Voltammetric Study of Some 3-Aryl-quinoxaline-2-carbonitrile 1,4-di-N-oxide Derivatives with Anti-Tumor Activities

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
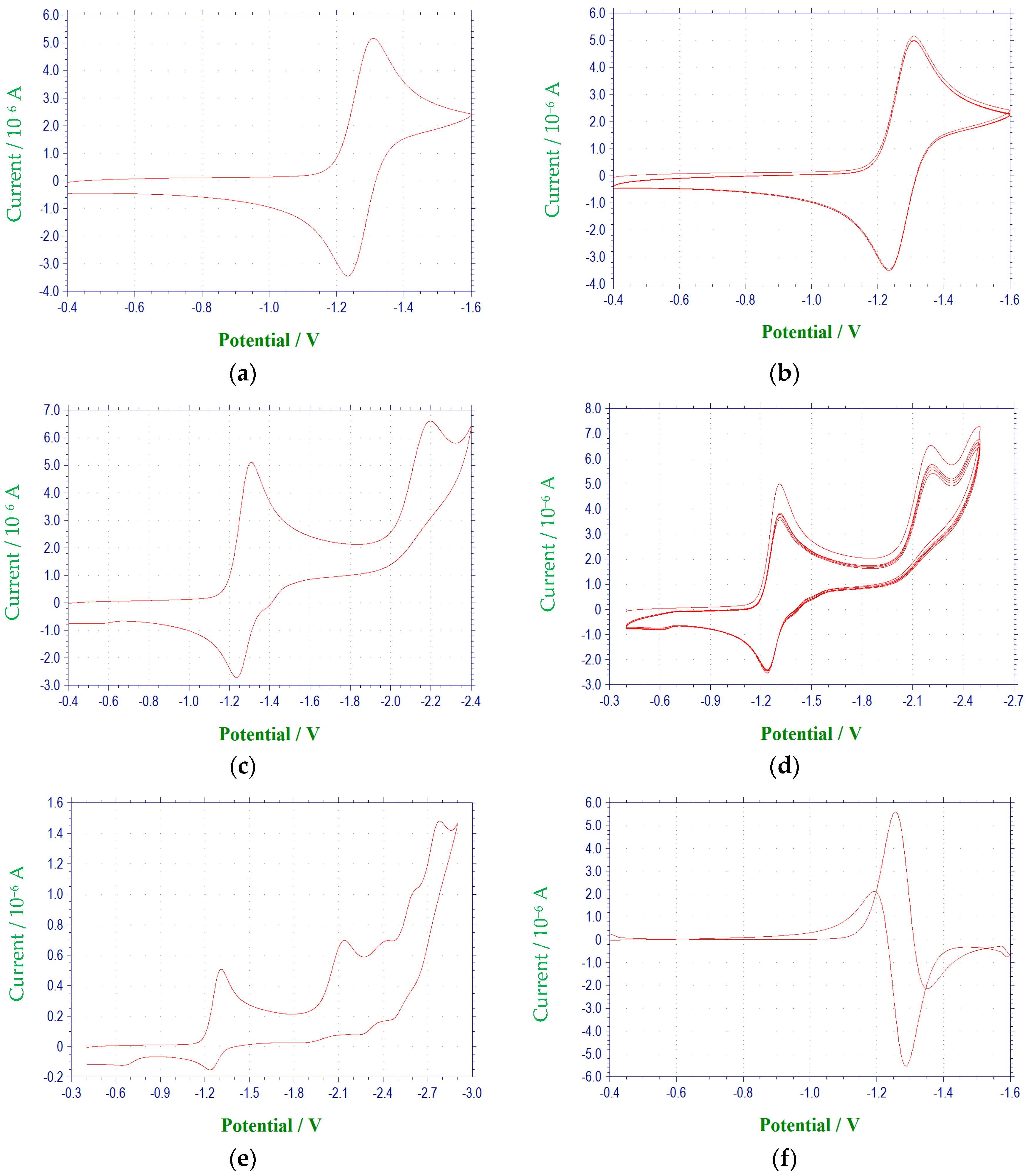
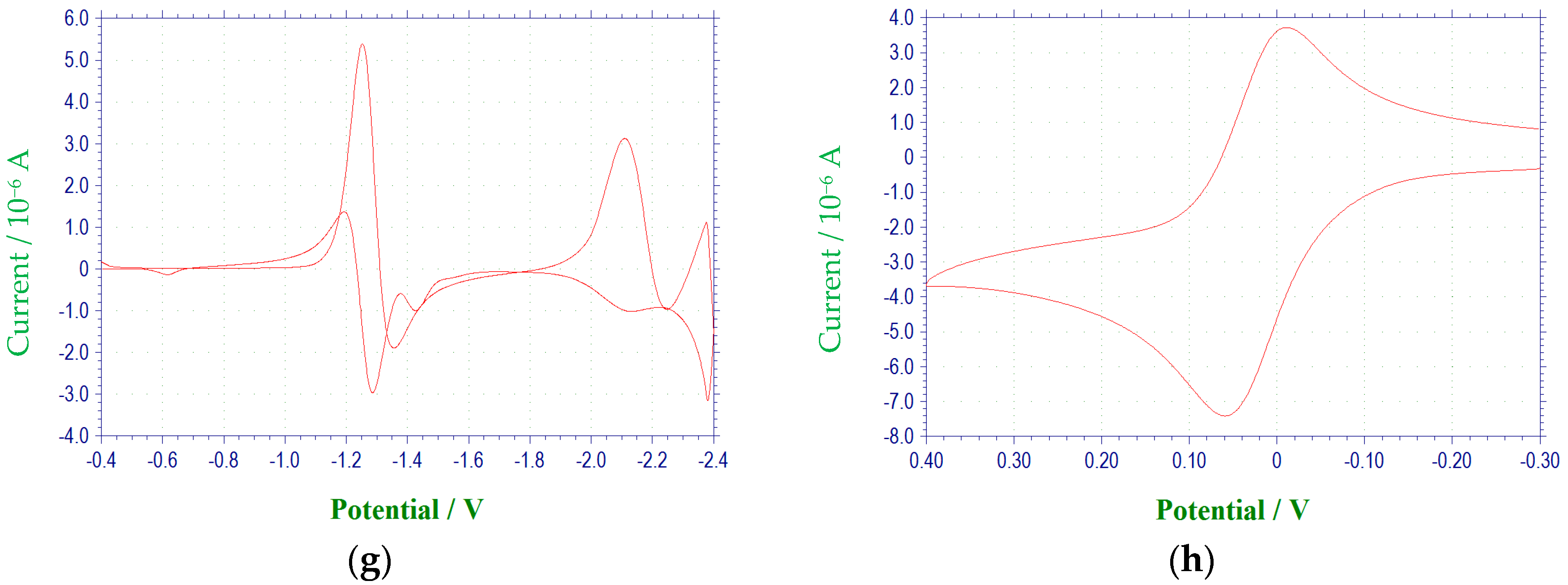
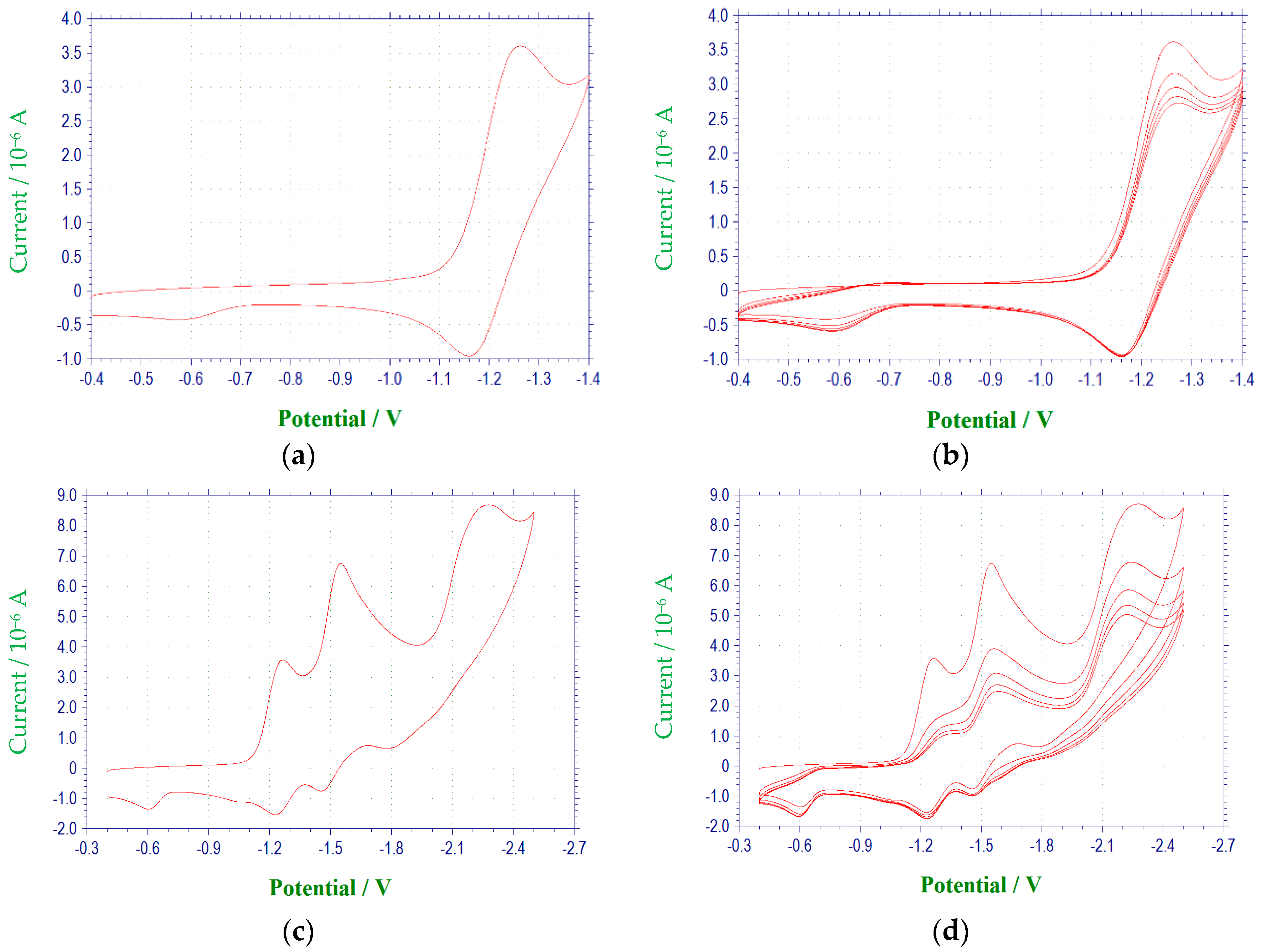
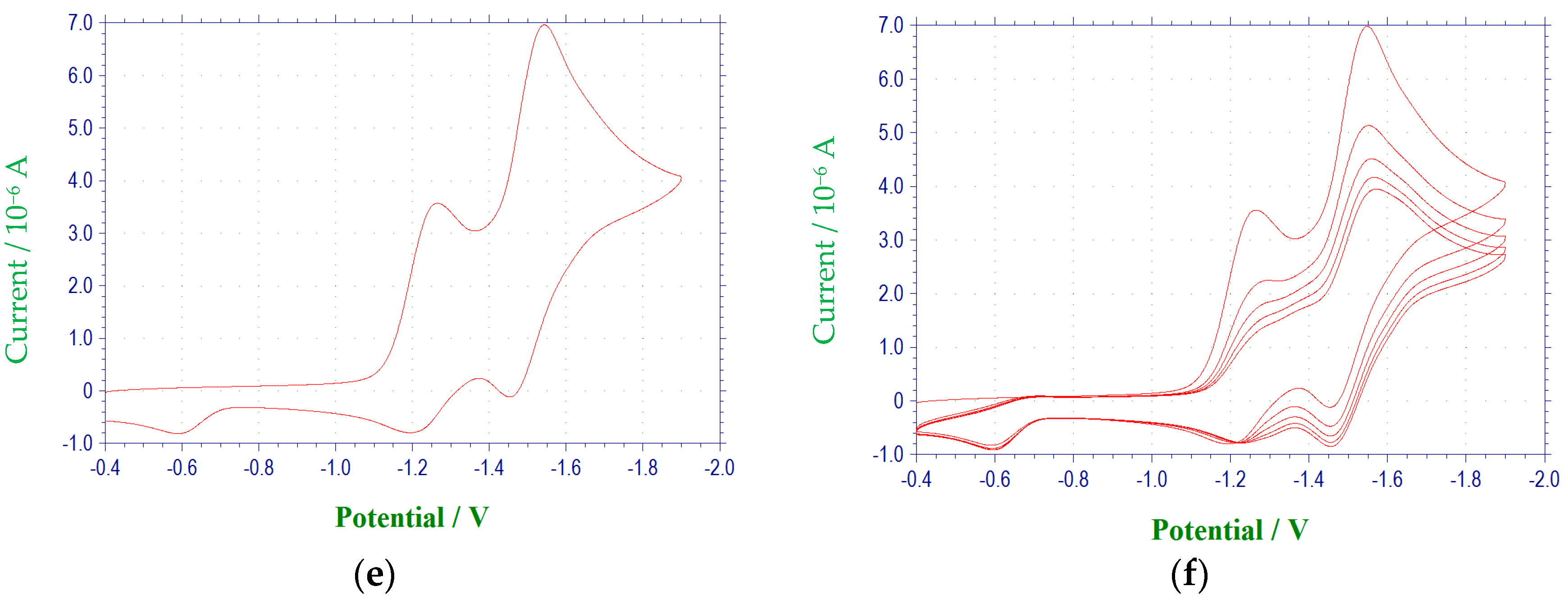
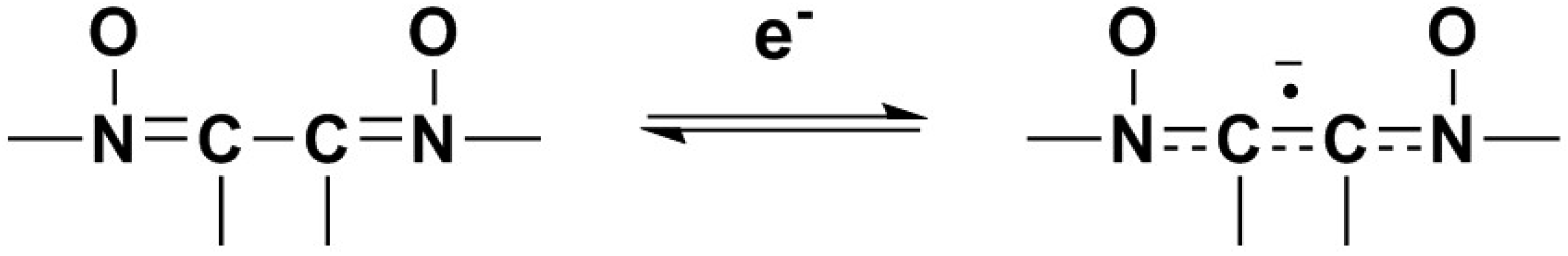
2.1. Electrochemical Behavior
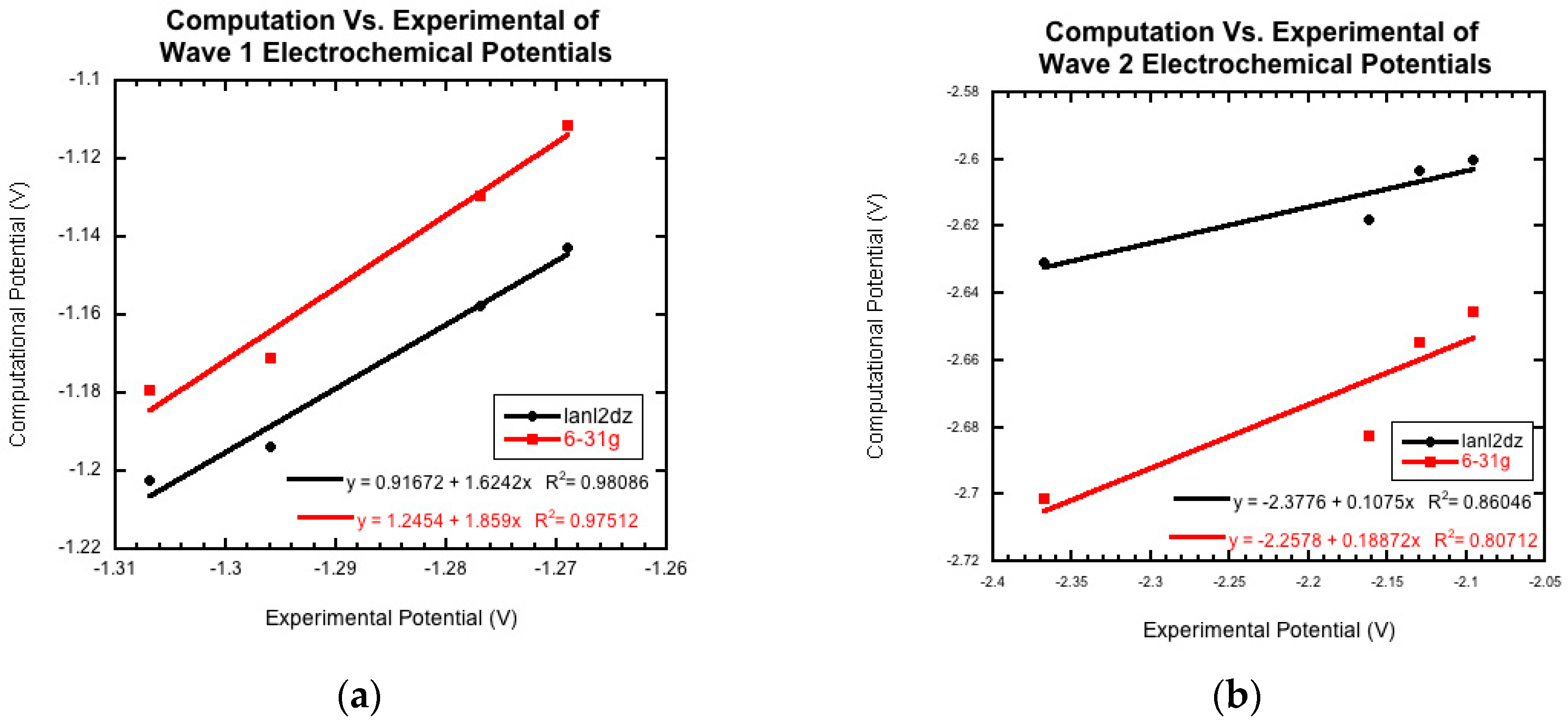
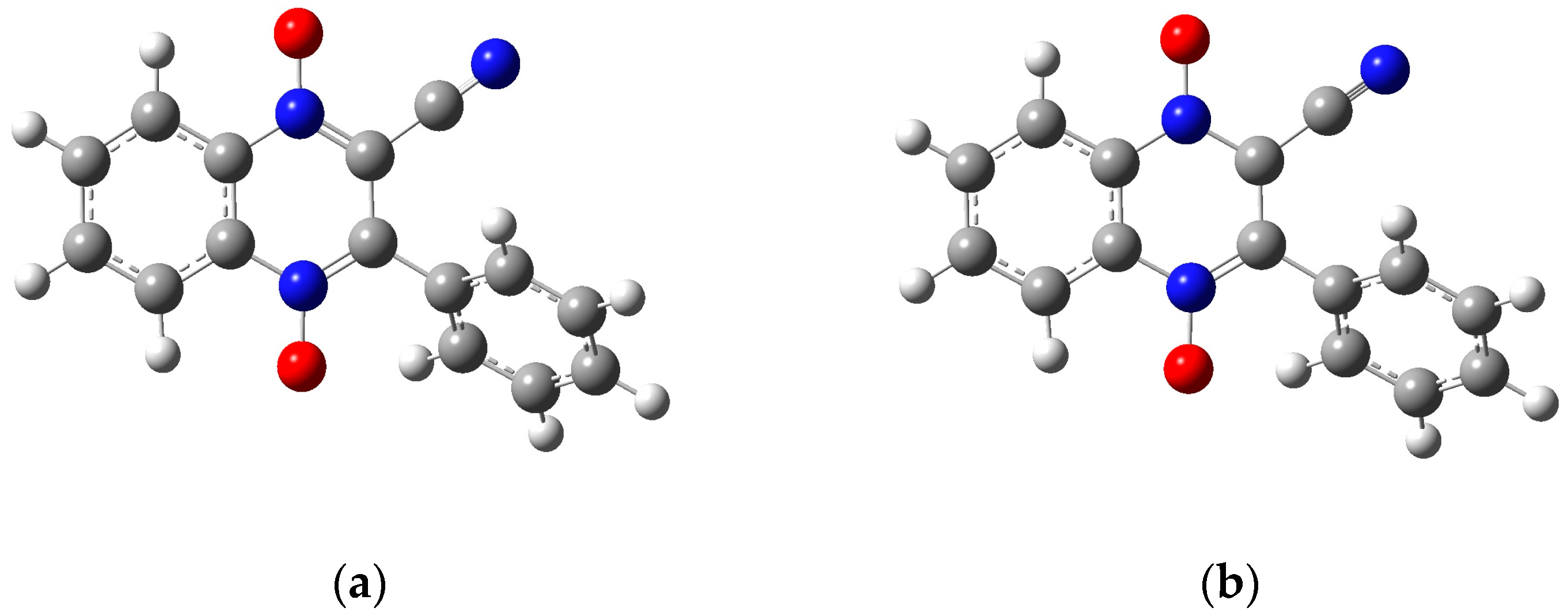
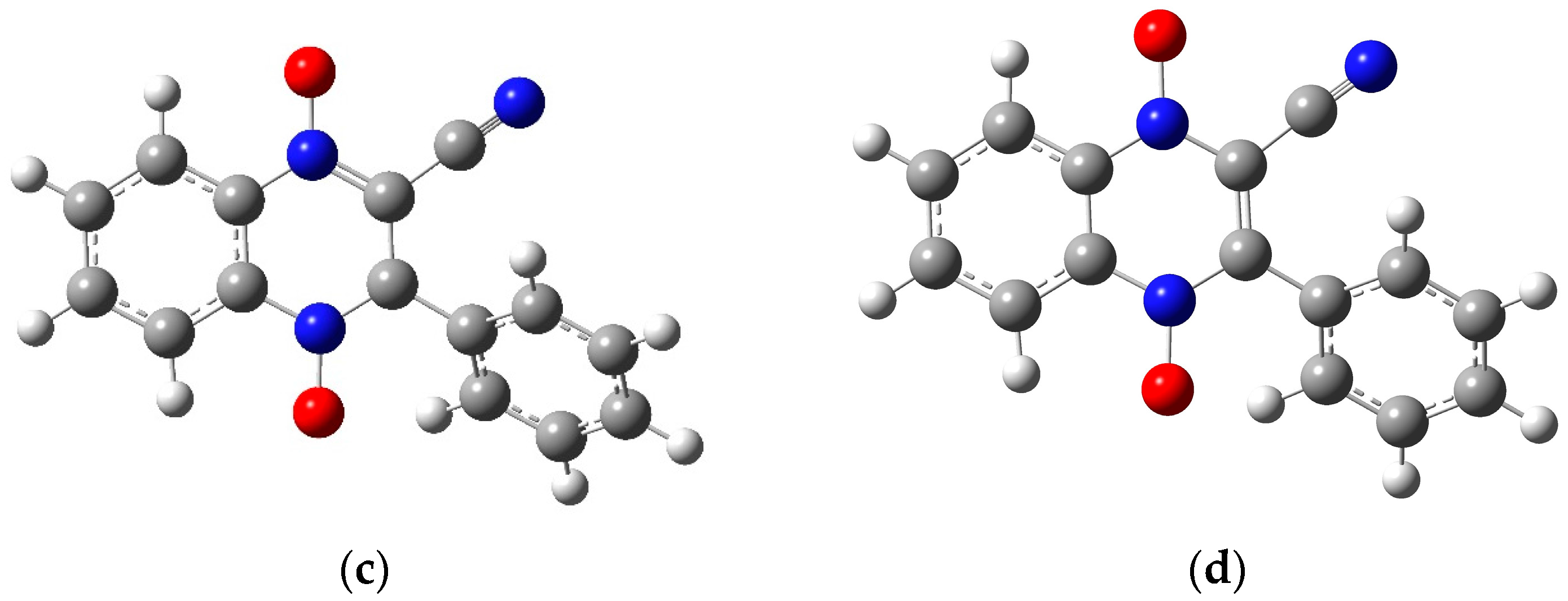
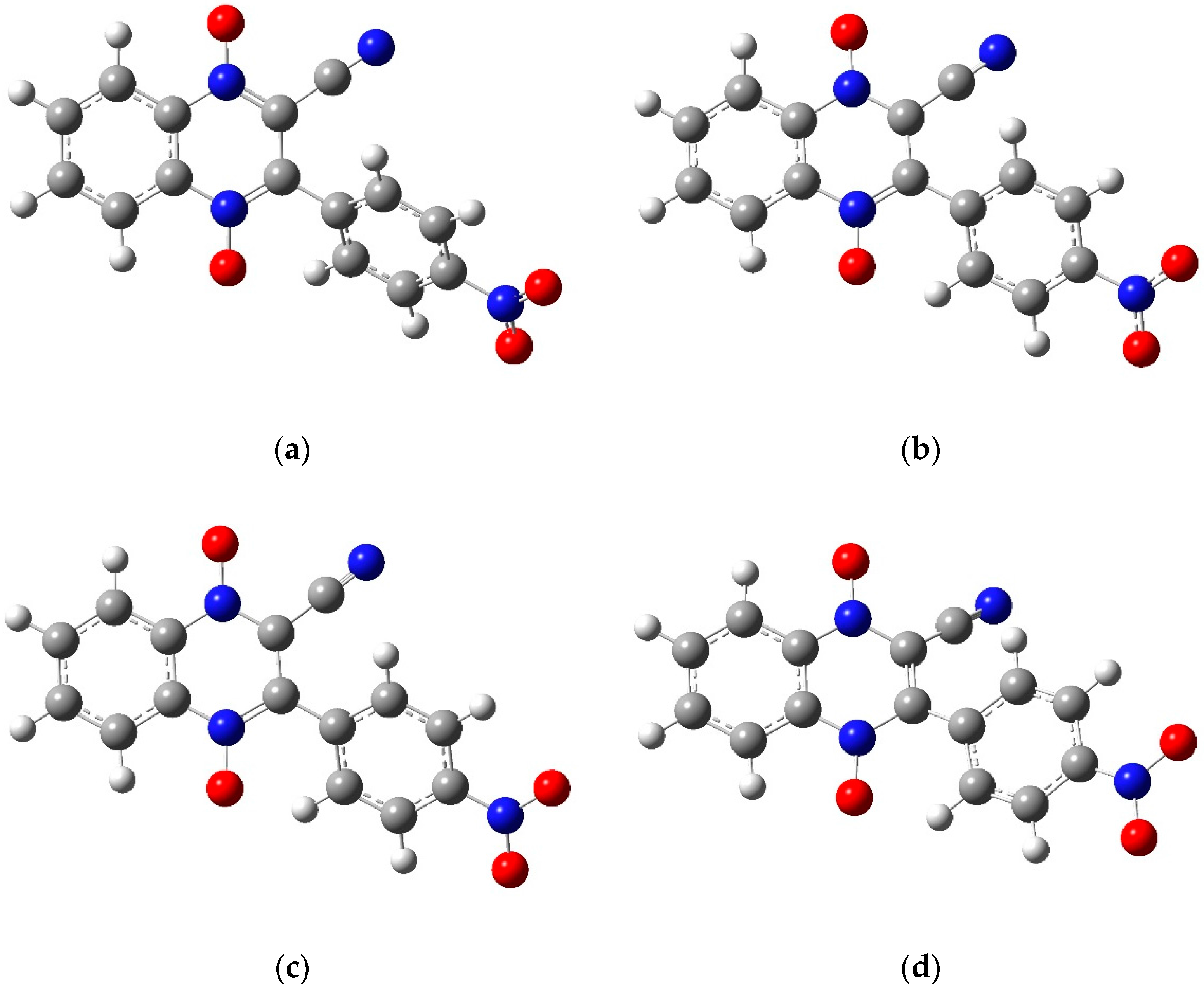
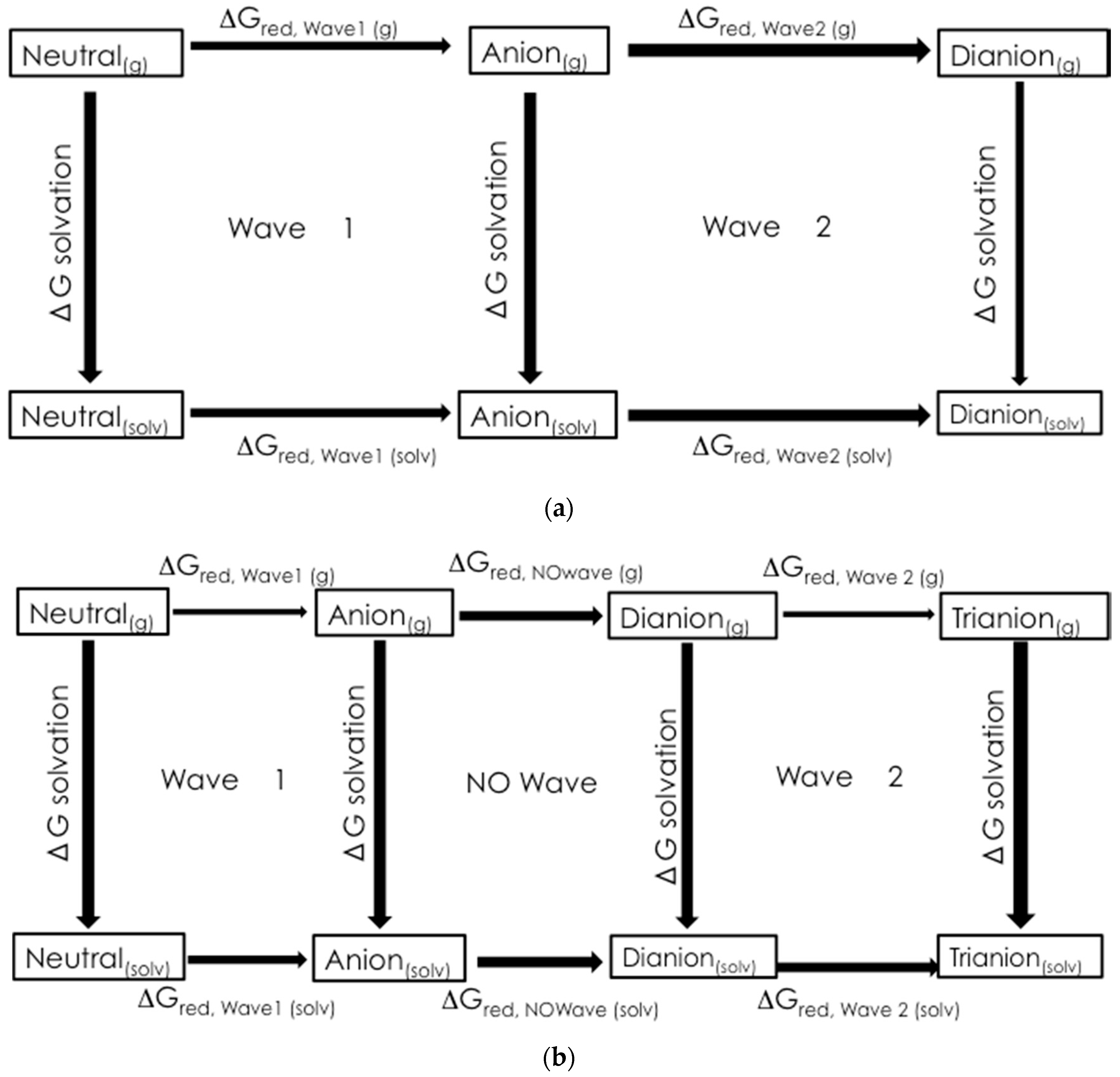
2.2. Preliminary Computational Study
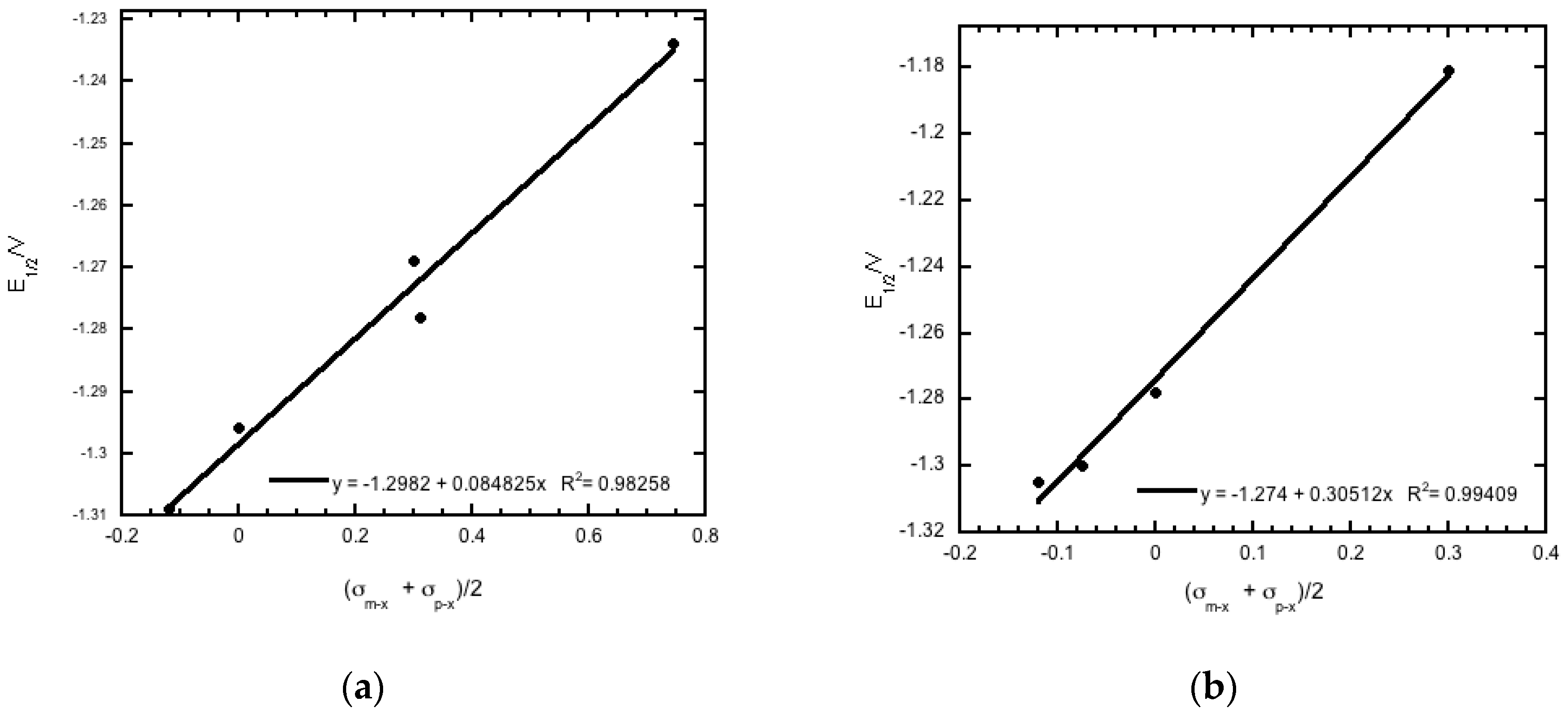
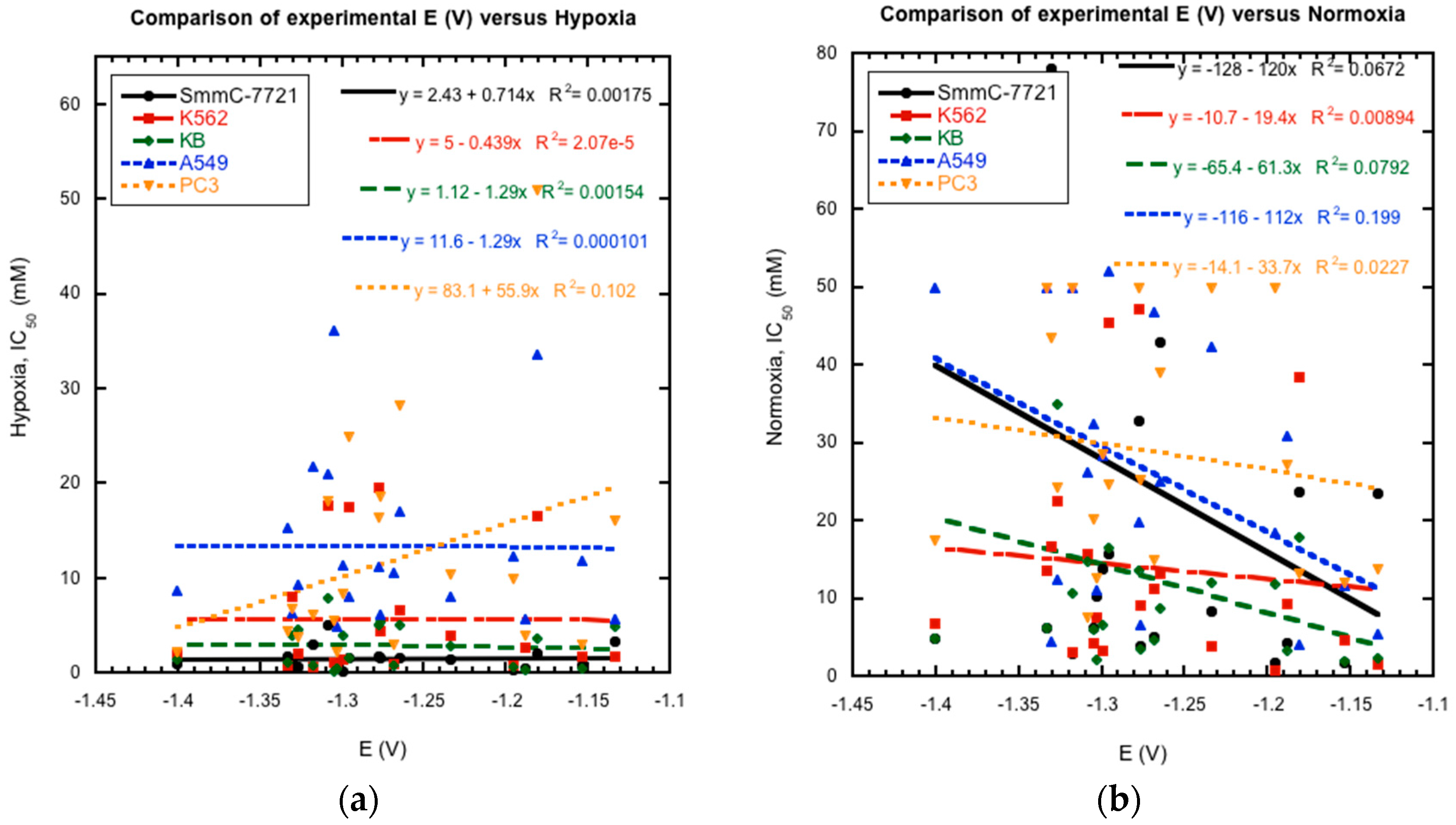
2.3. Reduction Potentials versus Cytotoxicity
3. Materials and Methods
3.1. Chemical Synthesis
3.2. Electrochemistry
3.3. Computational Chemistry
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Dimethylformamide | DMF |
| half-wave potential | E1/2 |
| anodic peak potential | Epa |
| cathodic peak potential | Epc |
| difference in peak potentials | ΔEp |
| anodic peak current | ipa |
| cathodic peak current | ipc |
| ratio of peak currents | ipa/ipc |
| reaction constant | ρπ,R |
| total polar substituent constant | σx |
| substituent constant for meta substituent | σm−x |
| substituent constant for para substituent | σp−x |
References
- Gonzalez, M.; Cerecetto, H.; Monge, A. Quinoxaline 1,4-dioxide and phenazine 5,10-dioxide chemistry and biology. Top. Heterocycl. Chem. 2007, 11, 179–211. [Google Scholar]
- Carta, A.; Corona, P.; Loriga, M. Quinoxaline 1,4-dioxide: A versatile scaffold endowed with manifold activities. Curr. Med. Chem. 2005, 12, 2259–2272. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Sa, W.; Cao, C.; Guo, L.; Hao, H.; Liu, Z.; Wang, X.; Yuan, Z. Quinoxaline 1,4-di-N-oxides: Biological activities and mechanisms of action. Front. Pharmacol. 2016, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Suter, W.; Rosselet, A.; Knusel, F. Mode of action of quindoxin and substituted quinoxaline-di-N-oxides on escherichia coli. Antimicrob. Agents Chemother. 1978, 13, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Soliman, D.H. Anti-bacterial and anti-fungal activities of new quinoxaline 1,4-di-N-oxide derivatives. Int. J. Organ. Chem. 2013, 3, 65–72. [Google Scholar] [CrossRef]
- Vicente, E.; Pérez-Silanes, S.; Lima, L.M.; Ancizu, S.; Burguete, A.; Solano, B.; Villar, R.; Aldana, I.; Monge, A. Selective activity against mycobacterium tuberculosis of new quinoxaline 1,4-di-N-oxides. Biorgan. Med. Chem. 2009, 17, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Estevez, T.; Quilano, M.; Burguete, A.; Cabanillas, B.; Zimic, M.; Malaga, E.; Verastegui, M.; Pérez-Silanes, S.; Aldana, I.; Monge, A. Trypanocidal properties, structure-activity relationship and computational studies of quinoxaline 1,4-di-N-oxide derivatives. Exp. Parasitol. 2011, 127, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Pabón, A.; Galiano, S.; Burguete, A.; Pérez-Silanes, S.; Deharo, E.; Monge, A.; Aldana, I. Synthesis, biological evaluation and structure-activity relationships of new quinoxaline derivatives as anti-plasmodium falciparum agents. Molecules 2014, 19, 2166–2180. [Google Scholar] [CrossRef] [PubMed]
- Ancizu, S.; Moreno, E.; Torres, E.; Burguete, A.; Pérez-Silanes, S.; Benítez, D.; Villar, R.; Solano, B.; Marín, A.; Aldana, I.; et al. Heterocyclic-2-carboxylic acid (3-cyano-1,4-di-N-oxidequinoxalin-2-yl)amide derivatives as hits for the development of neglected disease drugs. Molecules 2009, 14, 2256–2272. [Google Scholar] [CrossRef] [PubMed]
- Torres, E.; Moreno-Viguri, E.; Galiano, S.; Devarapally, G.; Crawford, P.W.; Azqueta, A.; Arbillaga, L.; Varela, J.; Birriel, E.; Di Maio, R.; et al. Novel quinoxaline 1,4-di-N-oxide derivatives as new potential antichagasic agents. Eur. J. Med. Chem. 2013, 66, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.; Paglietta, G.; Nikookar, M.E.R.; Sanna, P.; Sechi, L.; Zanetti, S. Novel substituted quinoxaline 1,4-dioxides with in vitro antimycobacterial and anticandida activity. Eur. J. Med. Chem. 2002, 37, 355–366. [Google Scholar] [CrossRef]
- Burguete, A.; Pontiki, E.; Hadjipavlou-Litina, D.; Ancizu, S.; Villar, R.; Solano, B.; Moreno, E.; Torres, E.; Perez, S.; Aldana, I.; et al. Synthesis and biological evaluation of new quinoxaline derivatives as antioxidant and anti-inflammatory agents. Chem. Biol. Drug Des. 2011, 77, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.F.; Yadav, P.; Shinde, S.S.; Hong, C.R.; Pullen, S.M.; Reynisson, J.; Wilson, W.R.; Hay, M.P. Radical chemistry and cytotoxicity of bioreductive 3-substituted quinoxaline di-N-oxides. Chem. Res. Toxicol. 2016, 29, 1310–1324. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, B.; Jaso, A.; Aldana, I.; Monge, A. Synthesis and anticancer activity evaluation of new 2-alkylcarbonyl and 2-benzoyl-3-trifluoromethyl-quinoxaline 1,4-di-N-oxide derivatives. Biorgan. Med. Chem. 2004, 12, 3711–3721. [Google Scholar] [CrossRef] [PubMed]
- Amin, K.M.; Ismail, M.F.; Noaman, D.; Soliman, D.H.; Ammar, Y.A. New quinoxaline 1,4-di-N-oxides. Part 1: Hypoxia-selective cytotoxins and anticancer agents derived from quinoxaline 1,4-di-N-oxides. Bioorgan. Med. Chem. 2006, 14, 6917–6923. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Viguri, E.; Pérez-Silanes, S. Quinoxaline 1,4-di-N-oxide derivatives: Interest in the treatment of chagas disease. Rev. Virtual Quím. 2013, 5, 1101–1119. [Google Scholar] [CrossRef]
- Crawford, P.W.; Scamehorn, R.G.; Hollstein, U.; Ryan, M.D.; Kovacic, P. Cyclic voltammetry of phenazines and quinoxalines including mono- and di-N-oxides. Relation to structure and antimicrobial activity. Chemico-Biol. Interact. 1986, 60, 67–84. [Google Scholar] [CrossRef]
- Perez-Silanes, S.; Devarapally, G.; Torres, E.; Moreno, E.; Aldana, I.; Monge, A.; Crawford, P.W. Cyclic voltammetric study of some anti-chagas active quinoxaline 1,4-di-N-oxide-2-ketone derivatives. Helv. Chim. Acta 2013, 96, 217–227. [Google Scholar] [CrossRef]
- Moreno, E.; Pérez-Silanes, S.; Gouravaram, S.; Macharam, A.; Ancizu, S.; Torres, E.; Aldana, I.; Monge, A.; Crawford, P.W. 1,4-Di-N-oxide quinoxaline-2-carboxamide: Cyclic voltammetry and relationship between electrochemical behavior, structure, and anti-tuberculosis activity. Electrochim. Acta 2011, 56, 3270–3275. [Google Scholar] [CrossRef]
- Ryan, M.D.; Scamehorn, R.G.; Kovacic, P. Charge transfer in the mechanism of drug action involving quinoxaline di-N-oxides. J. Pharm. Sci. 1985, 74, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xia, Q.; Shangguan, S.; Liu, X.; Hu, Y.; Sheng, R. Synthesis and biological evaluation of 3-aryl-quinoxaline-2-carbonitrile 1,4-di-N-oxide derivatives as hypoxic selective anti-tumor agents. Molecules 2012, 17, 9683–9696. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.; Jämstorp, E.; Strømme, M.; Sjödin, M. Computational electrochemistry study of 16 isoindole-4,7-diones as candidates for organic cathode materials. J. Phys. Chem. C 2012, 116, 3793–3801. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Rieger, P.H. Electrochemistry, 2nd ed.; Chapman and Hall: New York, NY, USA, 1994. [Google Scholar]
- Ames, J.R.; Houghtaling, M.A.; Terrian, D.L. Cyclic voltammetry of some quinoxaline di-N-oxides and quinoxalines in dimethylformamide. Electrochim. Acta 1992, 37, 1433–1436. [Google Scholar] [CrossRef]
- Barqawi, K.R.; Atfah, M.A. A cyclic voltammetric study of some quinoxaline di-N-oxides and quinoxalines in acetonitrile: Substituent effect on the cathodic reduction. Electrochimim. Acta 1987, 32, 597–599. [Google Scholar] [CrossRef]
- Miyazaki, H.; Matsuhisa, Y.; Kubota, T. Cyclic voltammetry of aromatic amine n-oxides in nonaqueous solvents and the stability of the free radicals produced. Bull. Chem. Soc. Jpn. 1981, 54, 3850–3853. [Google Scholar] [CrossRef]
- Zuman, P. Substituent Effects in Organic Polarography; Plenum Press: New York, NY, USA, 1967. [Google Scholar]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Strier, M.P.; Cavagnol, J.C. The polarography of quinoxaline. II. 6-substituted derivatives. J. Am. Chem. Soc. 1958, 80, 1565–1568. [Google Scholar] [CrossRef]
- Tocher, J.H.; Edwards, D.I. Electrochemical characteristics of nitroheterocyclic compounds of biological interest: I. The influence of solvent. Free Radic. Res. Commun. 1988, 4, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Tocher, J.H.; Edwards, D.I. Electrochemical characteristics of nitro-heterocyclic compounds of biological interest: IV. Lifetime of the metronidazole radical anion. Free Radic. Res. Commun. 1989, 6, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Tocher, J.H.; Edwards, D.I. Electrochemical characteristics of nitroheterocyclic compounds of biological interest: VIII. Stability of nitro radical anions from cyclic voltammetric studies. Free Radic. Res. Commun. 1992, 16, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Bollo, S.; Nunez-Vergara, L.J.; Martinez, C.; Chauviere, G.; Perie, J.; Squella, J.A. Voltammetric study of nitro radical anion generated from some nitrofuran compounds of pharmacological significance. Electroanalysis 2003, 15, 19–25. [Google Scholar] [CrossRef]
- Kunz, K.R.; Iyengar, B.S.; Dorr, R.T.; Alberts, D.S.; Remers, W.A. Structure activity relationship for mitomycin c and mitomycin a analogs. J. Med. Chem. 1991, 34, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Hoekman, D. Exploring Qsar: Fundamentals and Applications in Chemistry and Biology; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Gritzner, G.; Kuta, J. Recommendations on reporting electrode potentials in nonaqueous solvents. Pure Appl. Chem. 1984, 56, 461–466. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision 5.0.9; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. Gaussview; Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Palma, J.L.; Batista, V.S. Tutorial on ab Initio Redox Potential Calculations; Department of Chemistry, Yale University: New Haven, CT, USA, 2016. [Google Scholar]
Sample Availability: Not available. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
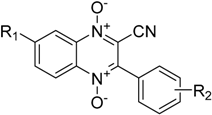
| Compound | R1 | R2 |
|---|---|---|
| 1a | H | H |
| 1b | H | 3-CH3 |
| 1c | H | 3-Cl |
| 1d | H | 4-Br |
| 1e | H | 4-NO2 |
| 2a | CH3 | H |
| 2b | CH3 | 3-CH3 |
| 2c | CH3 | 3-Cl |
| 2d | CH3 | 4-Br |
| 2e | CH3 | 4-NO2 |
| 3a | OCH3 | H |
| 3b | OCH3 | 3-CH3 |
| 3c | OCH3 | 3-Cl |
| 3d | OCH3 | 4-Br |
| 3e | OCH3 | 4-NO2 |
| 4a | Cl | H |
| 4b | Cl | 3-CH3 |
| 4c | Cl | 3-Cl |
| 4d | Cl | 4-Br |
| 4e | Cl | 4-NO2 |
| 1st N-oxide Reduction | Nitro Group Reduction | 2nd N-oxide Reduction b | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | E1/2 (V) | ΔEp (V) | Epc − E1/2 (V) | ipc (μA) | ipa/ipc | E1/2 (V) | ΔEp (V) | Epc − E1/2 (V) | ipc (μA) | ipa/ipc | Epc (V) | ipc (μA) |
| 1a | −1.296 | 0.076 | −0.038 | 4.883 | 0.918 | −2.163 | 3.869 | |||||
| 1b | −1.309 | 0.082 | −0.041 | 2.135 | 0.348 | −2.56 (sh) c | ||||||
| 1c | −1.269 | 0.070 | −0.035 | 2.846 | 0.761 | −2.097 | 3.753 | |||||
| 1d | −1.278 | 0.069 | −0.035 | 1.177 | 0.689 | −2.06 (sh) c | ||||||
| 1e | −1.234 | 0.103 | −0.051 | 3.220 | 0.297 | −1.518 | 0.088 | −0.044 | 3.826 | 0.630 | −2.306 | 3.808 |
| 2a | −1.327 | 0.097 | −0.049 | 3.488 | 0.830 | −2.310 | 1.501 | |||||
| 2b | −1.318 | 0.103 | −0.051 | 3.375 | 0.847 | −2.377 | 3.395 | |||||
| 2c | −1.303 | 0.089 | −0.045 | 4.119 | 0.835 | −2.125 | 2.020 | |||||
| 2d | −1.305 | 0.099 | −0.050 | 4.411 | 0.864 | −2.326 | 5.656 | |||||
| 2e | −1.265 | 0.083 | −0.042 | 5.261 | 0.270 | −1.539 | 0.076 | −0.038 | 5.280 | 0.641 | −2.352 | 7.525 |
| 3a | −1.331 | 0.077 | −0.039 | 3.675 | 0.798 | −2.166 | 2.518 | |||||
| 3b | −1.333 | 0.079 | −0.039 | 4.253 | 0.768 | −2.216 | 3.049 | |||||
| 3c | −1.401 b | 7.460 | ||||||||||
| 3d | −1.300 | 0.075 | −0.037 | 4.601 | 0.767 | −1.995 | 3.303 | |||||
| 3e | −1.277 | 0.095 | −0.047 | 3.392 | 0.239 | −1.566 | 0.096 | −0.044 | 2.546 | 0.699 | −2.372 | 3.816 |
| 4a | −1.188 | 0.076 | −0.038 | 4.237 | 0.871 | −1.973 | 3.650 | |||||
| 4b | −1.196 | 0.093 | −0.047 | 3.753 | 0.859 | −2.115 | 2.118 | |||||
| 4c | −1.154 | 0.129 | −0.065 | 2.899 | 0.841 | −2.080 | 1.342 | |||||
| 4d | −1.181 | 0.107 | −0.053 | 4.103 | 0.930 | −2.132 | 2.866 | |||||
| 4e | −1.134 | 0.072 | −0.036 | 3.071 | 0.483 | −1.514 | 0.063 | −0.031 | 3.775 | 0.641 | −2.141 | 3.904 |
| 1st N-oxide Reduction | Nitro Group Reduction | 2nd N-oxide Reduction c | ||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Epc (V) | Epa (V) | E1/2 (V) | Epc (V) | Epa (V) | E1/2 (V) | ΔEp (V) | Epc (V) |
| 1a | −1.335 | −1.256 | −1.296 | −2.162 | ||||
| 1b | −1.350 | −1.264 | −1.307 | −2.368 | ||||
| 1c | −1.305 | −1.232 | −1.269 | −2.096 | ||||
| 1d | −1.314 | −1.240 | −1.277 | −2.130 | ||||
| 1e | −1.287 | −1.179 | −1.233 | −1.574 | −1.467 | −1.521 | 0.107 | −2.309 |
| 2a | −1.375 | −1.276 | −1.326 | −2.308 | ||||
| 2b | −1.369 | −1.266 | −1.318 | −2.366 | ||||
| 2c | −1.348 | −1.258 | −1.303 | −2.122 | ||||
| 2d | −1.356 | −1.253 | −1.305 | −2.279 | ||||
| 2e | −1.308 | −1.221 | −1.265 | −1.577 | −1.499 | −1.538 | 0.078 | −2.355 |
| 3a | −1.372 | −1.290 | −1.331 | −2.168 | ||||
| 3b | −1.374 | −1.292 | −1.332 | −2.216 | ||||
| 3c | −1.402 | −1.402 c | −2.466 | |||||
| 3d | −1.339 | −1.26 | −1.299 | −1.996 | ||||
| 3e | −1.328 | −1.226 | −1.277 | −1.612 | −1.510 | −1.562 | 0.101 | −2.371 |
| 4a | −1.227 | −1.148 | −1.188 | −1.974 | ||||
| 4b | −1.243 | −1.146 | −1.195 | −2.114 | ||||
| 4c | −1.219 | −1.086 | −1.154 | −2.082 | ||||
| 4d | −1.236 | −1.126 | −1.181 | −2.130 | ||||
| 4e | −1.172 | −1.094 | −1.133 | −1.544 | −1.476 | −1.510 | 0.068 | −2.142 |
| Compound | 1st N-oxide Reduction | Nitro Group Reduction | 2nd N-oxide Reduction | |||
|---|---|---|---|---|---|---|
| Lanl2dz | 6-31g | Lanl2dz | 6-31g | Lanl2dz | 6-31g | |
| 1a | 3.6006 | 3.2891 | 2.1763 | 1.7774 | ||
| 1b | 3.5920 | 3.2807 | 2.1636 | 1.7586 | ||
| 1c | 3.6516 | 3.3488 | 2.1944 | 1.8146 | ||
| 1d | 3.6364 | 3.3307 | 2.1910 | 1.8055 | ||
| 1e | 3.8605 | 3.5518 | 2.6292 | 2.2985 | 2.9601 | 2.6072 |
| 1st N-oxide Reduction | Nitro Group Reduction | 2nd N-oxide Reduction | ||||
|---|---|---|---|---|---|---|
| Hydrogen Half−Cell Reduction Set to Zero | ||||||
| Compound | Lanl2dz | 6-31g | Lanl2dz | 6-31g | Lanl2dz | 6-31g |
| 1a | −1.9136 | −1.8908 | −3.3379 | −3.4025 | ||
| 1b | −1.9223 | −1.8993 | −3.3507 | −3.4213 | ||
| 1c | −1.8626 | −1.8312 | −3.3199 | −3.3654 | ||
| 1d | −1.8778 | −1.8492 | −3.3233 | −3.3744 | ||
| 1e | −1.6538 | −1.6281 | −2.8851 | −2.8814 | −2.5542 | −2.5728 |
| Ferrocene Half−Cell Reduction Set to Zero | ||||||
| Compound | Lanl2dz | 6-31g | Lanl2dz | 6-31g | Lanl2dz | 6-31g |
| 1a | −1.1936 | −1.1708 | −2.6179 | −2.6825 | ||
| 1b | −1.2023 | −1.1793 | −2.6307 | −2.7013 | ||
| 1c | −1.1426 | −1.1112 | −2.5999 | −2.6454 | ||
| 1d | −1.1578 | −1.1292 | −2.6033 | −2.6544 | ||
| 1e | −0.9338 | −0.9081 | −2.1651 | −2.1614 | −1.8342 | −1.8528 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, E.M.; Xia, Q.; Cella, M.E.; Nenninger, A.W.; Mruzik, M.N.; Brillos-Monia, K.A.; Hu, Y.Z.; Sheng, R.; Ragain, C.M.; Crawford, P.W. Voltammetric Study of Some 3-Aryl-quinoxaline-2-carbonitrile 1,4-di-N-oxide Derivatives with Anti-Tumor Activities. Molecules 2017, 22, 1442. https://doi.org/10.3390/molecules22091442
Miller EM, Xia Q, Cella ME, Nenninger AW, Mruzik MN, Brillos-Monia KA, Hu YZ, Sheng R, Ragain CM, Crawford PW. Voltammetric Study of Some 3-Aryl-quinoxaline-2-carbonitrile 1,4-di-N-oxide Derivatives with Anti-Tumor Activities. Molecules. 2017; 22(9):1442. https://doi.org/10.3390/molecules22091442
Chicago/Turabian StyleMiller, Eric M., Qing Xia, Mariah E. Cella, Austin W. Nenninger, Monica N. Mruzik, Krystina A. Brillos-Monia, Yong Zhou Hu, Rong Sheng, Christina M. Ragain, and Philip W. Crawford. 2017. "Voltammetric Study of Some 3-Aryl-quinoxaline-2-carbonitrile 1,4-di-N-oxide Derivatives with Anti-Tumor Activities" Molecules 22, no. 9: 1442. https://doi.org/10.3390/molecules22091442
APA StyleMiller, E. M., Xia, Q., Cella, M. E., Nenninger, A. W., Mruzik, M. N., Brillos-Monia, K. A., Hu, Y. Z., Sheng, R., Ragain, C. M., & Crawford, P. W. (2017). Voltammetric Study of Some 3-Aryl-quinoxaline-2-carbonitrile 1,4-di-N-oxide Derivatives with Anti-Tumor Activities. Molecules, 22(9), 1442. https://doi.org/10.3390/molecules22091442