Efficiency of Dinucleosides as the Backbone to Pre-Organize Multi-Porphyrins and Enhance Their Stability as Sandwich Type Complexes with DABCO


 ,
, 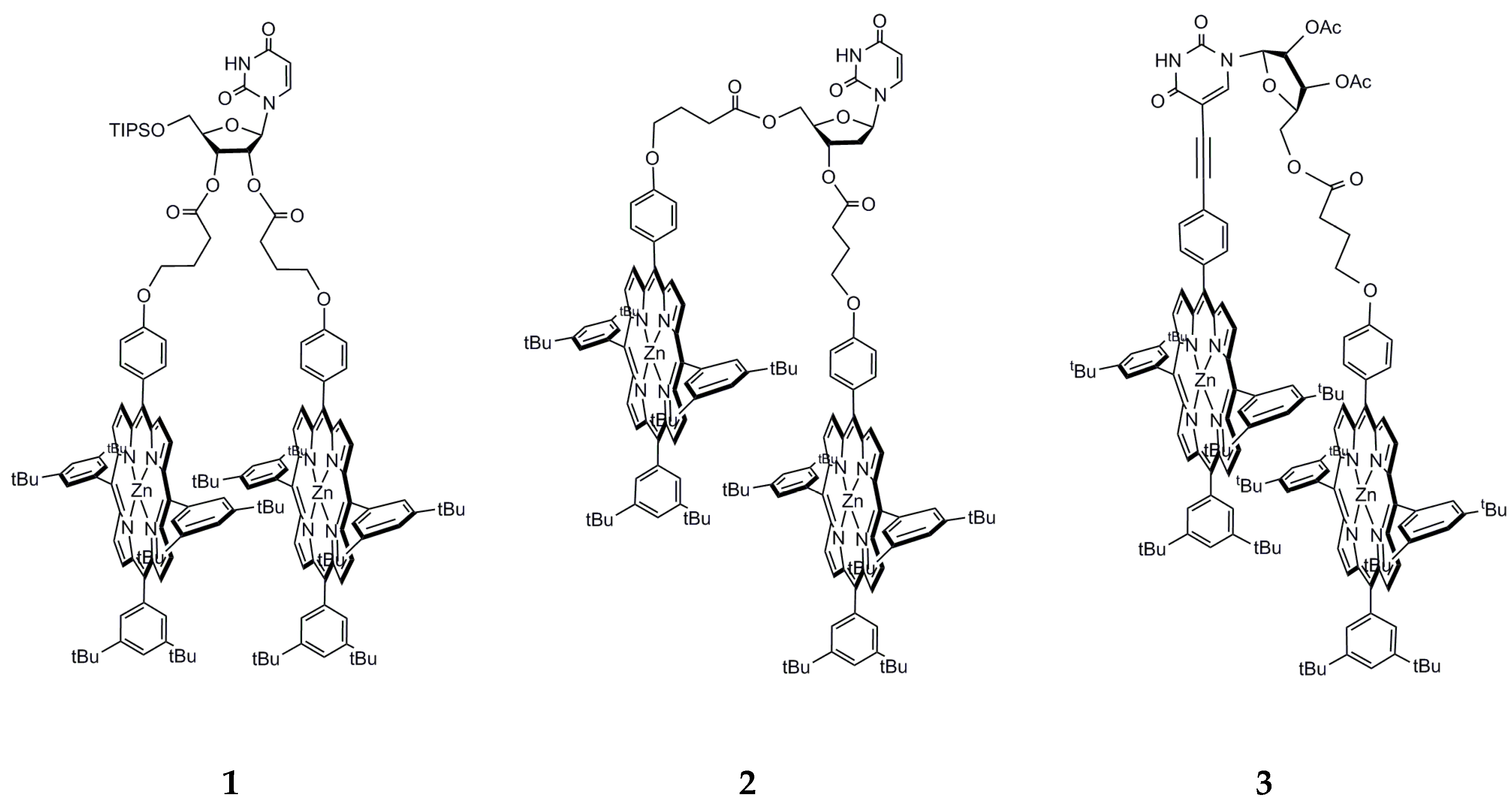{kind=link}
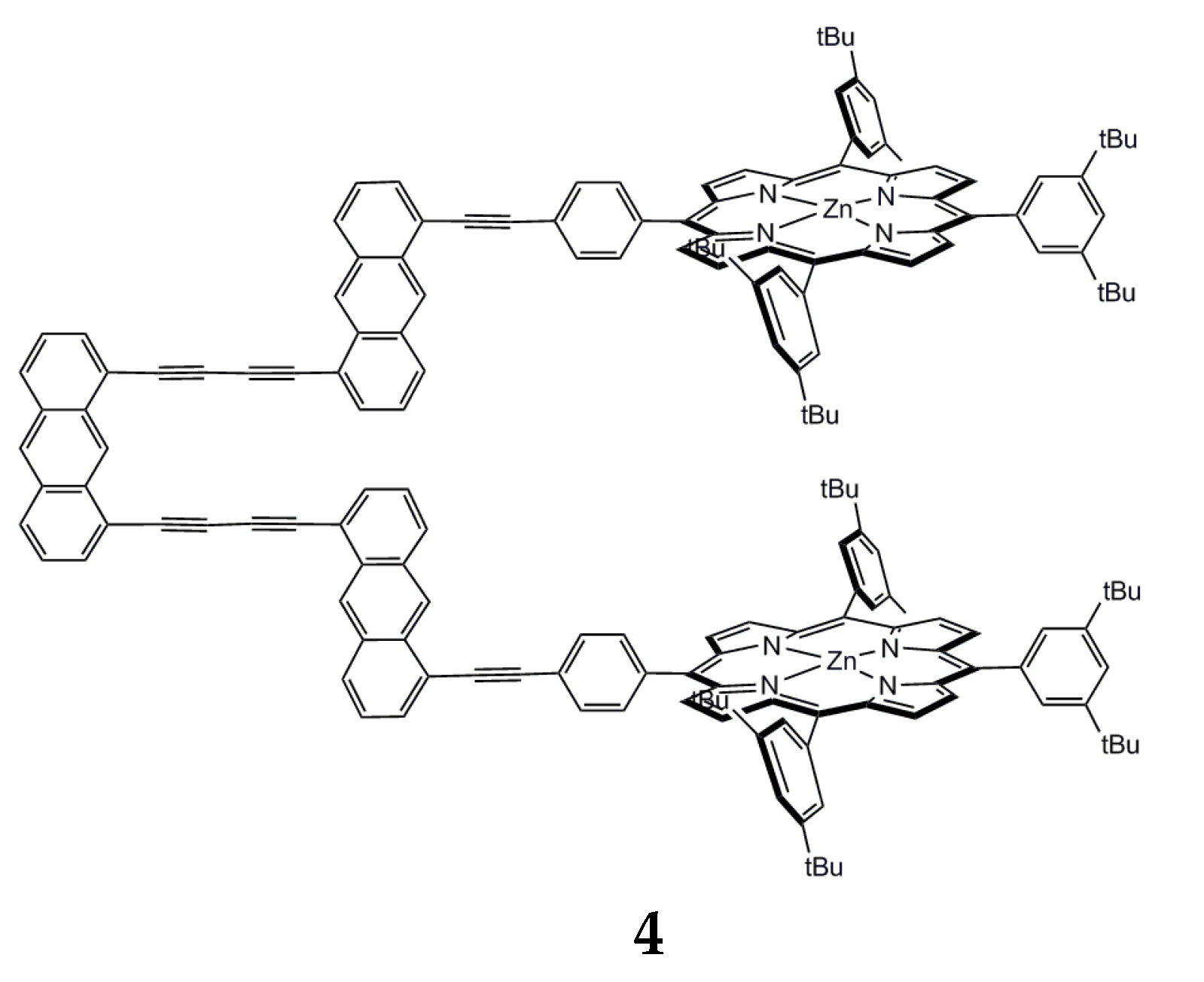{kind=link}
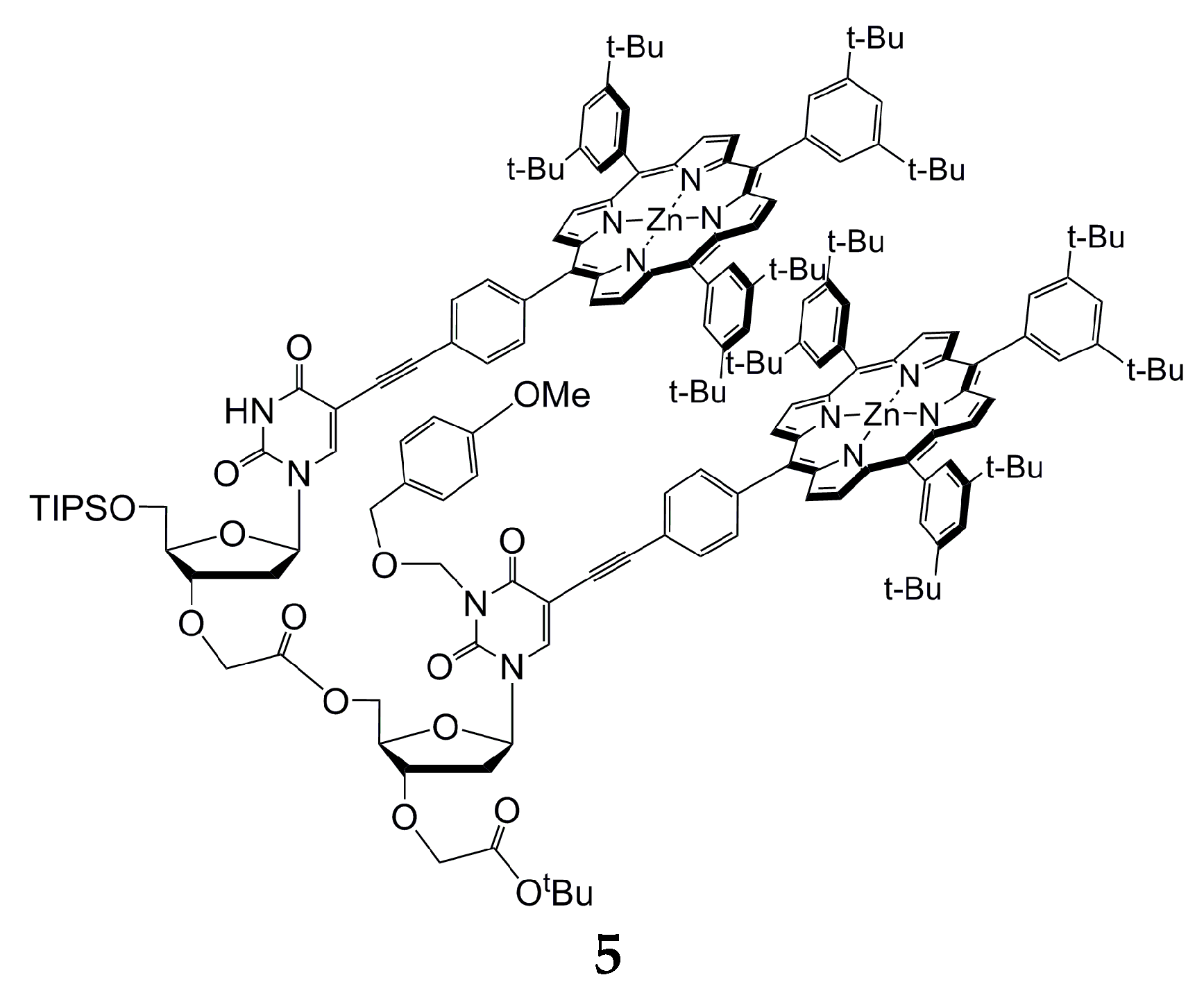{kind=link}
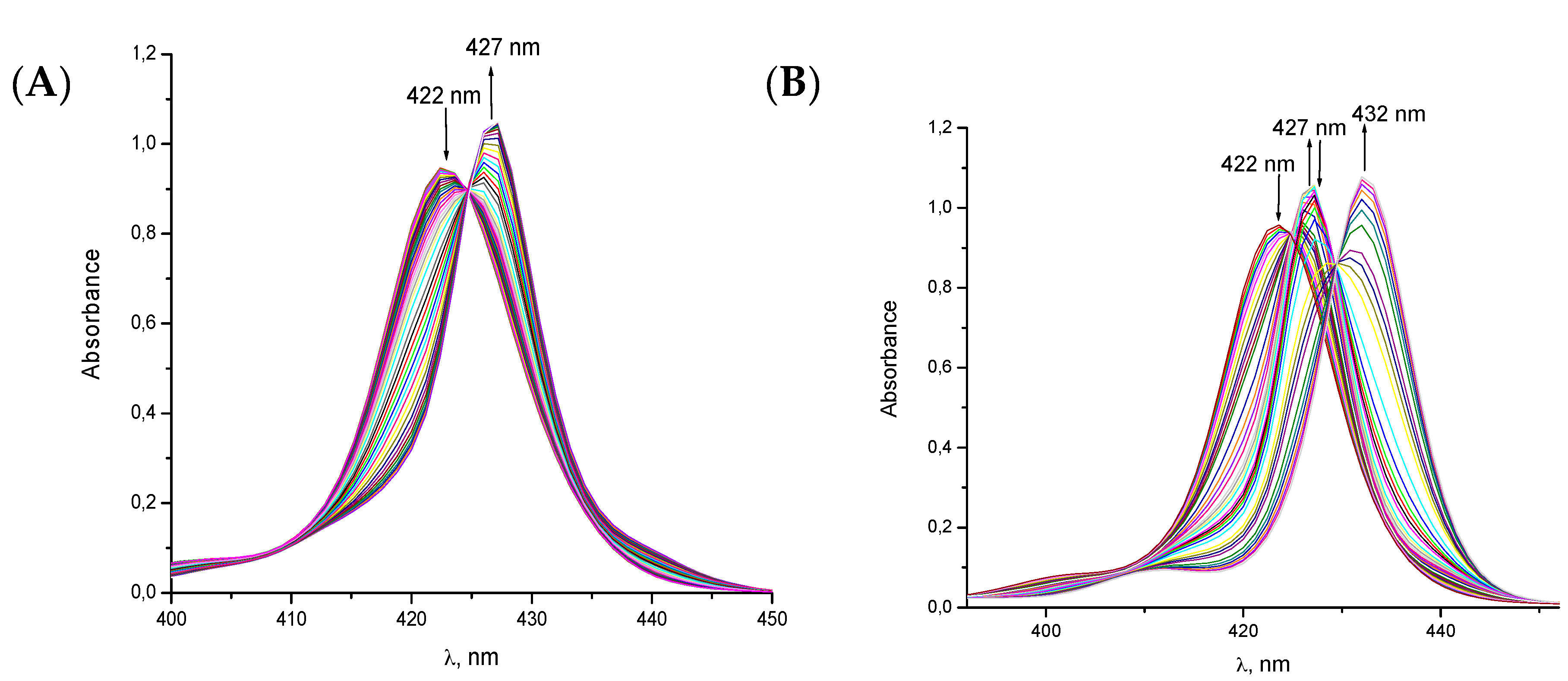{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
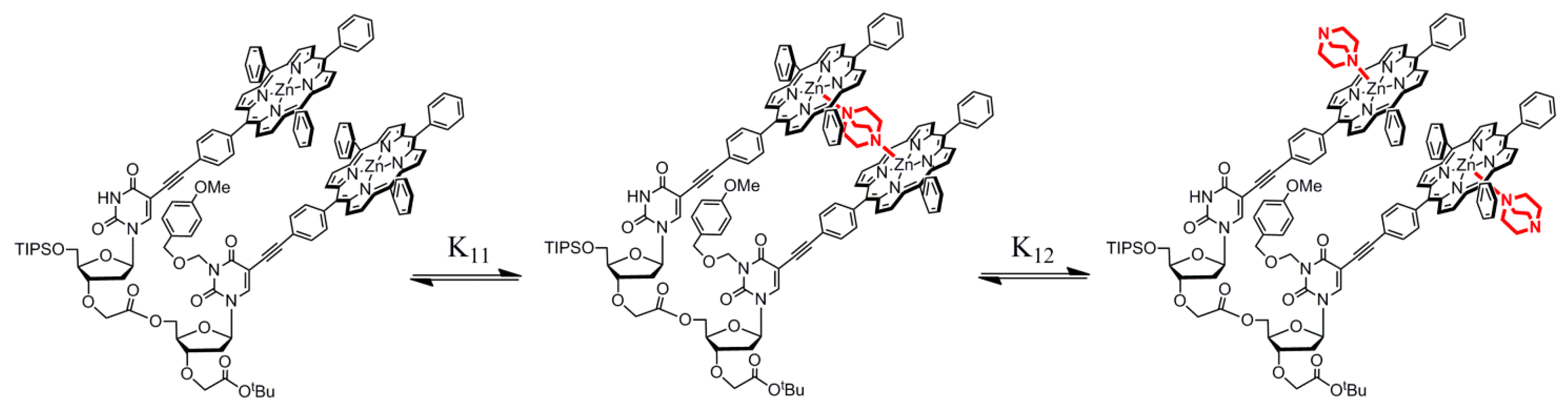
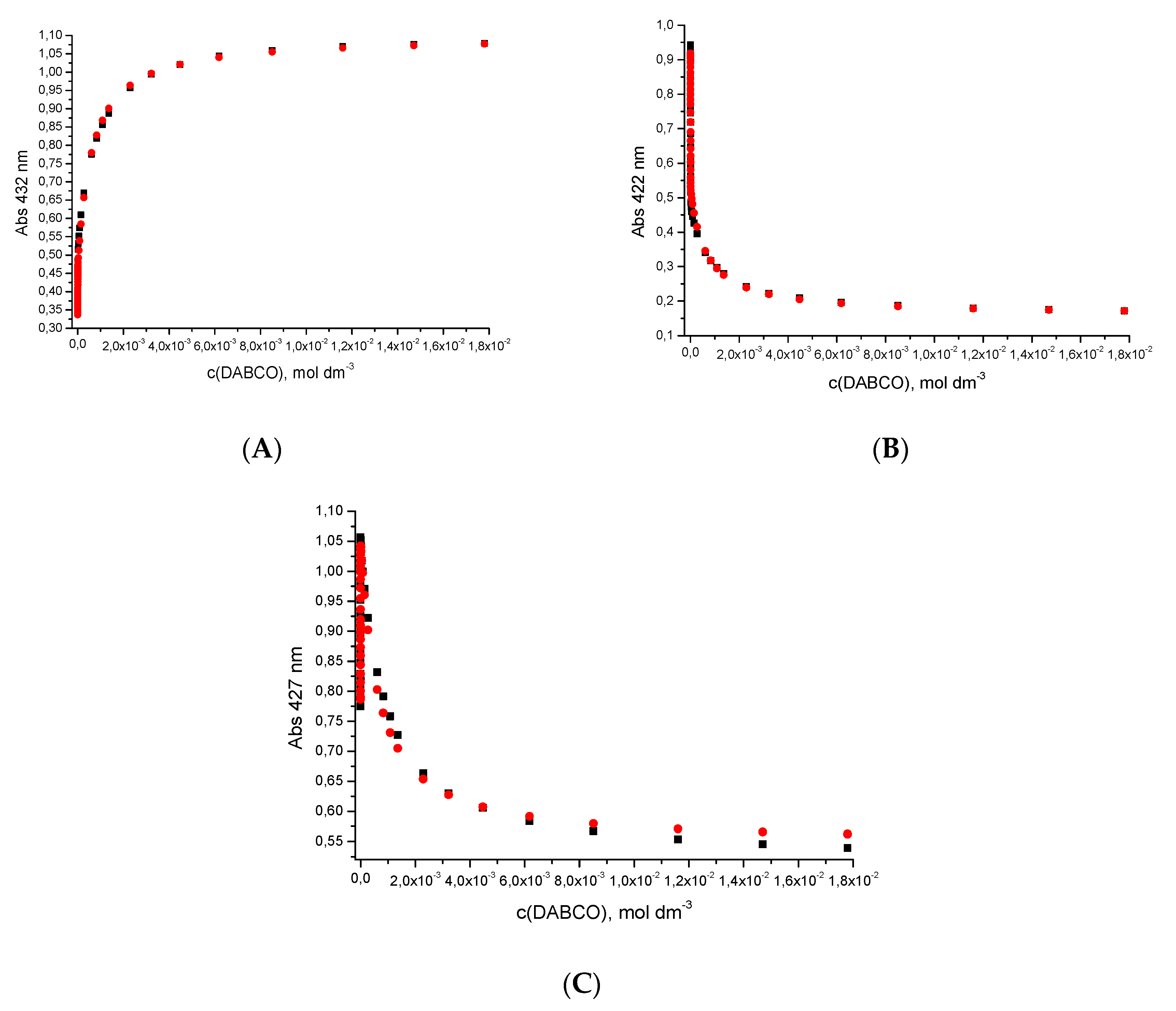
2.1. Advantage of Nucleosidic Linkers for the Pre-Organization of Dimers and Formation of Highly Stable Complexes with DABCO
2.2. Comparison with the Pre-Organization Obtained When Rigid Linkers Are Used
2.3. Dinucleotide: Formation of a Sandwich Type Molecular Complex with a Particularly High Association Constant
3. Materials and Methods
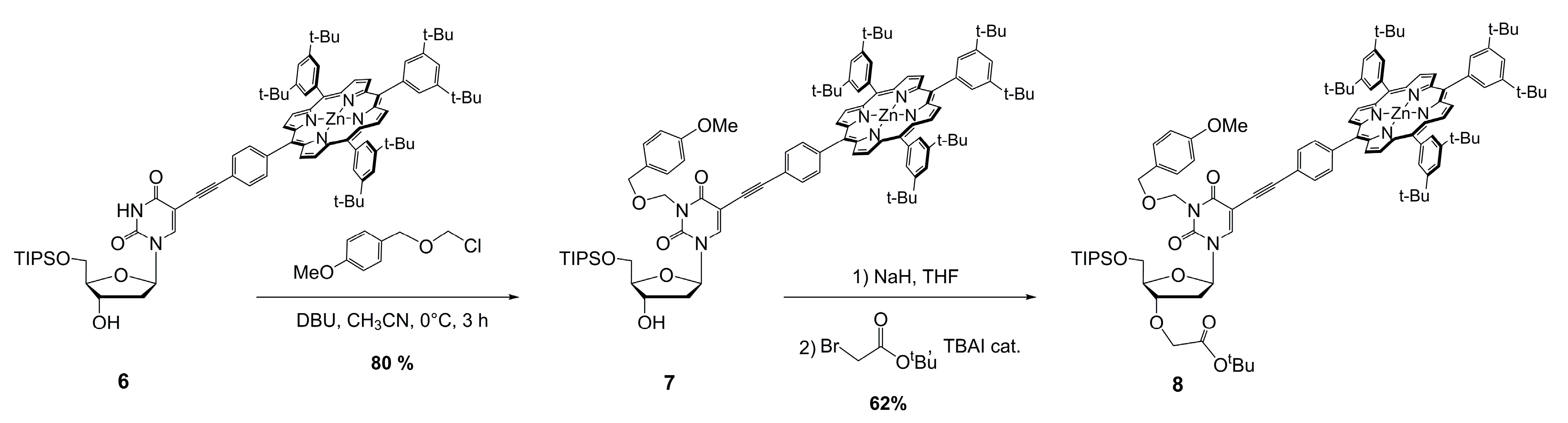
3.1. Preparation of Porphyrin–Uridine Conjugate 6
3.2. Preparation of Porphyrin–Uridine Conjugate Protected at N-3 7
3.3. Preparation of Monomer 8
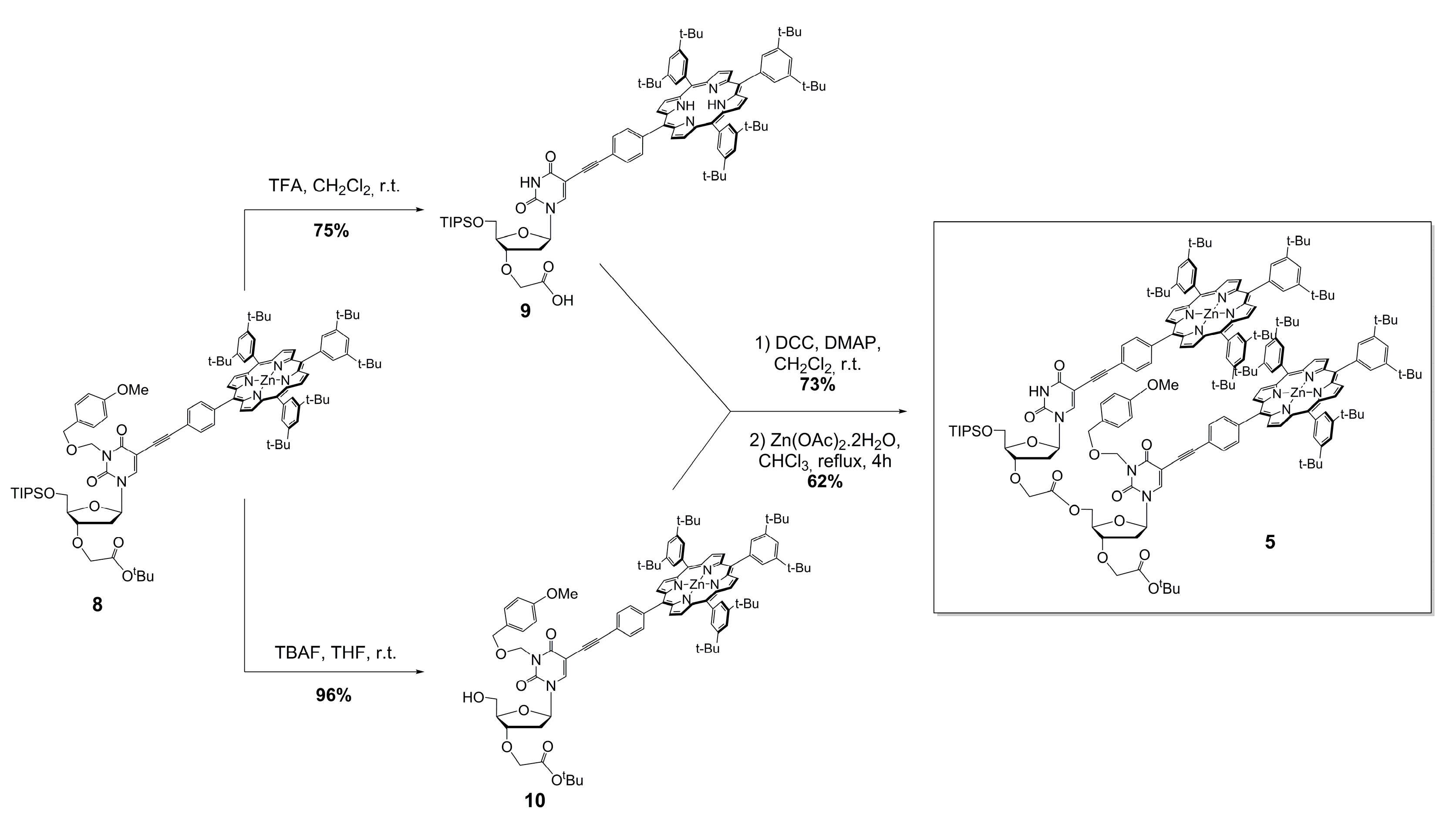
3.4. Preparation of Monomer-Acid 9
3.5. Preparation of Monomer-Alcohol 10
3.6. Preparation of Free-Base Dimer
3.7. Preparation of Zinc(II) Porphyrin Dimer 5
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lehn, J.-M. Supramolecular Chemistry—Scope and Perspectives Molecules, Supermolecules, and Molecular Devices (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1988, 27, 89–112. [Google Scholar] [CrossRef]
- Lehn, J.-M. Toward self-organization and complex matter. Science 2002, 295, 2400–2403. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, S.; Saito, K.; Ohkubo, K.; Troiani, V.; Qiu, H.; Gadde, S.; D’Souza, F.; Solladié, N. Multiple photosynthetic reaction centres using zinc porphyrinic oligopeptide-fulleropyrrolidine supramolecular complexes. Phys. Chem. Chem. Phys. 2011, 13, 17019–17023. [Google Scholar] [CrossRef] [PubMed]
- Piet, J.J.; Taylor, P.N.; Anderson, H.L.; Osuka, A.; Warman, J.M. Excitonic Interactions in the Singlet and Triplet Excited States of Covalently Linked Zinc Porphyrin Dimers. J. Am. Chem. Soc. 2000, 122, 1749–1757. [Google Scholar] [CrossRef]
- Satake, A.; Kobuke, Y. Dynamic supramolecular porphyrin systems. Tetrahedron 2005, 61, 13–41. [Google Scholar] [CrossRef]
- Choi, M.; Yamazaki, T.; Yamazaki, I.; Aida, T. Bioinspired Molecular Design of Light-Harvesting Multiporphyrin Arrays. Angew. Chem. Int. Ed. 2004, 43, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Tomizaki, K.; Yu, L.; Wei, L.; Bocian, D.F.; Lindsey, J.S. Synthesis of Cyclic Hexameric Porphyrin Arrays. Anchors for Surface Immobilization and Columnar Self-Assembly. J. Org. Chem. 2003, 68, 8199–8207. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.W.; Kamada, T.; Ahn, T.K.; Ko, D.M.; Nakamura, T.; Tsuda, A.; Osuka, A.; Kim, D. Porphyrin Boxes Constructed by Homochiral Self-Sorting Assembly: Optical Separation, Exciton Coupling, and Efficient Excitation Energy Migration. J. Am. Chem. Soc. 2004, 126, 16187–16198. [Google Scholar] [CrossRef] [PubMed]
- Mak, C.C.; Bampos, N.; Darling, S.L.; Montalti, M.; Prodi, L.; Sanders, J.K.M. A Strategy for the Assembly of Multiple Porphyrin Arrays Based on the Coordination Chemistry of Ru-Centered Porphyrin Pentamers. J. Org. Chem. 2001, 66, 4476–4486. [Google Scholar] [CrossRef] [PubMed]
- Haycock, R.A.; Yartsev, A.; Michelsen, U.; Sundström, V.; Hunter, C.A. Self-Assembly of Pentameric Porphyrin Light-Harvesting Antennae Complexes. Angew. Chem. Int. Ed. 2000, 39, 3616–3619. [Google Scholar] [CrossRef]
- Bretar, J.; Gieselbrecht, J.-P.; Gross, M.; Solladié, N. Tweezers hosts for intercalation of Lewis base guests: Tuning physico-chemical properties of cofacial porphyrin dimers. Chem. Commun. 2001, 733–734. [Google Scholar] [CrossRef]
- Rein, R.; Gross, M.; Solladié, N. Adjustable cavity for host-guest recognition in cofacial bis-porphyrinic tweezer. Chem. Commun. 2004, 1992–1993. [Google Scholar] [CrossRef] [PubMed]
- Flamigni, L.; Talarico, A.M.; Ventura, B.; Rein, R.; Solladié, N. A Versatile Bis-Porphyrin Tweezer Host for the Assembly of Noncovalent Photoactive Architectures: A Photophysical Characterization of the Tweezers and Their Association with Porphyrins and Other Guests. Chem. Eur. J. 2006, 12, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, C.; Tanaka, Y.; Fujihara, T.; Ishii, Y.; Ushiyama, T.; Yamamoto, K.; Yoshioka, N.; Inoue, H. Self-Assembly of a Porphyrin Array via the Molecular Recognition Approach: Synthesis and Properties of a Cyclic Zinc(II) Porphyrin Trimer Based on Coordination and Hydrogen Bonding. Inorg. Chem. 2001, 40, 3395–3405. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, C.; Satake, A.; Kobuke, Y. Proofs of Macrocyclization of Gable Porphyrins as Mimics of Photosynthetic Light-Harvesting Complexes. Org. Lett. 2003, 5, 4935–4938. [Google Scholar] [CrossRef] [PubMed]
- Kameyama, K.; Satake, A.; Kobuke, Y. Light-harvesting composites of directly connected porphyrin–phthalocyanine dyads and their coordination dimers. Tetrahedron Lett. 2004, 45, 7617–7620. [Google Scholar] [CrossRef]
- Schmittel, M.; Kishore, R.S.K. Tetrakis-heteroleptic Complexation at Porphyrins: A Convenient Route to Diversely Functionalized Aggregates. Org. Lett. 2004, 6, 1923–1926. [Google Scholar] [CrossRef] [PubMed]
- Balaban, T.S.; Goddard, R.; Linke-Schaetzel, M.; Lehn, J.-M. 2-Aminopyrimidine Directed Self-Assembly of Zinc Porphyrins Containing Bulky 3,5-Di-tert-butylphenyl Groups. J. Am. Chem. Soc. 2003, 125, 4233–4239. [Google Scholar] [CrossRef] [PubMed]
- Iengo, E.; Zangrando, E.; Alessio, E. Discrete Supramolecular Assemblies of Porphyrins Mediated by Coordination Compounds. Eur. J. Inorg. Chem. 2003, 2371–2384. [Google Scholar] [CrossRef]
- Iengo, E.; Zangrando, E.; Alessio, E.; Chambron, J.-C.; Heitz, V.; Flamigni, L.; Sauvage, J.-P. A Functionalized Noncovalent Macrocyclic Multiporphyrin Assembly from a Dizinc(II) Bis-Porphyrin Receptor and a Free-Base Dipyridylporphyrin. Chem. Eur. J. 2003, 9, 5879–5887. [Google Scholar] [CrossRef] [PubMed]
- Sugou, K.; Sasaki, K.; Kitajima, K.; Iwaki, T.; Kuroda, Y. Light-Harvesting Heptadecameric Porphyrin Assemblies. J. Am. Chem. Soc. 2002, 124, 1182–1183. [Google Scholar] [CrossRef] [PubMed]
- Wojaczynski, J.; Latos-Grazynski, L. Poly- and oligometalloporphyrins associated through coordination. Coord. Chem. Rev. 2000, 204, 113–171. [Google Scholar] [CrossRef]
- Holliday, B.J.; Mirkin, C.A. Strategies for the Construction of Supramolecular Compounds through Coordination Chemistry. Angew. Chem. Int. Ed. 2001, 40, 2022–2043. [Google Scholar] [CrossRef]
- Robertson, A.; Shinkai, S. Cooperative binding in selective sensors, catalysts and actuators. Coord. Chem. Rev. 2000, 205, 157–199. [Google Scholar] [CrossRef]
- Toma, H.E.; Araki, K. Supramolecular assemblies of ruthenium complexes and porphyrins. Coord. Chem. Rev. 2000, 196, 307–329. [Google Scholar] [CrossRef]
- Leininger, S.; Oleynuk, B.; Stang, P.J. Self-Assembly of Discrete Cyclic Nanostructures Mediated by Transition Metals. Chem. Rev. 2000, 100, 853–908. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.L.; Sanders, J.K.M. Enzyme mimics based on cyclic porphyrin oligomers: Strategy, design and exploratory synthesis. J. Chem. Soc. Perkin Trans. 1995, 2223–2229. [Google Scholar] [CrossRef]
- Vidal-Ferran, A.; Bampos, N.; Sanders, J.K.M. Stepwise Approach to Bimetalic Porphyrin Hosts: Spatially Enforced Coordination of a Nickel(II) Porphyrin. Inorg. Chem. 1997, 36, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Anderson, H.L. Conjugated Porphyrin Ladders. Inorg. Chem. 1994, 33, 972–981. [Google Scholar] [CrossRef]
- Taylor, P.N.; Anderson, H.L. Cooperative Self-Assembly of Double-Strand Conjugated Porphyrin Ladders. J. Am. Chem. Soc. 1999, 121, 11538–11545. [Google Scholar] [CrossRef]
- Wilson, G.S.; Anderson, H.L. A conjugated triple strand porphyrin array. Chem. Commun. 1999, 1539–1540. [Google Scholar] [CrossRef]
- Michelsen, U.; Hunter, C.A. Self-Assembled Porphyrin Polymers. Angew. Chem. Int. Ed. 2000, 39, 764–767. [Google Scholar] [CrossRef]
- Gardner, M.; Guerin, A.J.; Hunter, C.A.; Michelsen, U.; Rotger, C. Self-assembly of zinc aminoporphyrins. New. J. Chem. 1999, 309–316. [Google Scholar] [CrossRef]
- Fleischer, E.B.; Shachter, A.M. Coordination oligomers and a coordination polymer of zinc tetraarylporphyrins. Inorg. Chem. 1991, 30, 3763–3769. [Google Scholar] [CrossRef]
- Slone, R.V.; Hupp, J.H. Synthesis, Characterization, and Preliminary Host—Guest Binding Studies of Porphyrinic Molecular Squares Featuring fac-Tricarbonylrhenium(I) Chloro Corners. Inorg. Chem. 1997, 36, 5422–5423. [Google Scholar] [CrossRef]
- Drain, C.M.; Nifiatis, F.; Vasenko, A.; Batteas, J.D. Porphyrin Tessellation by Design: Metal-Mediated Self-Assembly of Large Arrays and Tapes. Angew. Chem. Int. Ed. 1998, 37, 2344–2347. [Google Scholar] [CrossRef]
- Milic, T.N.; Chi, N.; Yablon, D.G.; Flynn, G.W.; Batteas, J.D.; Drain, C.M. Controlled Hierarchical Self-Assembly and Deposition of Nanoscale Photonic Materials. Angew. Chem. Int. Ed. 2002, 41, 2117–2119. [Google Scholar] [CrossRef]
- Reek, J.N.H.; Schenning, A.P.H.J.; Bosman, A.W.; Meijer, E.W.; Crossley, M.J. Templated assembly of a molecular capsule. Chem. Commun. 1998, 11–12. [Google Scholar] [CrossRef]
- Baldini, L.; Ballester, P.; Casnati, A.; Gomila, R.M.; Hunter, C.A.; Sansone, F.; Ungaro, R. Molecular Acrobatics: Self-Assembly of Calixarene-Porphyrin Cages. J. Am. Chem. Soc. 2003, 125, 14181–14189. [Google Scholar] [CrossRef] [PubMed]
- Solladié, N.; Hamel, A.; Gross, M. Towards multiporphyrinic alpha-helices with a polypeptidic backbone as system endowed with light harvesting capabilities. Chirality 2001, 13, 736–738. [Google Scholar] [CrossRef] [PubMed]
- Solladié, N.; Aziat, F.; Bouatra, S.; Rein, R. Bis-porphyrin tweezers: Rigid or flexible linkers for better adjustment of the cavity to bidentate bases of various size. J. Porphyr. Phtalocyanines 2008, 12, 1250–1260. [Google Scholar] [CrossRef]
- Barber, J.; Andersson, B. Revealing the blueprint of photosynthesis. Nature 1994, 370, 31–34. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Many wheels make light work. Nature 1994, 374, 497–498. [Google Scholar] [CrossRef]
- McDermott, G.; Prince, S.M.; Freer, A.A.; Hawthornthwaite-Lawless, A.M.; Papiz, M.Z.; Cogdell, R.J.; Isaacs, N.W. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature 1995, 374, 517–521. [Google Scholar] [CrossRef]
- Pullerits, T.; Sundström, V. Photosynthetic Light-Harvesting Pigment–Protein Complexes: Toward Understanding How and Why. Acc. Chem. Res. 1996, 29, 381–389. [Google Scholar] [CrossRef]
- Hirose, K. A Practical Guide for the Determination of Binding Constants. J. Incl. Phenom. Macrocycl. Chem. 2001, 39, 193–209. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of equilibrium constants from multiwavelength spectroscopic data—IV: Model-free least-squares refinement by use of evolving factor analysis. Talanta 1986, 33, 943–951. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of equilibrium constants from multiwavelength spectroscopic data—I: Mathematical considerations. Talanta 1985, 32, 95–101. [Google Scholar] [CrossRef]
- Mak, C.C.; Bampos, N.; Sanders, J.K.M. Metalloporphyrin Dendrimers with Folding Arms. Angew. Chem. Int. Ed. 1998, 37, 3020–3023. [Google Scholar] [CrossRef]
- Ballester, P.; Costa, A.; Castilla, A.M.; Deyà, P.M.; Frontera, A.; Gomila, R.M.; Hunter, C.A. DABCO-Directed Self-Assembly of Bisporphyrins (DABCO=1,4-Diazabicyclo[2.2.2]octane). Chem. Eur. J. 2005, 11, 2196–2206. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Meah, M.N.; Sanders, J.K.M. DABCO-metalloporphyrin binding: Ternary complexes, host-guest chemistry and the measurement of π–π interactions. J. Am. Chem. Soc. 1990, 112, 5773–5780. [Google Scholar] [CrossRef]
- Anderson, H.L.; Hunter, C.A.; Meah, M.N.; Sanders, J.K.M. Thermodynamics of induced-fit binding inside polymacrocyclic porphyrin hosts. J. Am. Chem. Soc. 1990, 112, 5780–5789. [Google Scholar] [CrossRef]
- Kim, H.J.; Bampos, N.; Sanders, J.K.M. Assembly of Dynamic Heterometallic Oligoporphyrins Using Cooperative Zinc–Nitrogen, Ruthenium–Nitrogen, and Tin–Oxygen Coordination. J. Am. Chem. Soc. 1999, 121, 8120–8121. [Google Scholar] [CrossRef]
- Hunter, C.A.; Tregonning, R. Modular assembly of porphyrin sandwiches as potential hosts. Tetrahedron 2002, 58, 691–697. [Google Scholar] [CrossRef]
- Mak, C.C.; Pomeranc, D.; Montelti, M.; Prodi, L.; Sanders, J.K.M. A versatile synthetic strategy for construction of large oligomers: Binding and photophysical properties of a nine-porphyrin array. Chem. Commun. 1999, 1083–1084. [Google Scholar] [CrossRef]
Sample Availability: Samples of all compounds are available from the authors. |








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merkaš, S.; Bouatra, S.; Rein, R.; Piantanida, I.; Zinic, M.; Solladié, N. Efficiency of Dinucleosides as the Backbone to Pre-Organize Multi-Porphyrins and Enhance Their Stability as Sandwich Type Complexes with DABCO. Molecules 2017, 22, 1112. https://doi.org/10.3390/molecules22071112
Merkaš S, Bouatra S, Rein R, Piantanida I, Zinic M, Solladié N. Efficiency of Dinucleosides as the Backbone to Pre-Organize Multi-Porphyrins and Enhance Their Stability as Sandwich Type Complexes with DABCO. Molecules. 2017; 22(7):1112. https://doi.org/10.3390/molecules22071112
Chicago/Turabian StyleMerkaš, Sonja, Souhaila Bouatra, Régis Rein, Ivo Piantanida, Mladen Zinic, and Nathalie Solladié. 2017. "Efficiency of Dinucleosides as the Backbone to Pre-Organize Multi-Porphyrins and Enhance Their Stability as Sandwich Type Complexes with DABCO" Molecules 22, no. 7: 1112. https://doi.org/10.3390/molecules22071112
APA StyleMerkaš, S., Bouatra, S., Rein, R., Piantanida, I., Zinic, M., & Solladié, N. (2017). Efficiency of Dinucleosides as the Backbone to Pre-Organize Multi-Porphyrins and Enhance Their Stability as Sandwich Type Complexes with DABCO. Molecules, 22(7), 1112. https://doi.org/10.3390/molecules22071112