [Google Scholar] [CrossRef] [PubMed]
- Ghassemian, A.; Vila-Farrés, X.; Alewood, P.F.; Durek, T. Solid phase synthesis of peptide-selenoesters. Bioorg. Med. Chem. 2013, 21, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Gais, H.-J. Synthesis of Thiol and Selenol Esters from Carboxylic Acids and Thiols or Selenols, Respectively. Angew. Chem. Int. Ed. Eng. 1977, 16, 244–246. [Google Scholar] [CrossRef]
- Lin, S.-M.; Zhang, J.-L.; Chen, J.-X.; Gao, W.-X.; Ding, J.-C.; Su, W.-K.; Wu, H.-Y. An approach to the synthesis of thioesters and selenoesters promoted by Rongalite®. J. Braz. Chem. Soc. 2010, 21, 1616–1620. [Google Scholar] [CrossRef]
- Grieco, P.A.; Jaw, J.Y.; Claremon, D.A.; Nicolaou, K.C. N-Phenylselenophthalimide. A useful reagent for the facile transformation of (1) carboxylic acids into either selenol esters or amides and (2) alcohols into alkyl phenyl selenides. J. Org. Chem. 1981, 46, 1215–1217. [Google Scholar] [CrossRef]
- Grieco, P.A.; Yokoyama, Y.; Williams, E. Aryl selenocyanates and aryl thiocyanates: Reagents for the preparation of activated esters. J. Org. Chem. 1978, 43, 1283–1285. [Google Scholar] [CrossRef]
- Gais, H.-J.; Lied, T. A New Method for Selective Activation of Amino, Hydroxy, and Mercapto Carboxylic Acids at the Carboxyl Group: Preparation of Thiol and Selenol Esters. Angew. Chem. Int. Ed. Eng. 1978, 17, 267–268. [Google Scholar] [CrossRef]
- Dan, W.; Deng, H.; Chen, J.; Liu, M.; Ding, J.; Wu, H. A new odorless one-pot synthesis of thioesters and selenoesters promoted by Rongalite®. Tetrahedron 2010, 66, 7384–7388. [Google Scholar] [CrossRef]
- Perin, G.; Silveira, M.B.; Barcellos, A.M.; Araujo, D.R.; Jacob, R.G.; Barcellos, T.; Lenardão, E.J. Rongalite®/PEG-400 as reducing system in the synthesis of new glycerol-derived selenol esters using anhydrides and bis-(2,2-dimethyl-1,3-dioxolanylmethyl)diselenide as substrates. Arkivoc 2016, 2017, 138. [Google Scholar]
- Temperini, A.; Piazzolla, F.; Minuti, L.; Curini, M.; Siciliano, C. General, Mild, and Metal-Free Synthesis of Phenyl Selenoesters from Anhydrides and Their Use in Peptide Synthesis. J. Org. Chem. 2017, 82, 4588–4603. [Google Scholar] [CrossRef] [PubMed]
- Sviridov, A.F.; Ermolenko, M.S.; Yashunskii, D.V.; Kochetkov, N.K. Selenoesters in organic synthesis. 1. Conversion of mixed carboxylic acid esters to selenoesters. Bull. Acad. Sci. USSR Div. Chem. Sci. 1985, 34, 1509–1513. [Google Scholar] [CrossRef]
- Maeda, H.; Fujiwara, S.; Shin-Ike, T.; Kambe, N.; Sonoda, N. Carbonylation of Acidic Hydrocarbons with Selenium and Carbon Monoxide. A Novel Method for Synthesis of Selenol Esters. J. Am. Chem. Soc. 1996, 118, 8160–8161. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Tokunaga, K.; Kawamatsu, H.; Sonoda, N. Synthesis of selenol esters: Palladium-catalyzed coupling of phenyl tributylstannyl selenide with aryl iodides and carbon monoxide. Tetrahedron Lett. 2002, 43, 1507–1509. [Google Scholar] [CrossRef]
- Inoue, T.; Takeda, T.; Kambe, N.; Ogawa, A.; Ryu, I.; Sonoda, N. Synthesis of Thiol, Selenol, and Tellurol Esters from Aldehydes by the Reaction with iBu2AlYR (Y=S, Se, Te). J. Org. Chem. 1994, 59, 5824–5827. [Google Scholar] [CrossRef]
- He, C.; Qian, X.; Sun, P. Syntheses of thiol and selenol esters by oxidative coupling reaction of aldehydes with RYYR (Y=S, Se) under metal-free conditions. Org. Biomol. Chem. 2014, 12, 6072. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.A.; Illyés, T.Z.; Kövér, K.E.; Szilágyi, L. Convenient syntheses of 1,2-trans selenoglycosides using isoselenuronium salts as glycosylselenenyl transfer reagents. Carbohydr. Res. 2012, 360, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Ranu, B.C.; Mandal, T. Indium(I) Iodide-Promoted Cleavage of Diaryl Diselenides and Disulfides and Subsequent Condensation with Alkyl or Acyl Halides. One-Pot Efficient Synthesis of Diorganyl Selenides, Sulfides, Selenoesters, and Thioesters. J. Org. Chem. 2004, 69, 5793–5795. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tripathi, S.K.; Singh, H.B.; Wolmershäuser, G. Synthesis, reactivity, electrochemical and crystallographic studies of diferrocenoyl diselenide and ferrocenoyl selenides. J. Organomet. Chem. 2004, 689, 3046–3055. [Google Scholar] [CrossRef]
- Movassagh, B.; Mirshojaei, F. Synthesis of Selenol Esters from Acid Chlorides and Organic Diselenides in the Presence of the Zn/AlCl 3 System. Monatsh. Chem. 2003, 134, 831–835. [Google Scholar] [CrossRef]
- Takido, T.; Toriyama, M.; Yamashita, K.; Suwa, T.; Seno, M. A Novel Synthesis of Selenides and Selenol Esters Using Liquid-Liquid Phase-Transfer Catalysis. Phosphorus Sulfur Silicon Relat. Elem. 2003, 178, 319–326. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Sigeev, A.S.; Peregudov, A.S.; Petrovskii, P.V. Synthesis of selenoesters. Mendeleev Commun. 2000, 10, 127–128. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Sigeev, A.S.; Peregudov, A.S.; Petrovskii, P.V. Tributylstannyl Aryl Selenides as Efficient Arylselenating Agents in the Synthesis of Seleno Esters. Russ. J. Org. Chem. 2001, 37, 1703–1709. [Google Scholar] [CrossRef]
- Koketsu, M.; Asada, H.; Ishihara, H. Synthesis of Selenol Esters Using Acyl Halides and a Novel Selenating Reagent, LiAlHSeH. Phosphorus. Sulfur. Silicon Relat. Elem. 2004, 179, 591–595. [Google Scholar] [CrossRef]
- Huang, X.; Wang, J.-H. A Convenient and Stereoselective Synthesis of (E)-Vinylseleno Zirconocenes and (E)-Vinylic Selenol Esters. Synlett 1999, 1999, 569–570. [Google Scholar] [CrossRef]
- Munbunjong, W.; Lee, E.H.; Ngernmaneerat, P.; Kim, S.J.; Singh, G.; Chavasiri, W.; Jang, D.O. Indium-mediated cleavage of diphenyl diselenide and diphenyl disulfide: Efficient one-pot synthesis of unsymmetrical diorganyl selenides, sulfides, and selenoesters. Tetrahedron 2009, 65, 2467–2471. [Google Scholar] [CrossRef]
- Marin, G.; Braga, A.L.; Rosa, A.S.; Galetto, F.Z.; Burrow, R.A.; Gallardo, H.; Paixão, M.W. Efficient synthesis of selenol esters from acid chlorides mediated by indium metal. Tetrahedron 2009, 65, 4614–4618. [Google Scholar] [CrossRef]
- Narayanaperumal, S.; Alberto, E.E.; Gul, K.; Kawasoko, C.Y.; Dornelles, L.; Rodrigues, O.E.D.; Braga, A.L. Zn in ionic liquid: An efficient reaction media for the synthesis of diorganyl chalcogenides and chalcogenoesters. Tetrahedron 2011, 67, 4723–4730. [Google Scholar] [CrossRef]
- Silveira, C.C.; Braga, A.L.; Larghi, E.L. Synthesis of Thiol, Selenol, and Tellurol Esters by the Reaction of Organochalcogeno Mercurials with Acid Chlorides. Organometallics 1999, 18, 5183–5186. [Google Scholar] [CrossRef]
- Gul, K.; Narayanaperumal, S.; Dornelles, L.; Rodrigues, O.E.D.; Braga, A.L. Bimetallic system for the synthesis of diorganyl selenides and sulfides, chiral β-seleno amines, and seleno- and thioesters. Tetrahedron Lett. 2011, 52, 3592–3596. [Google Scholar] [CrossRef]
- Abdo, M.; Knapp, S. Biomimetic Seleninates and Selenonates. J. Am. Chem. Soc. 2008, 130, 9234–9235. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Asai, A.; Shin-ike, T.; Kambe, N.; Sonoda, N. A New Synthesis of Selenol Esters via Carbophilic Addition of Organocopper Reagents to Carbonyl Selenide. J. Org. Chem. 1998, 63, 1724–1726. [Google Scholar] [CrossRef]
- Nanami, M.; Ando, H.; Kawai, Y.; Koketsu, M.; Ishihara, H. Stereoselective synthesis of various α-selenoglycosides using in situ production of α-selenolate anion. Tetrahedron Lett. 2007, 48, 1113–1116. [Google Scholar] [CrossRef]
- Sheng, S.; Liu, X. One-Pot Synthesis of Selenoesters from Alkynyl Aryl Selenides. Org. Prep. Proced. Int. 2002, 34, 499–502. [Google Scholar] [CrossRef]
- Braga, A.L.; Martins, T.L.; Silveira, C.C.; Rodrigues, O.E. Synthesis of chalcogenol esters from chalcogenoacetylenes. Tetrahedron 2001, 57, 3297–3300. [Google Scholar] [CrossRef]
- Godoi, M.; Ricardo, E.W.; Botteselle, G.V.; Galetto, F.Z.; Azeredo, J.B.; Braga, A.L. Synthesis of selenol esters from diorganyl diselenides and acyl chlorides under solvent-free conditions and microwave irradiation. Green Chem. 2012, 14, 456. [Google Scholar] [CrossRef]
- Santi, C.; Battistelli, B.; Testaferri, L.; Tiecco, M. On water preparation of phenylselenoesters. Green Chem. 2012, 14, 1277. [Google Scholar] [CrossRef]
- Santi, C.; Santoro, S.; Testaferri, L.; Tiecco, M. A Simple Zinc-Mediated Preparation of Selenols. Synlett 2008, 2008, 1471–1474. [Google Scholar] [CrossRef]
- Tidei, C.; Sancineto, L.; Bagnoli, L.; Battistelli, B.; Marini, F.; Santi, C. A recyclable biphasic system for stereoselective and easily handled hydrochalcogenations. Eur. J. Org. Chem. 2014, 2014, 5968–5975. [Google Scholar] [CrossRef]
- Salman, S.; Schwab, R.; Alberto, E.; Vargas, J.; Dornelles, L.; Rodrigues, O.; Braga, A. Efficient Ring Opening of Protected and Unprotected Aziridines Promoted by Stable Zinc Selenolate in Ionic Liquid. Synlett 2011, 2011, 69–72. [Google Scholar] [CrossRef]
- Flemer, S. Fmoc-Sec(Xan)-OH: Synthesis and utility of Fmoc selenocysteine SPPS derivatives with acid-labile sidechain protection. J. Pept. Sci. 2015, 21, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Flemer, S. A comprehensive one-pot synthesis of protected cysteine and selenocysteine SPPS derivatives. Protein Pept. Lett. 2014, 21, 1257–1264. [Google Scholar] [PubMed]
- Bellino, G.; Scisciani, M.; Vargas, J.P.; Sancineto, L.; Bagnoli, L.; Marini, F.; Lüdtke, D.S.; Lenardao, E.J.; Santi, C. Reaction of Acyl Chlorides with in situ Formed Zinc Selenolates: Synthesis of Selenoesters versus Ring-Opening Reaction of Tetrahydrofuran. J. Chem. 2016, 2016, 2849140. [Google Scholar] [CrossRef]
- Sancineto, L.; Tidei, C.; Bagnoli, L.; Marini, F.; Lippolis, V.; Arca, M.; Lenardão, E.J.; Santi, C. Synthesis of Thiol Esters Using PhSZnBr as Sulfenylating Agent: A DFT-Guided Optimization of Reaction Conditions. Eur. J. Org. Chem. 2016, 2016, 2999–3005. [Google Scholar] [CrossRef]
- Anantharaman, G.; Chandrasekhar, V.; Nehete, U.N.; Roesky, H.W.; Vidovic, D.; Magull, J. New Polyhedral Zinc Siloxanes: Synthesis and X-ray Crystal Structures of Zn8Me7(dioxane)2(O3SiR)3 and [Zn7Me2(THF)5(O3SiR)4][R = (2,6-i-Pr2C6H3)N(SiMe3)]. Organometallics 2004, 23, 2251–2256. [Google Scholar] [CrossRef]
- Chen, W.-T.; Zeng, X.-R.; Liu, D.-S.; Ying, S.-M.; Liu, J.-H. An Unprecedented 2D 4f-3d-5d Multimetal-Isonicotinic Acid Complex: Synthesis, Structural Characterization and Magnetic Properties. Chin. J. Chem. 2008, 26, 1678–1682. [Google Scholar] [CrossRef]
- Chen, W.-T.; Zeng, X.-R.; Liu, D.-S.; Ying, S.-M.; Liu, J.-H. A novel 2-D 5d-4f-3d trimetal-isonicotinic acid complex: Synthesis and characterisation. J. Chem. Res. 2009, 705–709. [Google Scholar] [CrossRef]
- Ellison, J.J.; Power, P.P. Synthesis and Characterization of New Thiolato Derivatives of Lithium, Magnesium, and Zinc: Examples of Two-Coordinate Lithium and Zinc Species Ligated by Sulfur. Inorg. Chem. 1994, 33, 4231–4234. [Google Scholar] [CrossRef]
- Nguyen, T.; Panda, A.; Olmstead, M.M.; Richards, A.F.; Stender, M.; Brynda, M.; Power, P.P. Synthesis and Characterization of Quasi-Two-Coordinate Transition Metal Dithiolates M(SAr*)2(M=Cr, Mn, Fe, Co, Ni, Zn; Ar*=C6H3-2,6(C6H2-2,4,6-Pri3)2. J. Am. Chem. Soc. 2005, 127, 8545–8552. [Google Scholar] [CrossRef] [PubMed]
- Ellison, J.J.; Ruhlandt-Senge, K.; Hope, H.H.; Power, P.P. Synthesis and Characterization of the New Selenolate Ligand -SeC6H3-2,6-Mes2 (Mes=C6H2-2,4,6-Me3) and Its Two-Coordinate Zinc and Manganese Derivatives: Factors Affecting Bending in Two-Coordinate Metal Complexes with Aryl-Substituted Ligands. Inorg. Chem. 1995, 34, 49–54. [Google Scholar] [CrossRef]
- Jun, Y.; Koo, J.-E.; Cheon, J. One-step synthesis of size tuned zinc selenide quantum dots via a temperature controlled molecular precursor approach. Chem. Commun. 2000, 1243–1244. [Google Scholar] [CrossRef]
- Kudelko, A.; Wróblowska, M. An efficient synthesis of conjugated 5-aryl-1,3,4-oxadiazoles from 3-heteroarylacrylohydrazides and acid chlorides. Tetrahedron Lett. 2014, 55, 3252–3254. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01 2009, Gaussian, Inc.: Wallingford, CT, USA.
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y. Water Accelerated Sm/TMSCl Reductive Cleavage of the Se-Se Bond: Synthesis of Selenoesters and Selenoformates. Synth. Commun. 1999, 29, 3107–3115. [Google Scholar] [CrossRef]
- Ren, K.; Wang, M.; Liu, P.; Wang, L. Iron-Catalyzed Synthesis of Selenoesters from Diselenides and Acyl Chlorides or Acid Anhydrides in the Presence of Magnesium Dust. Synthesis 2010, 2010, 1078–1082. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y. Reductive Cleavage of Se–Si Bond in Arylselenotrimethylsilane: A Novel Method for the Synthesis of Selenoesters. Synth. Commun. 1998, 28, 3999–4002. [Google Scholar] [CrossRef]
Sample Availability: Original samples (due to their instability) are not available but can be provided upon request. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Solvent | Additive | Time | T (°C) | Discoloration |
|---|---|---|---|---|---|
| 1 | H2O | none | 2 h | 70 | No |
| 2 | THF | none | 2 h | reflux | No |
| 3 | H2O | TFA 10 mol % | 1 h | 70 | No |
| 4 | THF | TFA 10 mol % | 20 min | reflux | Yes |
| 5 | H2O/THF | TFA 10 mol % | 20 min | 70 | Yes |

| Entry | Reagent | Medium | Time (h) | Yield (%) a | ae (%) b | Reference |
|---|---|---|---|---|---|---|
| 1 | PhSeZnCl | THF | 24 | 25 | 66 | [50] |
| 2 | PhSeZnBr | THF | 24 | 30 | 60 | [50] |
| 3 | [PhSeZnSePh] 1 | THF | 3 | 32 c | 79.6 | [56] |
| 4 | [PhSeZnSePh] 1 | THF | 3 | 40 d | 79.6 | – |
| 5 | PhSZnBr | THF | 24 | 86 | 54.6 | [57] |
| 6 | [PhSeZnSePh/PhSeH] | HClacq/Et2O | 4 | 38 | – | [56] |
| 7 | PhSeZnCl | H2O | 3 | 60 | 66 | [50] |
| 8 | PhSeZnBr | H2O | 3 | 70 | 60 | [50] |
| 9 | PhSZnBr | H2O | 3 | 65 | 54.6 | [57] |
| 10 | [PhSeZnSePh] 1 | H2O | 0.5 | 83 | 79.6 | – |
| 11 | [PhSeZnSePh]TMEDA 3 | H2O | 0.5 | 66 | 77 | – |
| 12 | [PhSZnSPh] 2 | H2O | 0.5 | 50 | 76 | – |

| Entry | Substrate 4 | Product 5 | Conv, % of 4 in 5 (Yield, %) of 5 Using 1 | Conv, % of 4 in 5 Using 3 |
|---|---|---|---|---|
| 1 |  |  | 84 (80) | 66 |
| 2 |  |  | 81 (53) | 80 |
| 3 |  |  | 71 (57) | 40 |
| 4 |  |  | 92 (90) | 80 |
| 5 |  |  | 83 (64) | 62 |
| 6 |  |  | 80 (65) | 35 |
| 7 |  |  | 8 | 76 |
| 8 |  |  | 75 (69) | 80 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sancineto, L.; Pinto Vargas, J.; Monti, B.; Arca, M.; Lippolis, V.; Perin, G.; Lenardao, E.J.; Santi, C. Atom Efficient Preparation of Zinc Selenates for the Synthesis of Selenol Esters under “On Water” Conditions. Molecules 2017, 22, 953. https://doi.org/10.3390/molecules22060953
Sancineto L, Pinto Vargas J, Monti B, Arca M, Lippolis V, Perin G, Lenardao EJ, Santi C. Atom Efficient Preparation of Zinc Selenates for the Synthesis of Selenol Esters under “On Water” Conditions. Molecules. 2017; 22(6):953. https://doi.org/10.3390/molecules22060953
Chicago/Turabian StyleSancineto, Luca, Jaqueline Pinto Vargas, Bonifacio Monti, Massimiliano Arca, Vito Lippolis, Gelson Perin, Eder Joao Lenardao, and Claudio Santi. 2017. "Atom Efficient Preparation of Zinc Selenates for the Synthesis of Selenol Esters under “On Water” Conditions" Molecules 22, no. 6: 953. https://doi.org/10.3390/molecules22060953
APA StyleSancineto, L., Pinto Vargas, J., Monti, B., Arca, M., Lippolis, V., Perin, G., Lenardao, E. J., & Santi, C. (2017). Atom Efficient Preparation of Zinc Selenates for the Synthesis of Selenol Esters under “On Water” Conditions. Molecules, 22(6), 953. https://doi.org/10.3390/molecules22060953