General
All chemical reagents were purchased from
J and K Chemical or Sigma-Aldrich Chemistry Co. (Darmstadt, Germany). Unless otherwise noted, all reactions were conducted under atmosphere. Thin-layer chromatography (TLC) analysis (Qingdao Haiyang Chemical Co., Ltd., Shandong, Qingdao, China) was used to monitor the reaction process. Column chromatography was carried out on silica gel (200–300 mesh). The products were eluted in an appropriate solvent mixture under air pressure. Concentration and evaporation of the solvent after reaction or extraction was carried out on a rotary evaporator. IR spectra were obtained on a Thermo Nicolet NEXUS-670 spectrometer (Thermo Nicolet Corporationcompany, Waltham, MA, USA) and recorded as KBr thin films and absorptions are reported in cm
−1. HRMS were obtained with a Bruker Daltonics APEX II 47e mass spectrometer. NMR spectra (Bruker Corporation, Billerica, MA, USA) were recorded on a Bruker-400 MHz spectrometer (Bruker Corporation, Billerica, MA, USA) in appropriate solvents. Chemical shifts (δ) were expressed in parts per million (ppm) relative to the tetramethylsilane. Multiplicities of NMR signals are designated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad), etc.
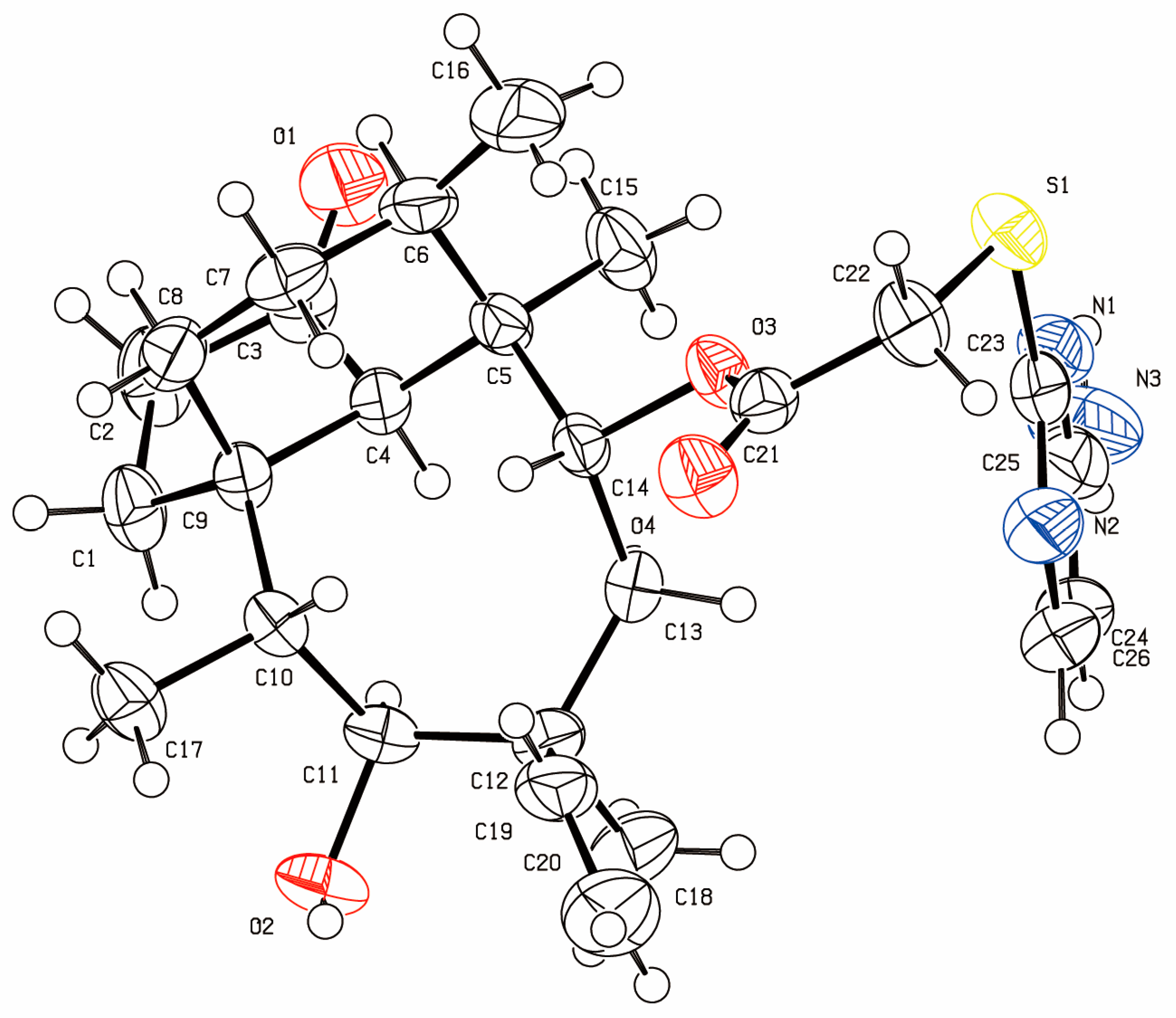
13C-NMR spectra were recorded on 100 MHz spectrometers. The single-crystal structure of the title compound was determined on a Bruker SMART APEX II X-diffractometer (Bruker Corporation, Billerica, MA, USA). All NMR, IR and HRMS datum were been added in
Supplementary Materials.
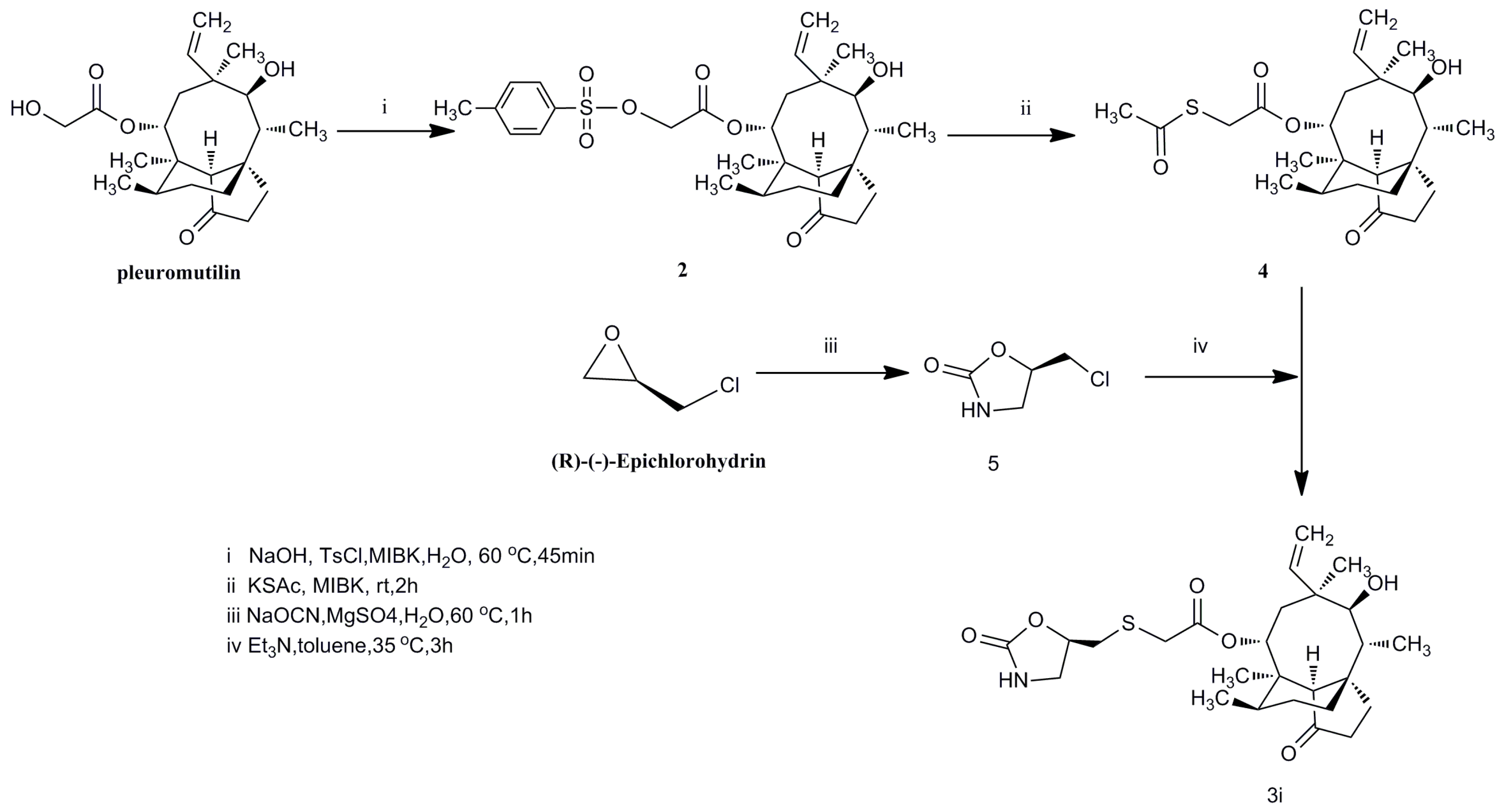
14-O-(p-Toluene sulfonyloxyacetyl)mutilin (2). A 5 mL of NaOH aqueous solution (2 g, 50 mmol) was added dropwise to a mixture of pleuromutilin (7.57 g, 20 mmol) and p-toluenesulfonyl chloride (4.2 g, 22 mmol) in methyl isobutyl ketone (10 mL) and water (5 mL). The mixture was vigorously stirred for 45 min at 60 °C, then the reaction mixture was cooled to 10 °C and separated. The organic layer was washed with 5 mL water and 5 mL saturated sodium carbonate solution. The organic phase was dried overnight with anhydrous sodium sulfate. After filteration, the solvent was concentrated in vacuo to give 10.56 g of yellow oil. It was used in the next step without further purification. Yield: 93%. IR (KBr): 3446 (OH), 2924 (CH2), 2863 (CH2), 1732 (C=O), 1633 (C-C), 1597 (C=C), 1456 (C=C), 1371 (CH3), 1297 (C-O-C), 1233 (CH), 1117 (C-(C=O)-C), 1035 (C-O-C), 832 (CH), 664 (CH2=), 560 (CH2=) cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.74 (d, J = 8.3 Hz, 2H), 7.26 (t, J = 13.3 Hz, 2H), 6.34 (dd, J = 17.4, 11.0 Hz, 1H), 5.70 (d, J = 8.5 Hz, 1H), 5.19 (dd, J = 55.1, 14.2 Hz, 2H), 4.47–4.33 (m, 2H), 3.28 (s, 1H), 2.38 (s, 3H), 2.25–2.09 (m, 3H), 2.01 (s, 1H), 1.99–1.88 (m, 1H), 1.71–1.62 (m, 1H), 1.61–1.51 (m, 2H), 1.46–1.38 (m, 2H), 1.35 (d, J = 7.8 Hz, 3H), 1.27 (d, J = 11.5 Hz, 1H), 1.18 (dd, J = 11.6, 4.5 Hz, 2H), 1.12–1.00 (m, 4H), 0.80 (d, J = 7.0 Hz, 3H), 0.55 (d, J = 7.0 Hz, 3H). 13C-NMR(100 MHz, CDCl3) δ215.71 (C=O), 163.87 (C=O), 144.29 (benzene-C), 137.70 (CH=), 131.63 (benzene-C), 128.91 (benzene-C), 127.09 (benzene-C), 116.38 (CH2=), 73.54 (CH), 69.29 (CH), 64.03 (CH), 57.02 (CH), 44.39 (C), 43.51 (CH2), 42.97 (C), 40.84 (C), 35.54 (CH), 35.03 (CH), 33.40 (CH2), 29.34 (CH3), 25.77(CH2), 25.39 (CH2), 23.81 (CH2), 20.68 (CH3), 15.53 (CH3), 13.76 (CH3), 10.47 (CH3). HRMS (ESI) calcd. [M + H]+ for C29H40O7S 533.250, found 533.2507.
14-O-(Acetic acidthioacetyl)mutilin (4). Compound 3 was prepared by stirring a mixing of compound 2 (10 mmol), potassium thioglycolate (20 mmol), and methyl isobutyl ketone (30 mL) in room temperature, and the mixture was stirred for 2 h. The mixture was extracted with water (10 mL). The organic phase were combined, dried over Na2SO4, and concentrated to give compound 3. Yield: 80%. IR (KBr): 3448 (OH), 2967 (CH2), 2924 (CH3), 2865 (CH2), 1731 (C=O), 1702 (C=O), 1453 (C-C), 1419, 1384 (CH3), 1295 (C-O-C), 1183 (CH), 1154 (C-O), 1115 (C-(C=O)-C), 1017 (C-O-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 6.58–6.30 (m, 1H), 5.72 (d, J = 8.4 Hz, 1H), 5.43–5.14 (m, 2H), 3.63 (s, 2H), 3.36 (d, J = 6.4 Hz, 1H), 2.36 (dd, J = 13.5, 3.5 Hz, 3H), 2.35–2.27 (m, 1H), 2.21 (dd, J = 17.4, 7.9 Hz, 1H), 2.13 (d, J = 14.9 Hz, 1H), 2.04 (dd, J = 26.1, 17.5 Hz, 1H), 1.73 (dd, J = 31.8, 10.6 Hz, 2H), 1.68–1.62 (m, 2H), 1.62–1.41 (m, 6H), 1.41–1.24 (m, 2H), 1.24–0.99 (m, 4H), 0.89 (t, J = 8.4 Hz, 3H), 0.78–0.61 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ 216.90 (C=O), 193.45 (C=O), 167.32 (C=O), 138.92 (CH=), 117.19 (CH2=), 74.60 (CH), 70.09 (CH), 58.13 (CH), 45.45 (C), 44.71 (CH2), 44.01 (C), 41.89 (C), 36.74 (CH), 36.00 (CH3), 34.45 (CH2), 32.20 (CH2), 30.42 (CH2), 30.06 (CH2), 26.84 (CH2), 26.41 (CH3), 24.82 (CH2), 16.74 (CH3), 14.81(CH3), 11.43 (CH3). HRMS (ESI) calcd. [M + H]+ for C24H36O5S 437.2356, found 437.2339.
(R)-5-ChloroMethyl-2-oxazolidinone (5). The title compound was prepared by stirring a mixing of magnesium sulphate (10 mmol), sodium cyanate (10 mmol) and water (50 mL) in room temperature. (R)-(−)-Epichlorohydrin to the solution (5 mmol) was added dropwise to the mixture. The result mixture was stirred under 60 °C for 1 h. The reaction mixture was concentrated in vacuo and extracted by ethyl acetate. The two layers were separated, and the organic layer was dried over Na2SO4, filtered, and concentrated to dryness. Yield: 57%. IR (KBr): 3365 (NH), 1744 (C=O), 1429 (C-N), 1240 (C-C(=O)-O), 736 (C-Cl) cm−1. 1H-NMR (400 MHz, DMSO) δ 7.60 (s, 1H), 4.84 (dd, J = 9.6, 4.7 Hz, 1H), 4.04–3.73 (m, 2H), 3.59 (t, J = 9.0 Hz, 1H), 3.25 (dd, J = 9.0, 6.2 Hz, 1H). 13C-NMR (101 MHz, d6-DMSO) δ 158.68 (C=O), 74.35 (CH), 46.66 (CH2), 43.03(CH2). HRMS (ESI) calcd. [M + H]+ for C4H6ClNO 136.0159, found 136.0150.
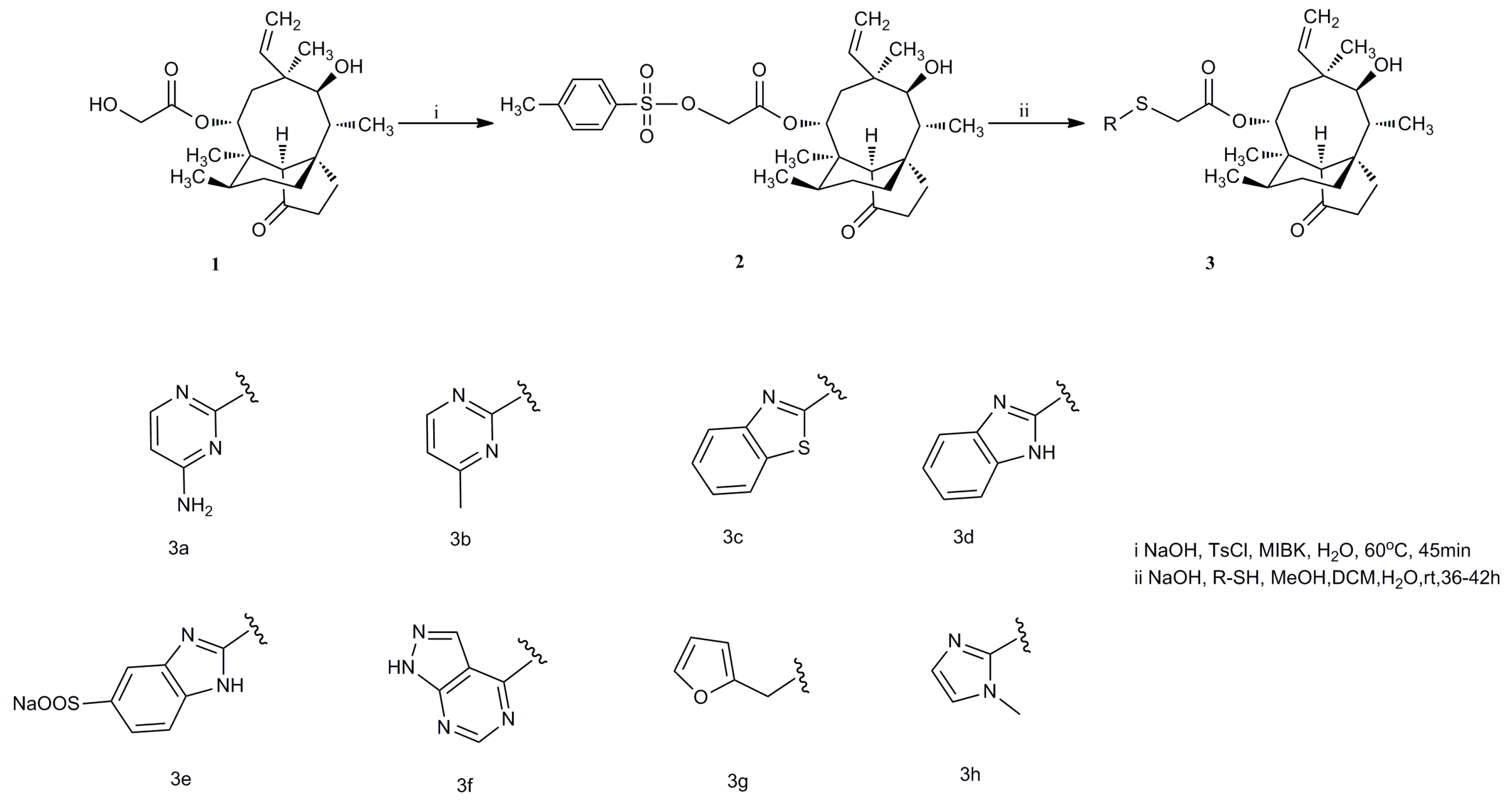
General Procedure for Synthesis of Compounds 3a–3g. A mixing of thiols (1 mmol), sodium hydroxide (1.1 mmol), water (0.5 mL) and methanol (3 mL) were stirred in room temperature. After 30 min, compound 2 (1.1 mmol) in 5 mL CH2Cl2 was added dropwise to the mixture for 36 h–42 h. The mixture was concentrated in vacuo. The residue was dissolved by CH2Cl2. The solution was extracted three times with water. The organic phase was dried overnight with anhydrous sodium sulfate. The solvent was concentrated in vacuo to give crude products. The crude product was purified by silica gel column chromatography.
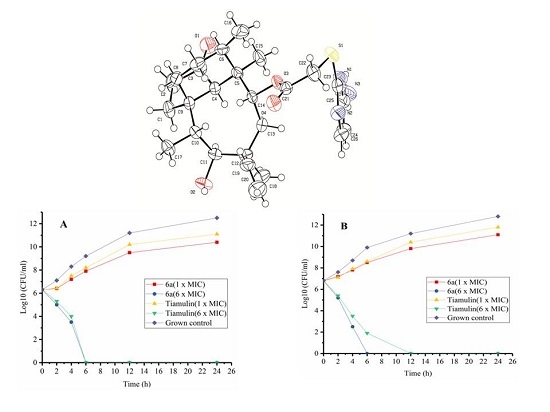
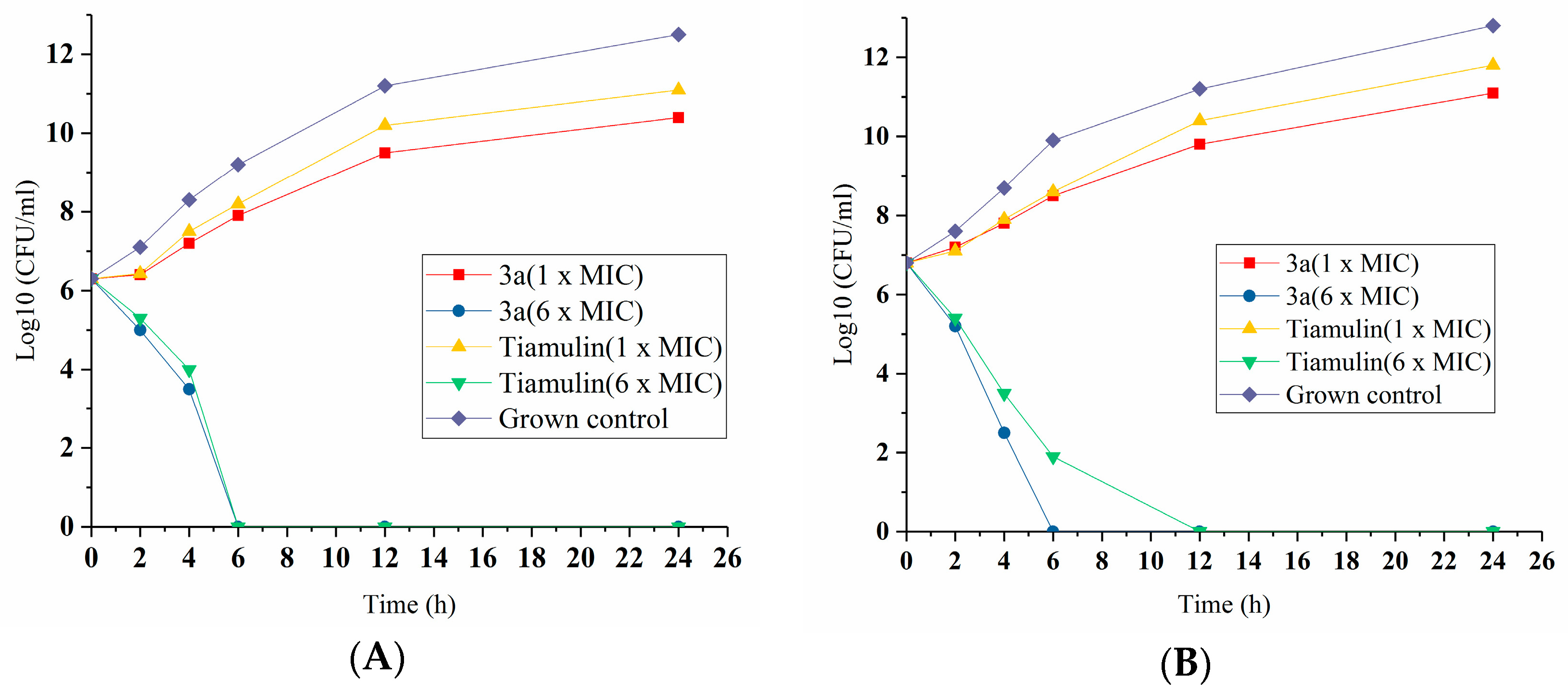
14-O-[(4-Amino-pyrimidinone-2-yl)thioacetyl]mutilin (3a). Compound 3a was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilinmutilin (2) and 4-amino-2-Pyrimidinone. The crude product was purified over silica gel column chromatography to give 4.09 g. Yield: 84%. IR (KBr): 3448 (OH), 2933 (CH2), 1730 (C=O), 1629 (C-C), 1583 (C=N), 1543 (C=C), 1467 (C=C), 1372 (CH3), 1249(C-C(=O)-O), 1153(C-O), 1117 (C-(C=O)-C), 1018 (C-O-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.89 (d, J = 5.1 Hz, 1H), 6.42 (dd, J = 17.1, 11.2 Hz, 1H), 6.04 (d, J = 5.2 Hz, 1H), 5.68 (d, J = 7.9 Hz, 1H), 5.17 (dd, J = 51.9, 14.0 Hz, 2H), 4.94 (s, 2H), 3.72 (dd, J = 32.8, 16.1 Hz, 2H), 3.28 (s, 1H), 2.24 (d, J = 6.5 Hz, 1H), 2.14 (dd, J = 14.8, 9.1 Hz, 2H), 2.02 (s, 1H), 1.94 (dd, J = 15.6, 8.5 Hz, 1H), 1.69 (d, J = 13.7 Hz, 1H), 1.61–1.52 (m, 2H), 1.48 (d, J = 12.0 Hz, 2H), 1.37 (s, 4H), 1.32–1.22 (m, 2H), 1.07 (s, 4H), 0.79 (d, J = 6.2 Hz, 3H), 0.68 (d, J = 6.2 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 216.11 (C=O), 168.72 (pyrimidine-C), 167.20 (C=O), 161.35 (pyrimidine-C), 154.88 (pyrimidine-C), 138.26 (CH=), 116.02 (CH2=), 100.18 (pyrimidine-C), 73.60 (CH), 68.51(CH), 57.36 (CH), 57.21(CH2), 44.46 (CH2), 43.47 (C), 42.93 (C), 40.87 (CH), 35.81 (CH), 35.02 (CH), 33.48 (CH2), 33.06 (CH2), 29.45(CH2), 25.89 (CH3), 23.84 (CH2), 15.74 (CH3), 13.92 (CH3), 10.44 (CH3). HRMS (ESI) calcd. [M + H]+ for C26H37N3O4S 488.2578, found 488.2570.
14-O-[(4-Methylpyrimidine-2-yl)thioacetyl]mutilin (3b). Compound 3b was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin(2) and 4-methy-2-pyrimidinone. The crude product was purified over silica gel column chromatography to give 3.55 g. Yield: 73%. IR (KBr): 3439 (OH), 2935 (CH2), 1733 (C=O), 1658 (C-C), 1580 (C=N), 1535 (C=C), 1458 (C=C), 1396 (CH3), 1285 (C-O-C), 1118 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 6.37 (dt, J = 33.7, 16.8 Hz, 1H), 5.99 (s, 1H), 5.69 (d, J = 8.4 Hz, 1H), 5.19 (dd, J = 56.5, 14.2 Hz, 2H), 3.88–3.73 (m, 2H), 3.29 (d, J = 5.6 Hz, 1H), 2.27–2.15 (m, 2H), 2.11 (d, J = 13.1 Hz, 3H), 2.02 (d, J = 6.4 Hz, 1H), 1.96 (d, J = 10.9 Hz, 1H), 1.69 (d, J = 14.2 Hz, 1H), 1.58 (dd, J = 21.0, 10.8 Hz, 2H), 1.49 (dd, J = 26.7, 13.3 Hz, 2H), 1.43–1.26 (m, 6H), 1.20 (dd, J = 17.5, 11.2 Hz, 2H), 1.05 (d, J = 20.9 Hz, 4H), 0.80 (d, J = 6.9 Hz, 3H), 0.67 (d, J = 6.9 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 215.94 (C=O), 165.80(C=O), 164.69 (pyrimidine-C), 164.11 (pyrimidine-C), 157.85 (pyrimidine-C), 137.90 (CH=), 116.27 (CH2=), 107.63 (pyrimidine-C), 73.55 (CH), 69.14 (CH), 57.08 (CH), 44.43 (C), 43.49 (CH2), 42.94 (C), 40.87 (C), 35.70 (CH), 35.00 (CH3), 33.44 (CH2), 32.23 (CH2), 29.39 (CH2), 25.84(CH3), 25.36(CH2), 23.82 (CH2), 23.09 (CH2), 15.84(CH3), 13.84 (CH3), 10.46 (CH3). HRMS (ESI) calcd. [M + H]+ for C27H38N2O4S 487.2625, found 487.2623.
14-O-[(Benzimidazole-2-yl)thioacetyl]mutilin (3c). Compound 3c was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and 2-mercaptobenzothiazole. The crude product was purified over silica gel column chromatography to give 3.54 g. Yield: 67%. IR (KBr): 3442 (OH), 2929 (CH2), 1731 (C=O), 1458 (C=C), 1429 (C-C), 1274 (C-O-C), 1153 (C-O), 1117 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.77 (dd, J = 19.9, 8.0 Hz, 2H), 7.39 (t, J = 7.7 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 6.42 (dd, J = 17.4, 11.0 Hz, 1H), 5.76 (d, J = 8.5 Hz, 1H), 5.19 (dd, J = 57.6, 14.2 Hz, 2H), 4.08 (dd, J = 41.4, 16.2 Hz, 2H), 3.31 (d, J = 6.4 Hz, 1H), 2.29 (dd, J = 14.1, 7.2 Hz, 1H), 2.20 (dd, J = 11.6, 6.3 Hz, 1H), 2.06 (s, 1H), 1.98 (dd, J = 16.0, 8.5 Hz, 1H), 1.82–1.64 (m, 2H), 1.64–1.51 (m, 2H), 1.50–1.33 (m, 6H), 1.24 (dd, J = 14.3, 7.4 Hz, 2H), 1.13–0.97 (m, 4H), 0.85 (d, J = 7.0 Hz, 3H), 0.77 (d, J = 6.9 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 215.94 (C=O), 165.84 (C=O), 163.54 (benzothiazole-C), 151.74 (benzothiazole-C), 137.76 (CH=), 134.49(benzothiazole-C), 125.02 (benzothiazole-C), 123.46 (benzothiazole-C), 120.68 (benzothiazole-C), 120.05 (benzothiazole-C), 116.21 (CH2=), 73.57 (CH), 69.20 (CH), 57.10 (CH), 44.42 (C), 43.41 (CH2), 42.90 (C), 40.86 (C), 35.74 (CH), 34.98 (CH), 34.67 (CH2), 33.43(CH2), 29.40 (CH2), 25.84 (CH2), 25.28 (CH3), 23.81 (CH2), 15.80(CH3), 13.81 (CH3), 10.44 (CH3). HRMS (ESI) calcd. [M + H]+ for C29H37NO4S 528.2237, found 528.2234.
14-O-[(Benzothiazole-2-yl)thioacetyl]mutilin (3d). Compound 3d was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and 2-mercaptobenzimidazole. The crude product was purified over silica gel column chromatography to give 3.68 g. Yield 72%. IR (KBr): 3423 (OH), 2925 (CH2), 1726 (C=O), 1458 (C=C), 1439 (C-C), 1271 (C-O-C), 1152 (C-O), 1117 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.50 (dd, J = 5.8, 3.1 Hz, 2H), 7.20 (dd, J = 6.0, 3.2 Hz, 2H), 6.41 (dd, J = 17.4, 11.0 Hz, 1H), 5.79 (d, J = 8.4 Hz, 1H), 5.18 (dd, J = 43.7, 14.2 Hz, 2H), 3.91 (s, 2H), 3.73 (q, J = 7.0 Hz, 1H), 3.35 (d, J = 6.4 Hz, 1H), 2.35–2.28 (m, 1H), 2.27–2.17 (m, 1H), 2.08 (s, 1H), 2.03 (dd, J = 16.1, 8.6 Hz, 1H), 1.82–1.65 (m, 2H), 1.64–1.52 (m, 2H), 1.49–1.32 (m, 6H), 1.24 (t, J = 7.0 Hz, 2H), 1.16–1.05 (m, 4H), 0.88 (d, J = 6.9 Hz, 3H), 0.72 (d, J = 7.0 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 215.89 (C=O), 167.78 (C=O), 147.20 (benzimidazole-C), 137.75 (CH=), 121.68 (benzimidazole-C), 116.28 (CH2=), 73.61 (CH), 69.82 (CH), 57.43 (CH), 57.09 (benzimidazole-C), 44.42 (C), 43.52 (CH2), 42.97 (C), 40.84 (C), 35.68 (CH), 35.04 (CH2), 34.16 (CH2), 33.42, 29.38 (CH2), 25.84 (CH2), 25.44 (CH3), 23.83 (CH2), 17.42 (CH2), 15.80 (CH3), 13.80 (CH3), 10.50 (CH3). HRMS (ESI) calcd. [M + H]+ for C29H38N2O4S 511.2526, found 511.2531.
14-O-[(5-Benzimidazolesulfonate-2-yl)thioacetyl]mutilin (3e). Compound 3e was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and sodium 2-mercapto-5-benzimidazolesulfonate dihydrate. The crude product was purified over silica gel column chromatography to give 3.2 g. Yield: 54.2%. IR (KBr): 3448 (OH), 2940 (CH2), 1732 (C=O) 1298 (C-O-C), 1190 (S=O), 1178 (S=O) cm−1. 1H-NMR (400 MHz, DMSO) δ 7.80 (s, 1H), 7.69 (d, J = 8.5 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 6.04 (dd, J = 17.8, 11.2 Hz, 1H), 5.50 (d, J = 8.1 Hz, 1H), 4.96 (dd, J = 38.2, 14.5 Hz, 2H), 4.38 (d, J = 4.4 Hz, 1H), 3.36 (d, J = 5.5 Hz, 1H), 2.51 (s, 1H), 2.35 (s, 1H), 2.13 (dd, J = 21.0, 10.7 Hz, 1H), 2.08–1.96 (m, 2H), 1.90 (dd, J = 16.0, 8.1 Hz, 1H), 1.60 (s, 2H), 1.44 (d, J = 6.9 Hz, 1H), 1.33 (d, J = 7.6 Hz, 2H), 1.27–1.12 (m, 6H), 1.10–1.02 (m, 1H), 1.02–0.90 (m, 4H), 0.78 (d, J = 6.8 Hz, 3H), 0.58 (t, J = 9.3 Hz, 3H). 13C-NMR (101 MHz, DMSO) δ 217.48 (C=O), 166.34 (C=O), 150.52 (benzimidazole-C), 145.67 (benzimidazole-C), 141.16 (benzimidazole-C), 133.41 (CH=), 128.59 (benzimidazole-C), 125.97 (benzimidazole-C), 123.22 (benzimidazole-C), 115.68 (CH2=), 113.22 (CH2), 110.71 (benzimidazole-C), 72.92 (CH), 71.44 (CH), 57.42 (CH), 45.35 (C), 44.58 (CH2), 41.95 (C), 36.89 (CH), 36.64 (CH), 35.03 (CH2), 34.40 (CH2), 30.53 (CH2), 29.06 (CH2), 26.99 (CH2), 24.85 (CH2), 16.47 (CH3), 14.64 (CH3), 11.96 (CH3). HRMS (ESI) calcd. [M + H]+ for C29H38N2O7S2 591.2193, found 591.2192.
14-O-[(Pyryrazolo[3,4d]pyrimidine-4-yl)thioacetyl]mutilin (3f). Compound 3f was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and 4-mercaptopyrazolo[3,4-d]pyrimidine. The crude product was purified over silica gel column chromatography to give 4.30 g. Yield: 84%. IR (KBr): 3431 (OH), 2934 (CH2), 1732 (C=O), 1567 (C=C), 1456 (C=C), 1406 (C-C), 1271 (C-O-C), 1152 (C-O), 1117 (C-(C=O)-C), 981 (CH) cm-1. 1H-NMR (400 MHz, CDCl3) δ 8.65 (d, J = 37.7 Hz, 1H), 8.29–8.08 (m, 1H), 6.43 (dt, J = 21.8, 10.9 Hz, 1H), 5.80 (d, J = 8.4 Hz, 1H), 5.38–5.29 (m, 1H), 5.24 (dd, J = 33.0, 14.5 Hz, 2H), 4.18–4.02 (m, 2H), 3.38 (d, J = 6.3 Hz, 1H), 2.31 (dd, J = 13.9, 7.1 Hz, 1H), 2.22 (dd, J = 13.0, 7.5 Hz, 1H), 2.11 (t, J = 8.4 Hz, 1H), 2.04 (d, J = 7.9 Hz, 1H), 1.73 (dd, J = 31.9, 9.2 Hz, 2H), 1.65 (dd, J = 17.1, 7.1 Hz, 2H), 1.58–1.37 (m, 6H), 1.36–1.22 (m, 2H), 1.19–1.06 (m, 4H), 0.88 (t, J = 11.3 Hz, 3H), 0.79 (t, J = 12.6 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 216.06 (C=O), 166.18 (C=O), 162.56 (pyrimidine-C), 153.05 (pyrimidine-C), 151.46 (pyrimidine-C), 137.88 (CH=), 131.82 (pyrazolo-C), 116.28 (CH2=), 110.65 (pyrimidine-C), 73.59 (CH), 69.22 (CH), 57.14 (CH), 44.46 (C), 43.57 (CH2), 42.92 (C), 40.88 (C), 35.75 (CH), 35.01 (CH2), 33.46 (CH2), 31.11 (CH2), 29.41 (CH2), 25.44 (CH3), 23.83 (CH2), 17.40(CH2), 15.74 (CH3), 13.86 (CH3), 10.47 (CH3). HRMS (ESI) calcd. [M + H]+ for C27H36N4O4S 513.2530, found 513.2527.
14-O-[(Furfuryl-2-yl)thioacetyl]mutilin (3g). Compound 3g was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and furfuryl mercaptan. The crude product was purified over silica gel column chromatography to give 3.53 g. Yield: 74%. IR (KBr): 3547 (OH), 2933 (CH2), 2882 (CH2), 1731 (C=O), 1455 (C=C), 1281 (C-O-C), 1150 (C-O), 1115 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.29 (s, 1H), 6.42 (dd, J = 17.4, 11.0 Hz, 1H), 6.31–6.06 (m, 2H), 5.71 (d, J = 8.4 Hz, 1H), 5.23 (dd, J = 57.0, 14.2 Hz, 2H), 3.75 (s, 2H), 3.30 (s, 1H), 3.02 (s, 2H), 2.29 (dd, J = 13.7, 6.8 Hz, 1H), 2.15 (dt, J = 19.6, 8.8 Hz, 2H), 2.09–1.98 (m, 2H), 1.75–1.66 (m, 1H), 1.64–1.55 (m, 2H), 1.53–1.35 (m, 6H), 1.30 (t, J = 14.9 Hz, 2H), 1.17–1.05 (m, 4H), 0.82 (d, J = 7.0 Hz, 3H), 0.68 (d, J = 6.8 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 215.99 (C=O), 167.67 (C=O), 149.27 (furan-C), 141.51(furan-C), 138.12 (CH=), 116.19 (CH2=), 109.37 (furan-C), 107.39 (furan-C), 73.65 (CH), 68.31 (CH), 57.21 (CH), 44.46 (C), 43.85 (CH2), 42.94 (C), 40.77 (C), 35.78 (CH), 35.04 (CH), 33.45 (CH2), 32.11 (CH2), 29.44 (CH2), 27.36 (CH2), 25.85 (CH3), 25.42 (CH2), 23.86 (CH2), 15.81 (CH3), 13.92 (CH3), 10.49 (CH3). HRMS (ESI) calcd. [M + H]+ for C27H38O5S 475.2513, found 475.2522.
14-O-[(1-Methylimidazole-2-yl)thioacetyl]mutilin (3h). Compound 3h was prepared according to the general procedure from 14-O-(p-toluene sulfonyloxyacetyl) mutilin (2) and 2-mercapto-1-methylimidazole. The crude product was purified over silica gel column chromatography to give 3.46 g. Yield: 73%. IR (KBr): 3423 (OH), 2924 (CH2), 2863 (CH2), 1717 (C=O), 1455 (C=C), 1410 (C-C), 1280 (C-O-C), 1145 (C-O), 1117 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 6.88 (d, J = 49.1 Hz, 2H), 6.35 (dd, J = 17.4, 11.0 Hz, 1H), 5.63 (d, J = 8.5 Hz, 1H), 5.32–5.01 (m, 2H), 3.92–3.62 (m, 2H), 3.56 (d, J = 11.0 Hz, 3H), 3.27 (s, 1H), 2.22 (dt, J = 13.8, 7.0 Hz, 1H), 2.19–2.07 (m, 2H), 2.03 (d, J = 21.6 Hz, 1H), 1.93 (dd, J = 16.0, 8.6 Hz, 1H), 1.77–1.61 (m, 1H), 1.58–1.46 (m, 3H), 1.45–1.35 (m, 2H), 1.31 (d, J = 14.3 Hz, 3H), 1.26–1.13 (m, 2H), 1.12–0.97 (m, 4H), 0.80 (d, J = 7.0 Hz, 3H), 0.59 (d, J = 6.9 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 215.93 (C=O), 166.75 (C=O), 139.06 (imidazole-C), 138.01 (CH=), 128.55 (imidazole-C), 121.39 (imidazole-C), 116.07 (CH2=), 73.57 (CH), 68.77 (CH), 57.12 (CH), 44.42 (C), 43.48 (CH2), 42.94 (C), 40.77 (C), 36.08 (CH), 35.70 (CH2), 35.00 (CH3), 33.44 (CH2), 32.35 (CH2), 29.40 (CH2), 25.83 (CH2), 25.41(CH3), 23.82 (CH2), 15.61 (CH3), 13.79 (CH3), 10.44 (CH3). HRMS (ESI) calcd. [M + H]+ for C26H38N2O4S 475.2625, found 475.2630.
14-O-(2-oxazolidinone,5-(methyl)-)(thioacetyl)mutilin (3i). A mixing of (R)-5-chloroMethyl-2-oxazolidinone (5) (1 mmol), sodiumiodide (0.1 mmol), and acetone (10 mL) were stirred in room temperature. After 30 min, the reaction solution was filtered and concentrated. Compound 4 (1.1 mmol) and triethylamine (20 mL) was added under N2. The solvent was stirred in 40 °C for 10 h. The mixture was extracted with water (10ml) and HCl (2N, 10 mL). The organic layers were concentrated in vacuo to give crude products. The crude product was purified by silica gel column chromatography. Yield: 62%. IR (KBr): 3422 (O), 2933 (CH2), 1735 (C=O), 1686 (C=O), 1458 (C=C), 1420 (C-C), 1284 (C-O-C), 1151 (C-O), 1117 (C-(C=O)-C) cm−1. 1H-NMR (400 MHz, CDCl3) δ 6.63–6.35 (m, 1H), 6.17 (d, J = 8.2 Hz, 1H), 5.75 (d, J = 8.2 Hz, 1H), 5.27 (dd, J = 51.9, 14.2 Hz, 2H), 4.92–4.67 (m, 1H), 3.82–3.62 (m, 1H), 3.45–3.32 (m, 2H), 3.30–3.07 (m, 2H), 2.99 (ddd, J = 9.9, 8.9, 4.4 Hz, 1H), 2.87 (dt, J = 12.8, 5.2 Hz, 1H), 2.34 (s, 1H), 2.28–2.17 (m, 2H), 2.11 (s, 1H), 2.08 (d, J = 8.6 Hz, 1H), 1.77 (d, J = 14.3 Hz, 1H), 1.65 (d, J = 10.4 Hz, 2H), 1.52 (dd, J = 25.3, 6.7 Hz, 2H), 1.44 (d, J = 1.0 Hz, 4H), 1.39 (s, 1H), 1.35–1.26 (m, 1H), 1.16 (d, J = 14.9 Hz, 4H), 0.89 (d, J = 6.8 Hz, 3H), 0.73 (d, J = 6.8 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 216.99 (C=O), 168.63 (C=O), 159.41 (C=O), 139.16 (CH=), 117.14 (CH2=), 75.66 (CH), 74.61 (C), 69.70 (CH), 58.17 (CH), 45.46 (C), 45.06 (CH2), 44.88 (CH2), 43.95 (C), 41.78 (C), 36.75 (CH), 36.03 (CH), 35.99, 34.81 (CH), 34.45 (CH2), 30.42 (CH2), 26.86 (CH3), 26.41 (CH2), 24.84 (CH2), 16.82 (CH3), 14.88 (CH3), 11.48 (CH3). HRMS (ESI) calcd. [M + Na]+ for C26H39NO6S 516.2395, found 516.2394.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}