The Role of Metal Binding in the Amyotrophic Lateral Sclerosis-Related Aggregation of Copper-Zinc Superoxide Dismutase


{kind=link}
{kind=link}
Abstract
:1. Introduction
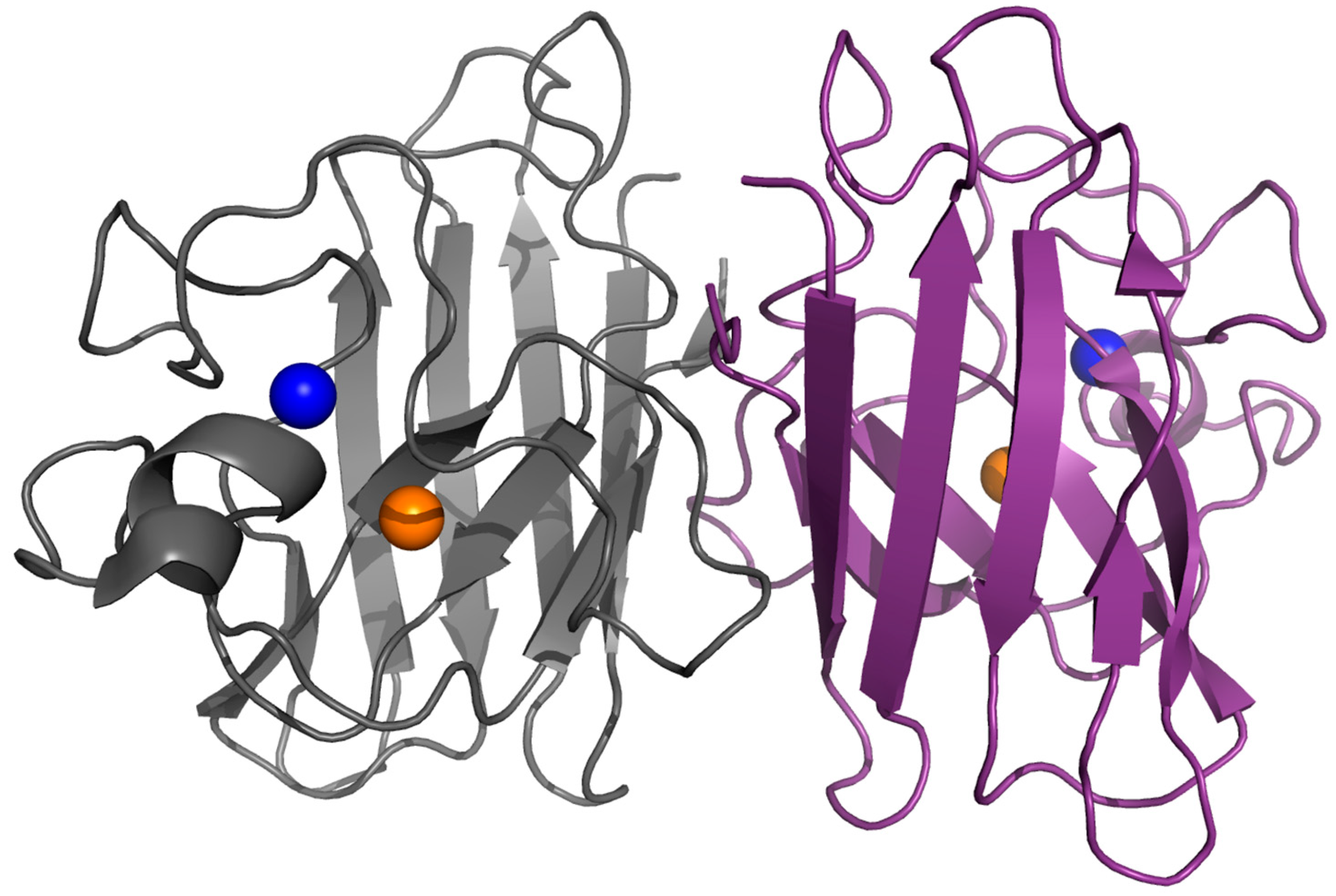
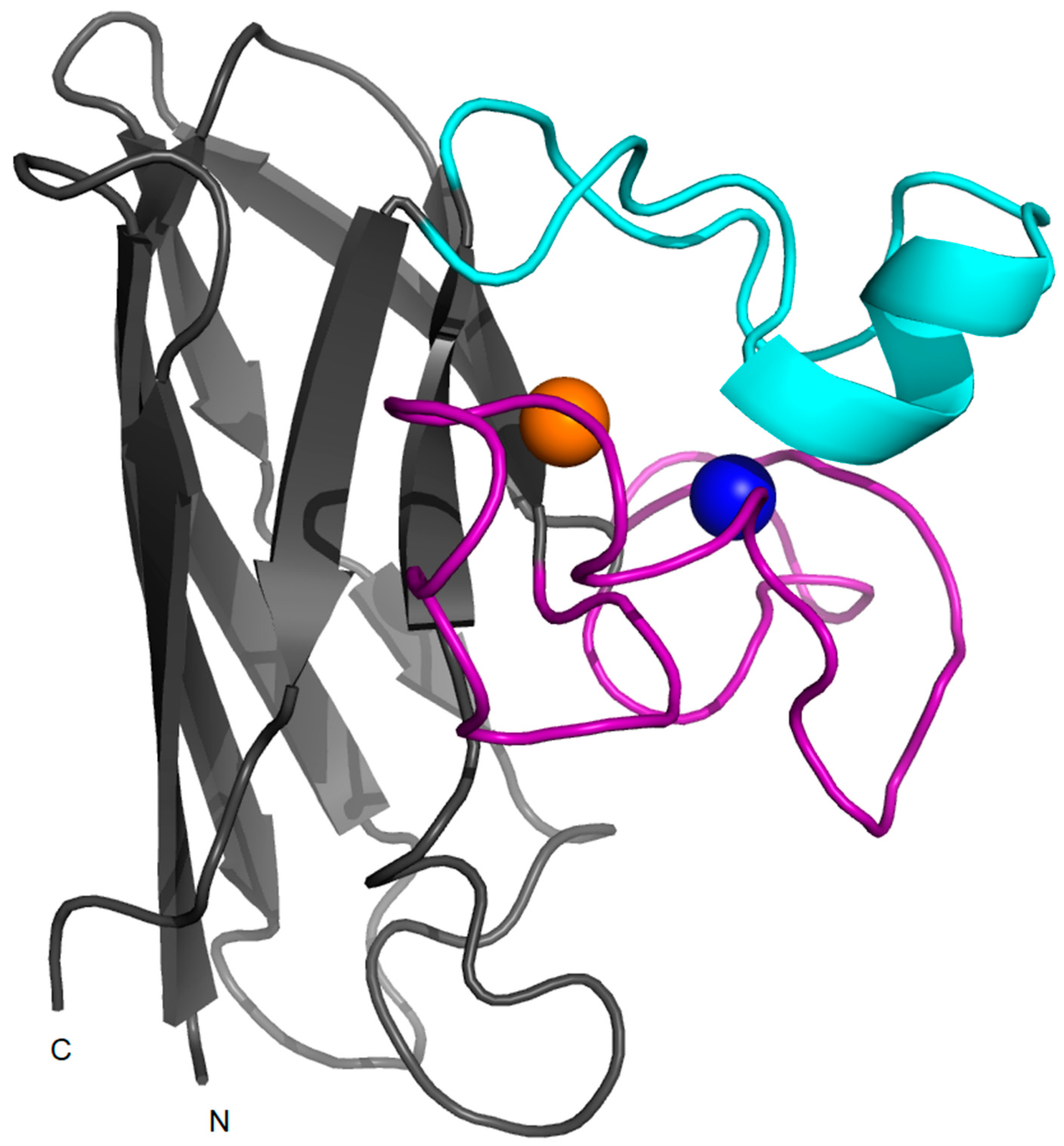
2. Structure, Folding and Stability in SOD1
3. Role of SOD1 Aggregation in ALS
4. Metal Binding and Aggregation of SOD1 in ALS
5. Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stefani, M. Protein misfolding and aggregation: New examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta 2004, 1739, 5–25. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, E.; Cascella, R.; Becatti, M.; Marrazza, G.; Dobson, C.M.; Chiti, F.; Stefani, M.; Cecchi, C. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci. Rep. 2016, 6, 32721. [Google Scholar] [CrossRef] [PubMed]
- Serpell, L.C. Alzheimer’s amyloid fibrils: Structure and assembly. Biochim. Biophys. Acta 2000, 1502, 16–30. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomicstructure and hierarchicalassembly of a cross-β amyloidfibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef] [PubMed]
- Gaggelli, E.; Kozlowski, H.; Valensin, D.; Valensin, G. Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem. Rev. 2006, 106, 1995–2044. [Google Scholar] [CrossRef] [PubMed]
- DeToma, A.S.; Salamekh, S.; Ramamoorthy, A.; Lim, M.H. Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem. Soc. Rev. 2012, 41, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Brender, J.R.; Salamekh, S.; Ramamoorthy, A. Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc. Chem. Res. 2012, 45, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Braymer, J.J.; Choi, J.S.; DeToma, A.S.; Wang, C.; Nam, K.; Kampf, J.W.; Ramamoorthy, A.; Lim, M.H. Development of bifunctional stilbene derivatives for targeting and modulating metal-amyloid-β species. Inorg. Chem. 2011, 50, 10724–10734. [Google Scholar] [CrossRef] [PubMed]
- Salamekh, S.; Brender, J.R.; Hyung, S.J.; Nanga, R.P.; Vivekanandan, S.; Ruotolo, B.T.; Ramamoorthy, A. A two-site mechanism for the inhibition of IAPP amyloidogenesis by zinc. J. Mol. Biol. 2011, 410, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Braymer, J.J.; Nanga, R.P.; Ramamoorthy, A.; Lim, M.H. Design of small molecules that target metal-Aβ species and regulate metal-induced Aβ aggregation and neurotoxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 21990–21995. [Google Scholar] [CrossRef] [PubMed]
- Manto, M. Abnormal copper homeostasis: Mechanisms and roles in neurodegeneration. Toxics 2014, 2, 327–345. [Google Scholar] [CrossRef]
- Valentine, J.S.; Doucette, P.A.; Potter, S.Z. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 2005, 74, 563–593. [Google Scholar] [CrossRef] [PubMed]
- Culotta, V.C.; Yang, M.; O’Halloran, T.V. Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta 2006, 1763, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Boca, M.; Girotto, S.; Martinelli, M.; Valentine, J.S.; Vieru, M. SOD1 and amyotrophic lateral sclerosis: Mutations and oligomerization. PLoS ONE 2008, 3, e1677. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Rouleau, G.A. Oxidized/misfolded superoxide dismutase-1: The cause of all amyotrophic lateral sclerosis? Ann. Neurol. 2007, 62, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Gruzman, A.; Wood, W.L.; Alpert, E.; Prasad, M.D.; Miller, R.G.; Rothstein, J.D.; Bowser, R.; Hamilton, R.; Wood, T.D.; Cleveland, D.W.; et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 12524–12529. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Valentine, J.S. Aggregation of copper-zinc superoxide dismutase in familial and sporadic ALS. Antioxid. Redox Signal. 2009, 11, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Rotunno, M.S.; Bosco, D.A. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front. Cell. Neurosci. 2013, 7, 253. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Andersen, P.M.; Marklund, S.L.; Brannstrom, T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011, 121, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y. Pathological roles of wild-type Cu, Zn-superoxide dismutase in amyotrophic lateral sclerosis. Neurol. Res. Int. 2012, 2012, 323261. [Google Scholar] [CrossRef] [PubMed]
- Guareschi, S.; Cova, E.; Cereda, C.; Ceroni, M.; Donetti, E.; Bosco, D.A.; Trotti, D.; Pasinelli, P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 2012, 109, 5074–5079. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Durazo, A.; Girotto, S.; Gralla, E.B.; Martinelli, M.; Valentine, J.S.; Vieru, M.; Whitelegge, J.P. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: A possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. USA 2007, 104, 11263–11267. [Google Scholar] [CrossRef] [PubMed]
- Oztug Durer, Z.A.; Cohlberg, J.A.; Dinh, P.; Padua, S.; Ehrenclou, K.; Downes, S.; Tan, J.K.; Nakano, Y.; Bowman, C.J.; Hoskins, J.L.; et al. Loss of metal ions, disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase. PLoS ONE 2009, 4, e5004. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Liba, A.; Sohn, S.H.; Seetharaman, S.V.; Bilsel, O.; Matthews, C.R.; Hart, P.J.; Valentine, J.S.; Hayward, L.J. Metal deficiency increases aberrant hydrophobicity of mutant superoxide dismutases that cause amyotrophic lateral sclerosis. J. Biol. Chem. 2009, 284, 27746–27758. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Chattopadhyay, M.; Whitelegge, J.; Valentine, J.S. SOD1 aggregation and ALS: Role of metallation states and disulfide status. Curr. Top. Med. Chem. 2012, 12, 2560–2572. [Google Scholar] [CrossRef] [PubMed]
- Sirangelo, I.; Vella, F.M.; Irace, G.; Manco, G.; Iannuzzi, C. Glycation in demetalated superoxide dismutase 1 prevents amyloid aggregation and produces cytotoxic ages adducts. Front. Mol. Biosci. 2016, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; McMahon, B.; Gill, S.; Lelie, H.L.; Fromholt, S.; Brown, H.; Valentine, J.S.; Whitelegge, J.P.; Borchelt, D.R. Relationship between mutant Cu/Zn superoxide dismutase 1 maturation and inclusion formation in cell models. J. Neurochem. 2017, 140, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, R.; Cunningham, P.; Furtos-Matei, A.; Dahan, S.; Qi, X.F.; Crow, J.P.; Cashman, N.R.; Kondejewski, L.H.; Chakrabartty, A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 47551–47556. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, G.; Gonzales, V.; Coonfield, M.; Fromholt, D.; Copeland, N.G.; Jenkins, N.A.; Borchelt, D.R. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol. Dis. 2002, 10, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Boca, M.; Calderone, V.; Cantini, F.; Girotto, S.; Vieru, M. Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc. Natl. Acad. Sci. USA 2009, 106, 6980–6985. [Google Scholar] [CrossRef] [PubMed]
- Shaw, B.F.; Valentine, J.S. How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem. Sci. 2007, 32, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Lelie, H.L.; Liba, A.; Bourassa, M.W.; Chattopadhyay, M.; Chan, P.K.; Gralla, E.B.; Miller, L.M.; Borchelt, D.R.; Valentine, J.S.; Whitelegge, J.P. Copper and zincmetallation status of copper-zincsuperoxidedismutase from amyotrophiclateralsclerosistransgenic mice. J. Biol. Chem. 2011, 286, 2795–2806. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.K.; Chattopadhyay, M.; Sharma, S.; Souda, P.; Gralla, E.B.; Borchelt, D.R.; Whitelegge, J.P.; Valentine, J.S. Structural similarity of wild-type and ALS-mutant superoxide dismutase-1 fibrils using limited proteolysis and atomic force microscopy. Proc. Natl. Acad. Sci. USA 2013, 110, 10934–10939. [Google Scholar] [CrossRef] [PubMed]
- Ming, L.J.; Valentine, J.S. Insights into SOD1-linked amyotrophic lateral sclerosis from NMR studies of Ni2+- and other metal-ion-substituted wild-type copper-zinc superoxide dismutases. J. Biol. Inorg. Chem. 2014, 19, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Almer, G.; Yamashita, S.; Guégan, C.; Nagai, M.; Xu, Z.; Sosunov, A.A.; McKhann, G.M.; Przedborski, S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. USA 2006, 103, 6025–6030. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2010, 1802, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Cramaro, F.; Del Conte, R.; Viezzoli, M.S. Solution structure of apo Cu, Zn superoxidedismutase: Role of metal ions in proteinfolding. Biochemistry 2003, 42, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Bordo, D.; Djinović, K.; Bolognesi, M. Conserved patterns in the Cu, Zn superoxide dismutase family. J. Mol. Biol. 1994, 238, 366–386. [Google Scholar] [CrossRef] [PubMed]
- Assfalg, M.; Banci, L.; Bertini, I.; Turano, P.; Vasos, P.R. Superoxide dismutase folding/unfolding pathway: Role of the metal ions in modulating structural and dynamical features. J. Mol. Biol. 2003, 330, 145–158. [Google Scholar] [CrossRef]
- Caruano-Yzermans, A.L.; Bartnikas, T.B.; Gitlin, J.D. Mechanisms of the copper-dependent turnover of the copper chaperone for superoxide dismutase. J. Biol. Chem. 2006, 281, 13581–13587. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Barbieri, L.; Bertini, I.; Cantini, F.; Luchinat, E. In-cell NMR in E. coli to monitor maturationsteps of hSOD1. PLoS ONE 2011, 6, e23561. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Cantini, F.; Kozyreva, T.; Massagni, C.; Palumaa, P.; Rubino, J.T.; Zovo, K. Human superoxide dismutase 1 (hSOD1) maturation through interaction with human copper chaperone for SOD1 (hCCS). Proc. Natl. Acad. Sci. USA 2012, 109, 13555–13560. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Torres, A.S.; O’Halloran, T.V. Oxygen-induced maturation of SOD1: A key role for disulfide formation by the copper chaperone CCS. EMBO J. 2004, 23, 2872–2881. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.C.; Girouard, J.B.; Ulloa, J.L.; Subramaniam, J.R.; Wong, P.C.; Valentine, J.S.; Culotta, V.C. Mechanisms for activating Cu- and Zn-containing superoxide dismutase in the absence of the CCS Cu chaperone. Proc. Natl. Acad. Sci. USA 2004, 101, 5964–5969. [Google Scholar] [CrossRef] [PubMed]
- Hörnberg, A.; Logan, D.T.; Marklund, S.L.; Oliveberg, M. The coupling between disulphide status, metallation and dimer interface strength in Cu/Zn superoxide dismutase. J. Mol. Biol. 2007, 365, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Arnesano, F.; Banci, L.; Bertini, I.; Martinelli, M.; Furukawa, Y.; O’Halloran, T.V. The unusually stable quaternary structure of human Cu, Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J. Biol. Chem. 2004, 279, 47998–48003. [Google Scholar] [CrossRef] [PubMed]
- Doucette, P.A.; Whitson, L.J.; Cao, X.; Schirf, V.; Demeler, B.; Valentine, J.S.; Hansen, J.C.; Hart, P.J. Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability. J. Biol. Chem. 2004, 279, 54558–54566. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, M.J.; Normark, J.; Holmgren, A.; Oliveberg, M. Folding of human superoxide dismutase: Disulfide reduction prevents dimerization and produces marginally stable monomers. Proc. Natl. Acad. Sci. USA 2004, 101, 15893–15898. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; O’Halloran, T.V. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 2005, 280, 17266–17274. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.Z.; Zhu, H.; Shaw, B.F.; Rodriguez, J.A.; Doucette, P.A.; Sohn, S.H.; Durazo, A.; Faull, K.F.; Gralla, E.B.; Nersissian, A.M.; et al. Binding of a single zinc ion to one subunit of copper-zinc superoxide dismutase apoprotein substantially influences the structure and stability of the entire homodimeric protein. J. Am. Chem. Soc. 2007, 129, 4575–4583. [Google Scholar] [CrossRef] [PubMed]
- Leal, S.S.; Cristóvão, J.S.; Biesemeier, A.; Cardoso, I.; Gomes, C.M. Aberrant zinc binding to immature conformers of metal-free copper-zinc superoxide dismutase triggers amorphous aggregation. Metallomics 2015, 7, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Luchinat, E.; Barbieri, L.; Rubino, J.T.; Kozyreva, T.; Cantini, F.; Banci, L. In-cell NMR reveals potential precursor of toxic species from SOD1 fALS mutants. Nat. Commun. 2014, 5, 5502. [Google Scholar] [CrossRef] [PubMed]
- Lamb, A.L.; Torres, A.S.; O’Halloran, T.V.; Rosenzweig, A.C. Heterodimeric structure of superoxide dismutase in complex with its metallochaperone. Nat. Struct. Biol. 2001, 8, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Cantini, F.; D’Onofrio, M.; Viezzoli, M.S. Structure and dynamics of copper-free SOD: The protein before binding copper. Protein Sci. 2002, 11, 2479–2492. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.S.; Pantoliano, M.W.; McDonnell, P.J.; Burger, A.R.; Lippard, S.J. pH-dependent migration of copper(II) to the vacant zinc-binding site of zinc-free bovine erythrocyte superoxide dismutase. Proc. Natl. Acad. Sci. USA 1979, 76, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Barbieri, L.; Luchinat, E.; Secci, E. Visualization of redox-controlled protein fold in living cells. Chem. Biol. 2013, 20, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Fridovich, I. On the stability of bovine superoxide dismutase: The effect of metals. J. Biol. Chem. 1973, 248, 2645–2649. [Google Scholar] [PubMed]
- Stathopulos, P.B.; Rumfeldt, J.A.; Karbassi, F.; Siddall, C.A.; Lepock, J.R.; Meiering, E.M. Calorimetric analysis of thermodynamic stability and aggregation for apo and holo amyotrophic lateral sclerosis-associated Gly-93 mutants of superoxide dismutase. J. Biol. Chem. 2006, 281, 6184–6193. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Miller, T.M.; Cleveland, D.W. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu. Rev. Neurosci. 2004, 27, 723–749. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R. Mutation in Cu, Zn superoxide dismutase gene are associated with familial amyotrophyc lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Velde, C.V.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Reaume, A.G.; Elliott, J.L.; Hoffman, E.K.; Kowall, N.W.; Ferrante, R.J.; Siwek, D.R.; Wilcox, H.M.; Flood, D.G.; Beal, M.F.; Brown, R.H. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 1996, 13, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Tu, P.H.; Raju, P.; Robinson, K.A.; Gurney, M.E.; Trojanowski, J.Q.; Lee, V.M. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc. Natl. Acad. Sci. USA 1996, 93, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Lee, M.K.; Slunt, H.S.; Guarnieri, M.; Xu, Z.; Wong, P.C.; Brown, R.H.; Price, D.L.; Sisodia, S.S.; Cleveland, D.W. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8292–8296. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.A.; Dalton, M.J.; Gurney, M.E.; Kopito, R.R. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2000, 97, 12571–12576. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, P.A.; Ernhill, K.; Andersen, P.M.; Bergemalm, D.; Brännström, T.; Gredal, O.; Nilsson, P.; Marklund, S.L. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain 2004, 127 Pt 1, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Zetterström, P.; Stewart, H.G.; Bergemalm, D.; Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brännström, T.; Oliveberg, M.; Marklund, S.L. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc. Natl. Acad. Sci. USA 2007, 104, 14157–14162. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, T.E.; Li, Y.; Glass, J.D. Cellular toxicity of mutant SOD1 protein is linked to an easily soluble, non-aggregated form in vitro. Neurobiol. Dis. 2013, 49, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Hayward, L.J.; Rodriguez, J.A.; Kim, J.W.; Tiwari, A.; Goto, J.J.; Cabelli, D.E.; Valentine, J.S.; Brown, R.H. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 15923–15931. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Shaw, B.F.; Durazo, A.; Sohn, S.H.; Doucette, P.A.; Nersissian, A.M.; Faull, K.F.; Eggers, D.K.; Tiwari, A.; Hayward, L.J.; et al. Destabilization of apoprotein is insufficient to explain Cu, Zn-superoxide dismutase-linked ALS pathogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 10516–10521. [Google Scholar] [CrossRef] [PubMed]
- Bergemalm, D.; Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brännström, T.; Rehnmark, A.; Marklund, S.L. Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models. J. Neurosci. 2006, 26, 4147–4154. [Google Scholar] [CrossRef] [PubMed]
- Elia, F.; Cantini, F.; Chiti, F.; Dobson, C.M.; Bemporad, F. Direct conversion of an enzyme from native-like to amyloid-like aggregates within inclusion bodies. Biophys. J. 2017, 112, 2540–2551. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Plotkin, S.S. SOD1 exhibits allosteric frustration to facilitate metal binding affinity. Proc. Natl. Acad. Sci. USA 2013, 110, 3871–3876. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.K.; Kopito, R.R. Impaired post-translational folding of familial ALS-linked Cu, Zn superoxide dismutase mutants. EMBO J. 2007, 26, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Teilum, K.; Smith, M.H.; Schulz, E.; Christensen, L.C.; Solomentsev, G.; Oliveberg, M.; Akke, M. Transient structural distortion of metal-free Cu/Zn superoxide dismutase triggers aberrant oligomerization. Proc. Natl. Acad. Sci. USA 2009, 106, 18273–18278. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.M.; Boswell, S.A.; Colón, W. Kinetic stability of Cu/Zn superoxide dismutase is dependent on its metal ligands: Implications for ALS. Biochemistry 2004, 43, 16525–16531. [Google Scholar] [CrossRef] [PubMed]
- Leinartaite, L.; Saraboji, K.; Nordlund, A.; Logan, D.T.; Oliveberg, M. Folding catalysis by transient coordination of Zn2+ to the Cu ligands of the ALS-associated enzyme Cu/Zn superoxide dismutase 1. J. Am. Chem. Soc. 2010, 132, 13495–13504. [Google Scholar] [CrossRef] [PubMed]
- Kayatekin, C.; Zitzewitz, J.A.; Matthews, C.R. Zinc binding modulates the entire folding free energy surface of human Cu, Zn superoxide dismutase. J. Mol. Biol. 2008, 384, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Rakhit, R.; Crow, J.P.; Lepock, J.R.; Kondejewski, L.H.; Cashman, N.R.; Chakrabartty, A. Monomeric Cu, Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J. Biol. Chem. 2004, 279, 15499–15504. [Google Scholar] [CrossRef] [PubMed]
- Shaw, B.F.; Durazo, A.; Nersissian, A.M.; Whitelegge, J.P.; Faull, K.F.; Valentine, J.S. Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J. Biol. Chem. 2006, 281, 18167–18176. [Google Scholar] [CrossRef] [PubMed]
- Durazo, A.; Shaw, B.F.; Chattopadhyay, M.; Faull, K.F.; Nersissian, A.M.; Valentine, J.S.; Whitelegge, J.P. Metal-free superoxide dismutase-1 and three different amyotrophic lateral sclerosis variants share a similar partially unfolded beta-barrel at physiological temperature. J. Biol. Chem. 2009, 284, 34382–34389. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Furukawa, Y.; Nukina, N.; Dokholyan, N.V. Local unfolding of Cu, Zn superoxide dismutase monomer determines the morphology of fibrillar aggregates. J. Mol. Biol. 2012, 421, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kaneko, K.; Yamanaka, K.; O’Halloran, T.V.; Nukina, N. Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J. Biol. Chem. 2008, 283, 24167–24176. [Google Scholar] [CrossRef] [PubMed]
- Bartnikas, T.B.; Gitlin, J.D. Mechanisms of biosynthesis of mammalian copper/zinc superoxide dismutase. J. Biol. Chem. 2003, 278, 33602–33608. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.J.; Nersissian, A.; Huang, H.; Yeom, H.; Nishida, C.R.; Graden, J.A.; Gralla, E.B.; Valentine, J.S. The metal binding properties of the zinc site of yeast copper-zinc superoxide dismutase: Implications for amyotrophic lateral sclerosis. J. Biol. Inorg. Chem. 2000, 5, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.P.; Sampson, J.B.; Zhuang, Y.; Thompson, J.A.; Beckman, J.S. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 1997, 69, 1936–1944. [Google Scholar] [CrossRef] [PubMed]
- Leal, S.S.; Cardoso, I.; Valentine, J.S.; Gomes, C.M. Calcium ions promote superoxide dismutase 1 (SOD1) aggregation into non-fibrillar amyloid: A link to toxic effects of calcium overload in amyotrophic lateral sclerosis (ALS)? J. Biol. Chem. 2013, 288, 25219–25228. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, D.D.; Schuermann, J.P.; Cao, X.; Holloway, S.P.; Borchelt, D.R.; Carroll, M.C.; Proescher, J.B.; Culotta, V.C.; Hart, P.J. Structural and biophysical properties of the pathogenic SOD1 variant H46R/H48Q. Biochemistry 2009, 48, 3436–3647. [Google Scholar] [CrossRef] [PubMed]
- Leal, S.S.; Botelho, H.M.; Gomes, C.M. Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev. 2012, 256, 2253–2270. [Google Scholar] [CrossRef]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar] [CrossRef] [PubMed]
- DuBay, K.F.; Pawar, A.P.; Chiti, F.; Zurdo, J.; Dobson, C.M.; Vendruscolo, M. Prediction of the absolute aggregation rates of amyloidogenic polypeptide chains. J. Mol. Biol. 2004, 341, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Kurlander, H.M.; Patten, B.M. Metals in spinal cord tissue of patients dying of motor neuron disease. Ann. Neurol. 1979, 6, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, E.; Furukawa, Y. Copper homeostasis as a therapeutic target in amyotrophic lateral sclerosis with SOD1 mutations. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, E.; Okawa, E.; Watanabe, S.; Ono, S.; Marklund, S.L. Dysregulation of intracellular copper homeostasis is common to transgenic mice expressing human mutant superoxide dismutase-1s regardless of their copper-binding abilities. Neurobiol. Dis. 2013, 54, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Bousser, M.G.; Malier, M. Penicillamine in amyotrophic lateral sclerosis. Lancet 1979, 1, 168. [Google Scholar] [CrossRef]
Sample Availability: Not available. |


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirangelo, I.; Iannuzzi, C. The Role of Metal Binding in the Amyotrophic Lateral Sclerosis-Related Aggregation of Copper-Zinc Superoxide Dismutase. Molecules 2017, 22, 1429. https://doi.org/10.3390/molecules22091429
Sirangelo I, Iannuzzi C. The Role of Metal Binding in the Amyotrophic Lateral Sclerosis-Related Aggregation of Copper-Zinc Superoxide Dismutase. Molecules. 2017; 22(9):1429. https://doi.org/10.3390/molecules22091429
Chicago/Turabian StyleSirangelo, Ivana, and Clara Iannuzzi. 2017. "The Role of Metal Binding in the Amyotrophic Lateral Sclerosis-Related Aggregation of Copper-Zinc Superoxide Dismutase" Molecules 22, no. 9: 1429. https://doi.org/10.3390/molecules22091429
APA StyleSirangelo, I., & Iannuzzi, C. (2017). The Role of Metal Binding in the Amyotrophic Lateral Sclerosis-Related Aggregation of Copper-Zinc Superoxide Dismutase. Molecules, 22(9), 1429. https://doi.org/10.3390/molecules22091429