Site-Specific PEGylated Adeno-Associated Viruses with Increased Serum Stability and Reduced Immunogenicity

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
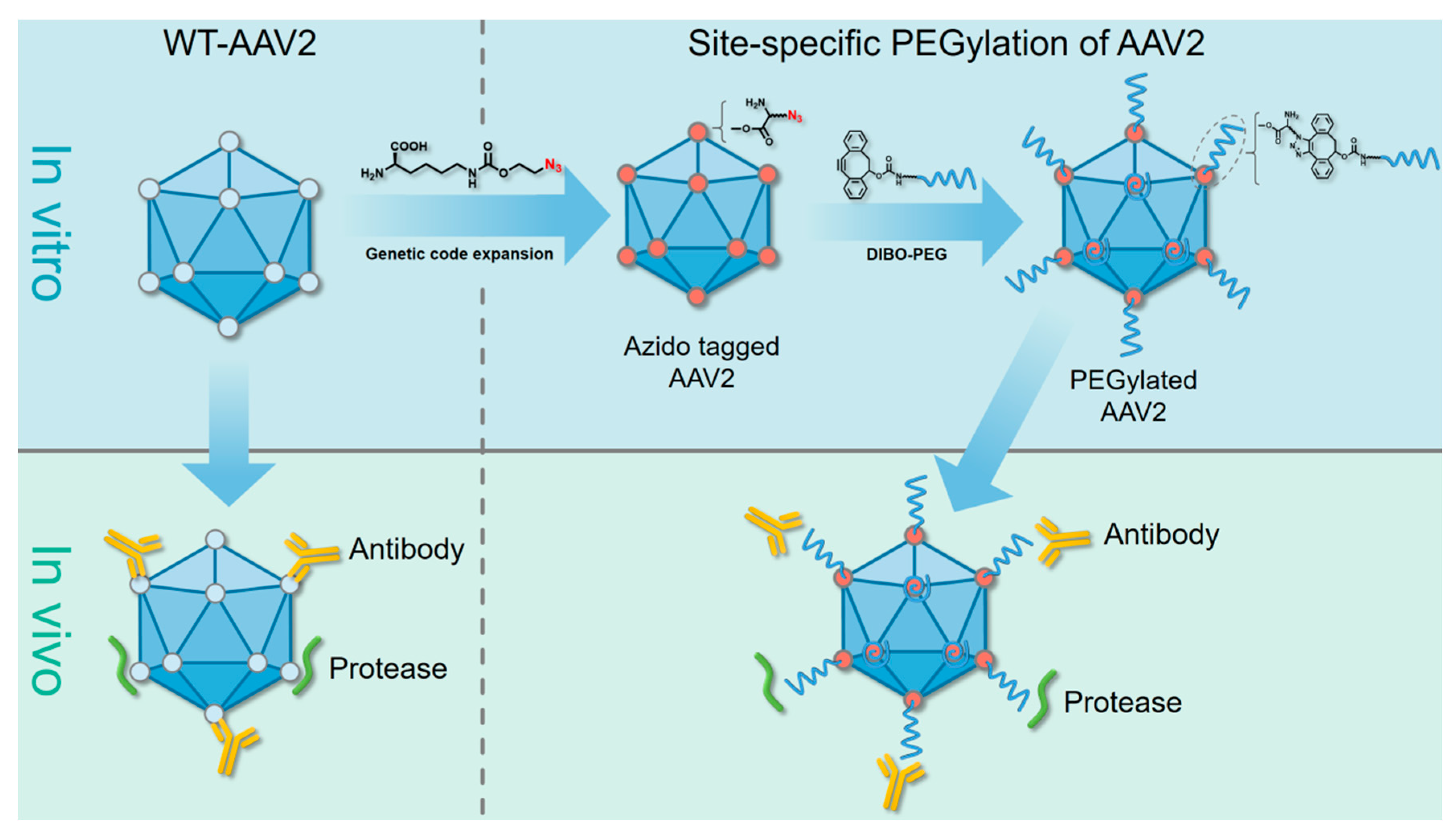
2.1. Site-Specific PEGylation of VP1 Proteins and rAAV-2 Vectors via a Bioorthogonal Reaction
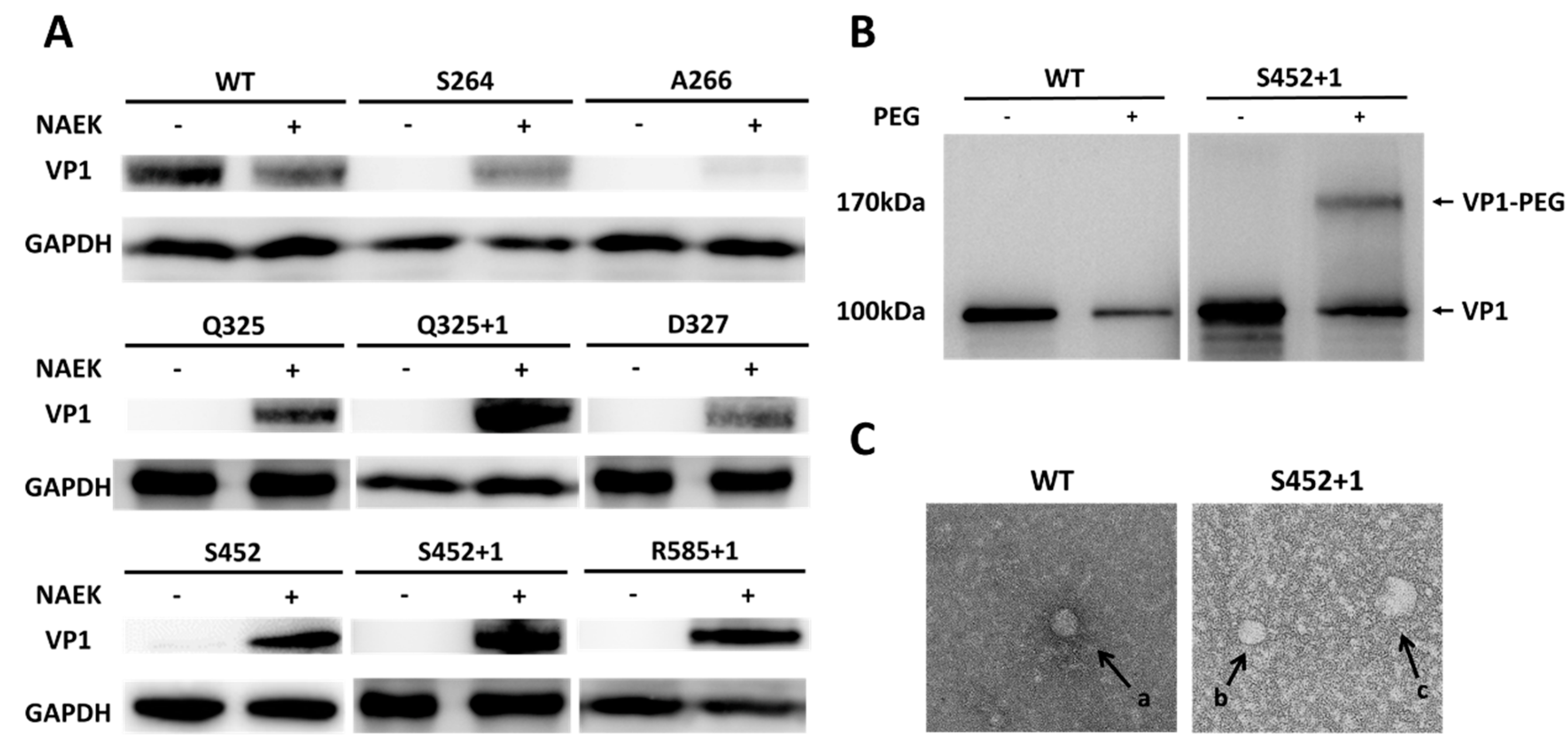
2.2. Successful Production of Site-specific Mutant rAAV2 Particles and Their Structural-Functional Relationships
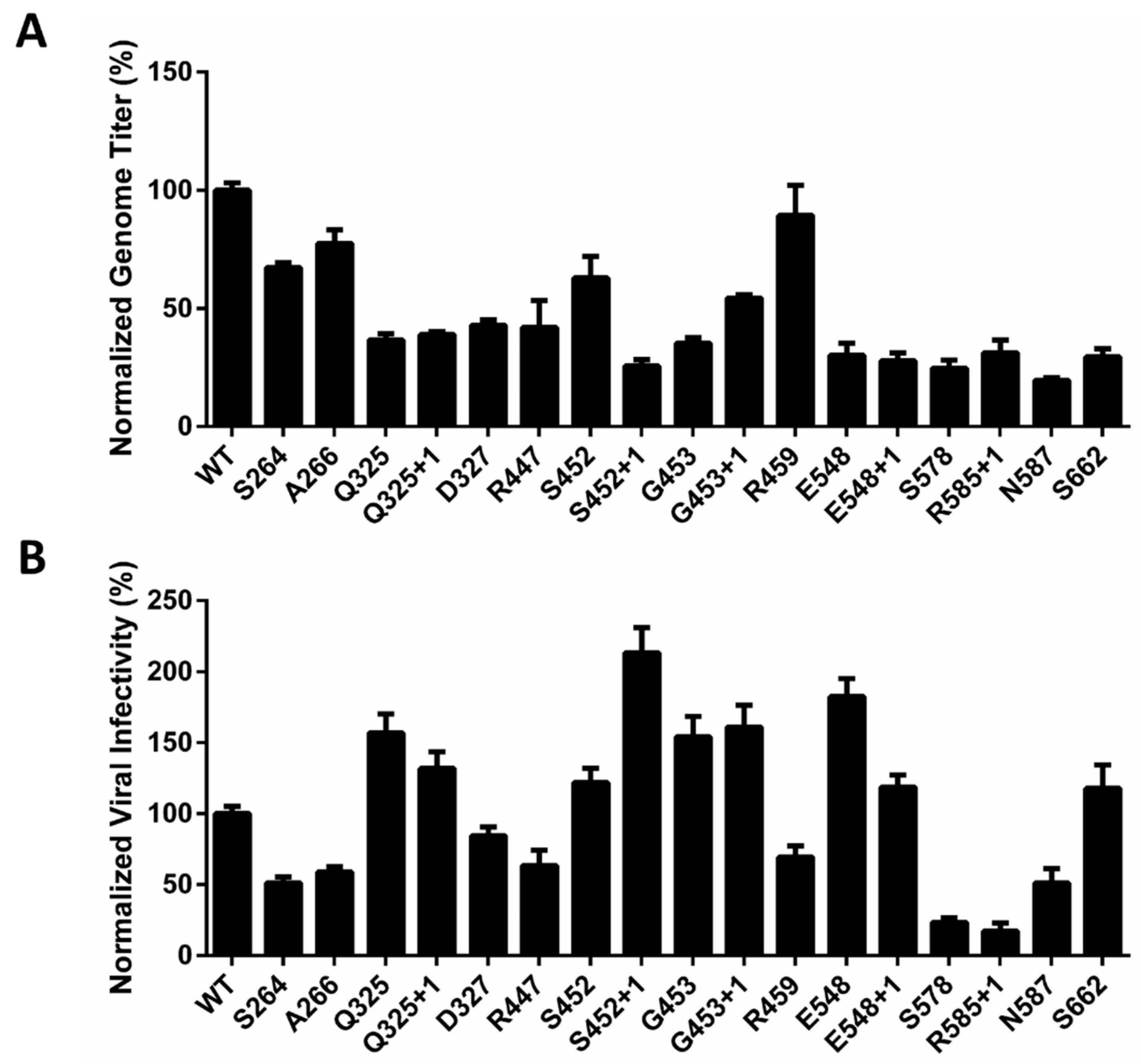
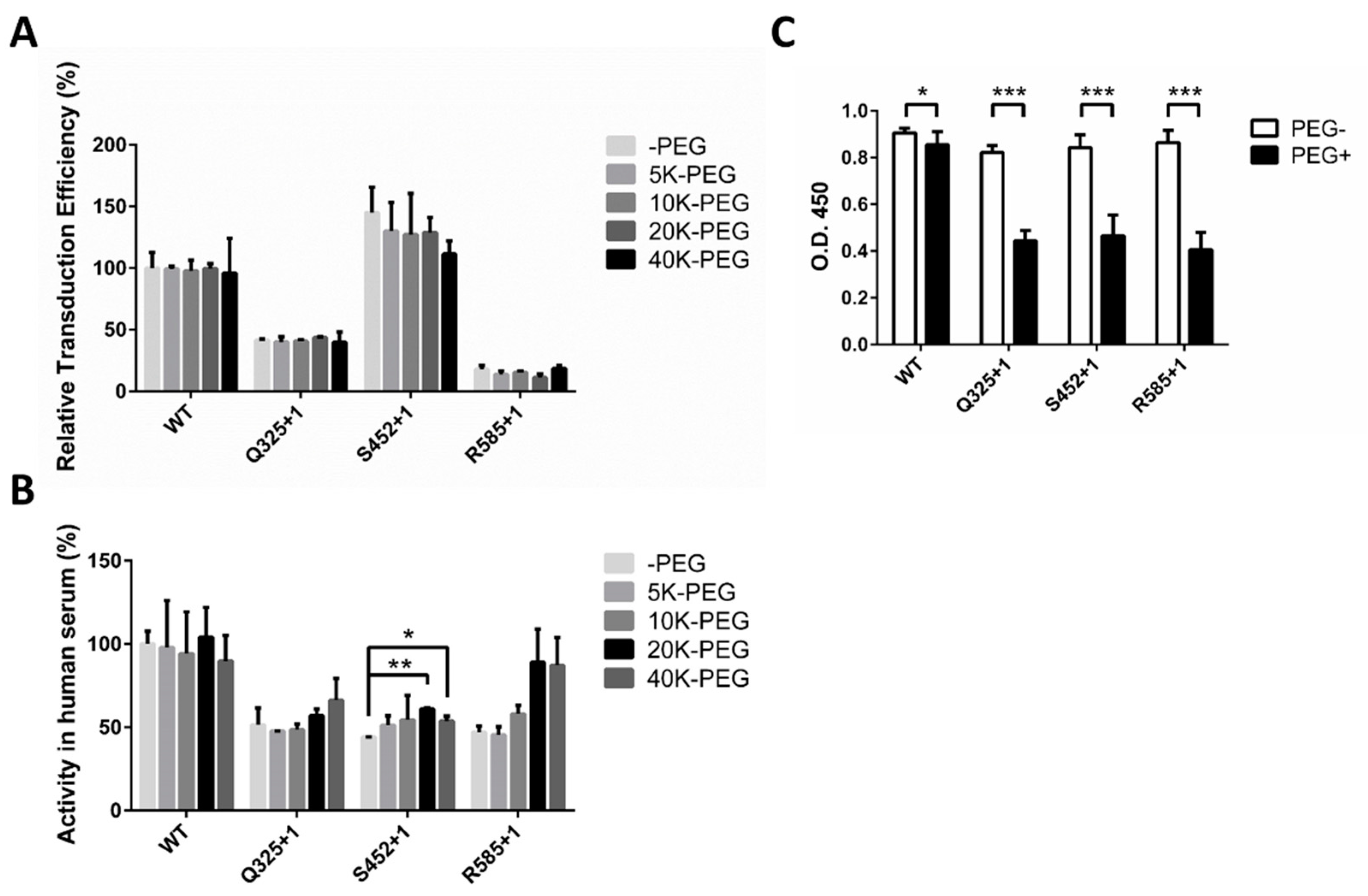
2.3. Optimized Sites for rAAV2 PEGylation
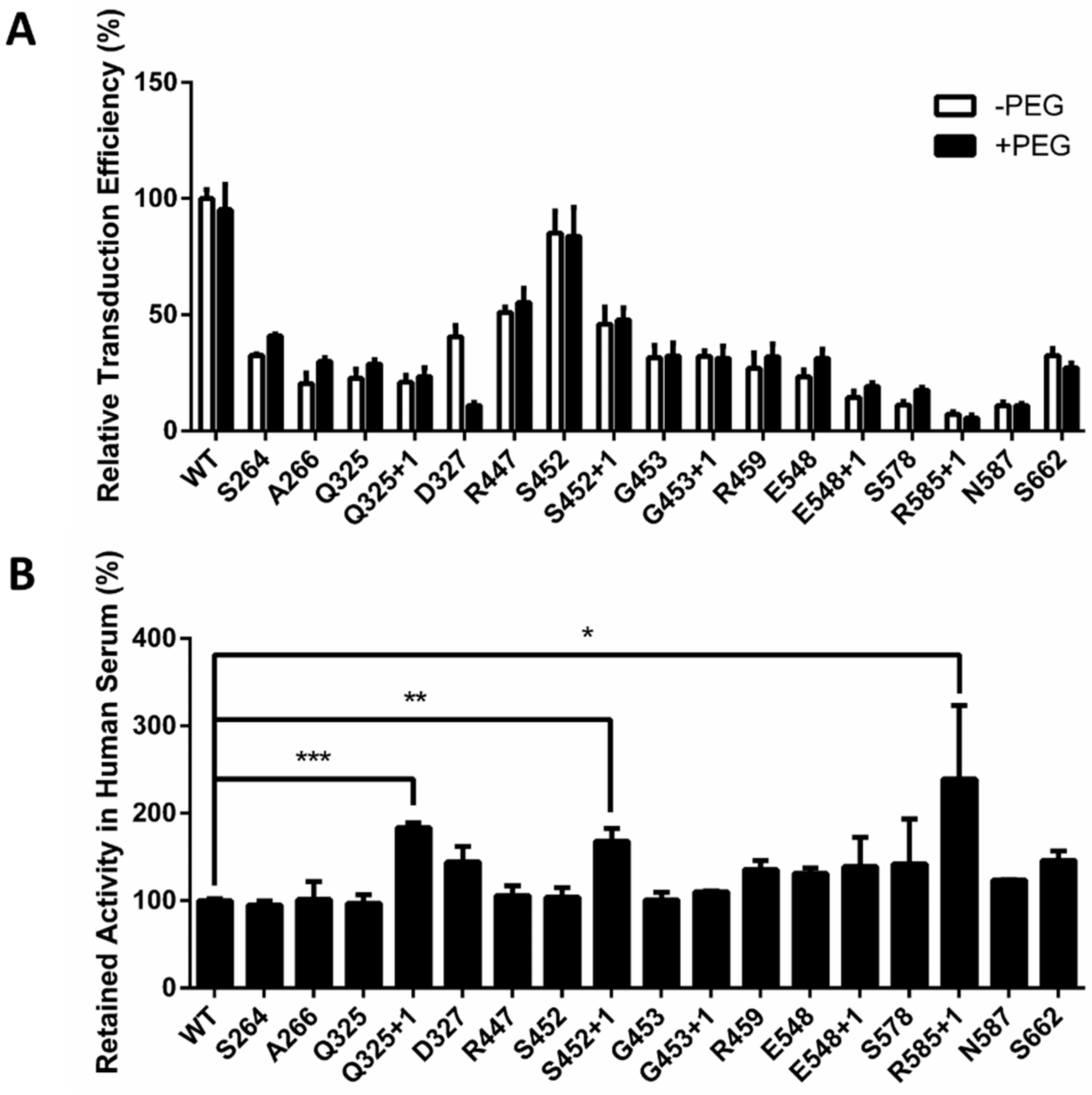
2.4. Optimal PEG Length for rAAV2 PEGylation
2.5. Decreased Antibody Recognition of PEGylated rAAV2 In Vitro
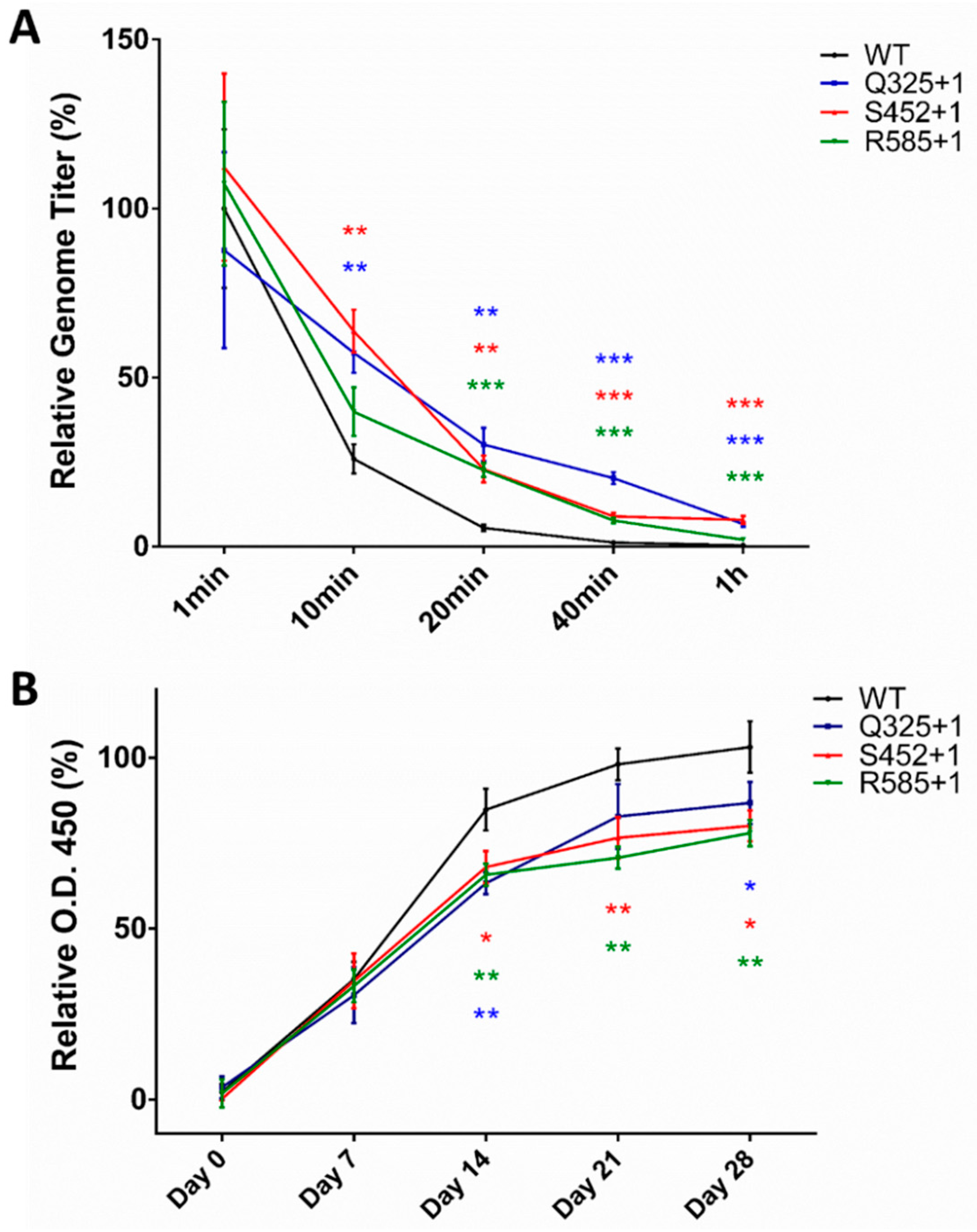
2.6. In Vivo Verification of Improved Stability and Decreased Immunogenicity of PEGylated AAV2
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Antibodies and Reagents
4.2. Plasmid Construction
4.3. rAAV2 Production and Purification
4.4. Quantification of Viral Genomic Titer by Real-time qPCR
4.5. Detection of Gene Transduction
4.6. PEG Conjugation
4.7. TEM of Negatively Stained rAAV2 Vectors
4.8. Human Serum Stability Assay
4.9. rAAV2 ELISA
4.10. Animal Research
4.11. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fields, B.; Knipe, D.; Howley, P.; Griffin, D. Fields Virology, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Muzyczka, N.; Berns, K. Parvoviridae: The viruses and their replication. Fields Virol. 2001, 2, 2327–2359. [Google Scholar]
- Vasileva, A.; Jessberger, R. Precise hit: Adeno-associated virus in gene targeting. Nat. Rev. Microbiol. 2005, 3, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Sehara, Y.; Fujimoto, K.-I.; Ikeguchi, K.; Katakai, Y.; Ono, F.; Takino, N.; Ito, M.; Ozawa, K.; Muramatsu, S.-I. Persistent expression of dopamine-synthesizing enzymes 15 years after gene transfer in a primate model of parkinson's disease. Hum. Gene Ther. Clin. Dev. 2017, 28, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Tarantal, A.F.; Lee, C.C.I.; Martinez, M.L.; Asokan, A.; Samulski, R.J. Systemic and persistent muscle gene expression in rhesus monkeys with a liver de-targeted adeno-associated virus vector. Hum. Gene Ther. 2017, 28, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Vidović, D.; Gijsbers, R.; Quiles-Jimenez, A.; Dooley, J.; Van den Haute, C.; Van der Perren, A.; Liston, A.; Baekelandt, V.; Debyser, Z.; Carlon, M.S. Noninvasive imaging reveals stable transgene expression in mouse airways after delivery of a nonintegrating recombinant adeno-associated viral vector. Hum. Gene Ther. 2015, 27, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Bortolussi, G.; Zentillin, L.; Vaníkova, J.; Bockor, L.; Bellarosa, C.; Mancarella, A.; Vianello, E.; Tiribelli, C.; Giacca, M.; Vitek, L. Life-long correction of hyperbilirubinemia with a neonatal liver-specific aav-mediated gene transfer in a lethal mouse model of crigler–najjar syndrome. Hum. Gene Ther. 2014, 25, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Mearini, G.; Stimpel, D.; Geertz, B.; Weinberger, F.; Krämer, E.; Schlossarek, S.; Mourot-Filiatre, J.; Stoehr, A.; Dutsch, A.; Wijnker, P.J. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat. Commun. 2014, 5, 5515. [Google Scholar] [CrossRef] [PubMed]
- Vil, L.; Roca, C.; Elias, I.; Casellas, A.; Lage, R.; Franckhauser, S.; Bosch, F. Aav-mediated sirt1 overexpression in skeletal muscle activates oxidative capacity but does not prevent insulin resistance. Mol. Ther.-Methods Clin. Dev. 2016, 3, 16072. [Google Scholar] [CrossRef] [PubMed]
- Boye, S.E.; Alexander, J.J.; Witherspoon, C.D.; Boye, S.L.; Peterson, J.J.; Clark, M.E.; Sandefer, K.J.; Girkin, C.A.; Hauswirth, W.W.; Gamlin, P.D. Highly efficient delivery of adeno-associated viral vectors to the primate retina. Hum. Gene Ther. 2016, 27, 580–597. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Harris, A.F.; Cabral, D.J.; Keeler, A.M.; Sapp, E.; Ferreira, J.S.; Gray-Edwards, H.L.; Johnson, J.A.; Johnson, A.K.; Su, Q. Widespread central nervous system gene transfer and silencing after systemic delivery of novel aav-as vector. Mol. Ther. 2016, 24, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Ortiz, J.L.; Schaffer, D.V. Adeno-associated virus (AAV) vectors in cancer gene therapy. J. Control. Release 2016, 240, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Moskalenko, M.; Chen, L.; van Roey, M.; Donahue, B.A.; Snyder, R.O.; McArthur, J.G.; Patel, S.D. Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: Implications for gene therapy and virus structure. J. Virol. 2000, 74, 1761–1766. [Google Scholar] [CrossRef] [PubMed]
- Boutin, S.; Monteilhet, V.; Veron, P.; Leborgne, C.; Benveniste, O.; Montus, M.F.; Masurier, C. Prevalence of serum igg and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using aav vectors. Hum. Gene Ther. 2010, 21, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Wang, Y.; Feng, Y.L.; Zhang, Y.N.; Li, J.; Hu, X.R.; Wang, L.N.; Zhong, M.F.; Zhai, X.F.; Zolotukhin, I.; et al. Prevalence of neutralizing antibodies against liver-tropic adeno-associated virus serotype vectors in 100 healthy chinese and its potential relation to body constitutions. J. Integr. Med. 2015, 13, 341–346. [Google Scholar] [CrossRef]
- Calcedo, R.; Vandenberghe, L.H.; Gao, G.; Lin, J.; Wilson, J.M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 2009, 199, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Ertl, H.C.J.; High, K.A. Impact of aav capsid-specific t-cell responses on design and outcome of clinical gene transfer trials with recombinant adeno-associated viral vectors: An evolving controversy. Hum. Gene Ther. 2017, 28, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Thwaite, R.; Pages, G.; Chillon, M.; Bosch, A. Aavrh.10 immunogenicity in mice and humans. Relevance of antibody cross-reactivity in human gene therapy. Gene Ther. 2015, 22, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Mimuro, J.; Mizukami, H.; Hishikawa, S.; Ikemoto, T.; Ishiwata, A.; Sakata, A.; Ohmori, T.; Madoiwa, S.; Ono, F.; Ozawa, K.; et al. Minimizing the inhibitory effect of neutralizing antibody for efficient gene expression in the liver with adeno-associated virus 8 vectors. Mol. Ther. 2013, 21, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Hardet, R.; Chevalier, B.; Dupaty, L.; Naïmi, Y.; Riou, G.; Drouot, L.; Jean, L.; Salvetti, A.; Boyer, O.; Adriouch, S. Oral-tolerization prevents immune responses and improves transgene persistence following gene transfer mediated by adeno-associated viral vector. Mol. Ther. 2016, 24, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, Z.; Gyorgy, B.; Skog, J.; Maguire, C.A. Extracellular vesicles as enhancers of virus vector-mediated gene delivery. Hum. Gene Ther. 2014, 25, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; Anguela, X.M.; Pavani, G.; Chen, Y.; Davidson, R.J.; Hui, D.J.; Yazicioglu, M.; Elkouby, L.; Hinderer, C.J.; Faella, A. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci. Transl. Med. 2013, 5, 194ra192. [Google Scholar] [CrossRef] [PubMed]
- Tse, L.V.; Klinc, K.A.; Madigan, V.J.; Rivera, R.M.C.; Wells, L.F.; Havlik, L.P.; Smith, J.K.; Agbandje-McKenna, M.; Asokan, A. Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc. Natl. Acad. Sci. USA 2017, 114, E4812–E4821. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Wonganan, P.; Clemens, C.C.; Brasky, K.; Pastore, L.; Croyle, M.A. Species differences in the pharmacology and toxicology of pegylated helper-dependent adenovirus. Mol. Pharm. 2010, 8, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Wonganan, P.; Croyle, M.A. Pegylated adenoviruses: From mice to monkeys. Viruses 2010, 2, 468–502. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.K.; Maheshri, N.; Kaspar, B.; Schaffer, D.V. Peg conjugation moderately protects adeno-associated viral vectors against antibody neutralization. Biotechnol. Bioeng. 2005, 92, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Le, H.T.; Yu, Q.C.; Wilson, J.M.; Croyle, M.A. Utility of pegylated recombinant adeno-associated viruses for gene transfer. J. Control. Release 2005, 108, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the genetic code of escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Deiters, A.; Cropp, T.A.; Summerer, D.; Mukherji, M.; Schultz, P.G. Site-specific pegylation of proteins containing unnatural amino acids. Bioorg. Med. Chem. Lett. 2004, 14, 5743–5745. [Google Scholar] [CrossRef] [PubMed]
- Chin, J.W.; Cropp, T.A.; Anderson, J.C.; Mukherji, M.; Zhang, Z.; Schultz, P.G. An expanded eukaryotic genetic code. Science 2003, 301, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Xiao, W.; Conlon, T.; Hughes, J.; Agbandje-McKenna, M.; Ferkol, T.; Flotte, T.; Muzyczka, N. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of aav2 vectors with altered tropism. J. Virol. 2000, 74, 8635–8647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yao, T.; Zheng, Y.; Li, Z.; Zhang, Q.; Zhang, L.; Zhou, D. Development of next generation adeno-associated viral vectors capable of selective tropism and efficient gene delivery. Biomaterials 2016, 80, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Bu, W.; Bhatia, S.; Hare, J.; Somasundaram, T.; Azzi, A.; Chapman, M.S. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 10405–10410. [Google Scholar] [CrossRef] [PubMed]
- Qu, G.; Bahr-Davidson, J.; Prado, J.; Tai, A.; Cataniag, F.; McDonnell, J.; Zhou, J.; Hauck, B.; Luna, J.; Sommer, J.M.; et al. Separation of adeno-associated virus type 2 empty particles from genome containing vectors by anion-exchange column chromatography. J. Virol. Methods 2007, 140, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Rohr, U.P.; Wulf, M.A.; Stahn, S.; Steidl, U.; Haas, R.; Kronenwett, R. Fast and reliable titration of recombinant adeno-associated virus type-2 using quantitative real-time pcr. J. Virol. Methods 2002, 106, 81–88. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds and plasmids are available from the authors. |






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, T.; Zhou, X.; Zhang, C.; Yu, X.; Tian, Z.; Zhang, L.; Zhou, D. Site-Specific PEGylated Adeno-Associated Viruses with Increased Serum Stability and Reduced Immunogenicity. Molecules 2017, 22, 1155. https://doi.org/10.3390/molecules22071155
Yao T, Zhou X, Zhang C, Yu X, Tian Z, Zhang L, Zhou D. Site-Specific PEGylated Adeno-Associated Viruses with Increased Serum Stability and Reduced Immunogenicity. Molecules. 2017; 22(7):1155. https://doi.org/10.3390/molecules22071155
Chicago/Turabian StyleYao, Tianzhuo, Xueying Zhou, Chuanling Zhang, Xiaojuan Yu, Zhenyu Tian, Lihe Zhang, and Demin Zhou. 2017. "Site-Specific PEGylated Adeno-Associated Viruses with Increased Serum Stability and Reduced Immunogenicity" Molecules 22, no. 7: 1155. https://doi.org/10.3390/molecules22071155
APA StyleYao, T., Zhou, X., Zhang, C., Yu, X., Tian, Z., Zhang, L., & Zhou, D. (2017). Site-Specific PEGylated Adeno-Associated Viruses with Increased Serum Stability and Reduced Immunogenicity. Molecules, 22(7), 1155. https://doi.org/10.3390/molecules22071155