Zeolites as Catalysts for Fuels Refining after Indirect Liquefaction Processes


Abstract
:1. Introduction
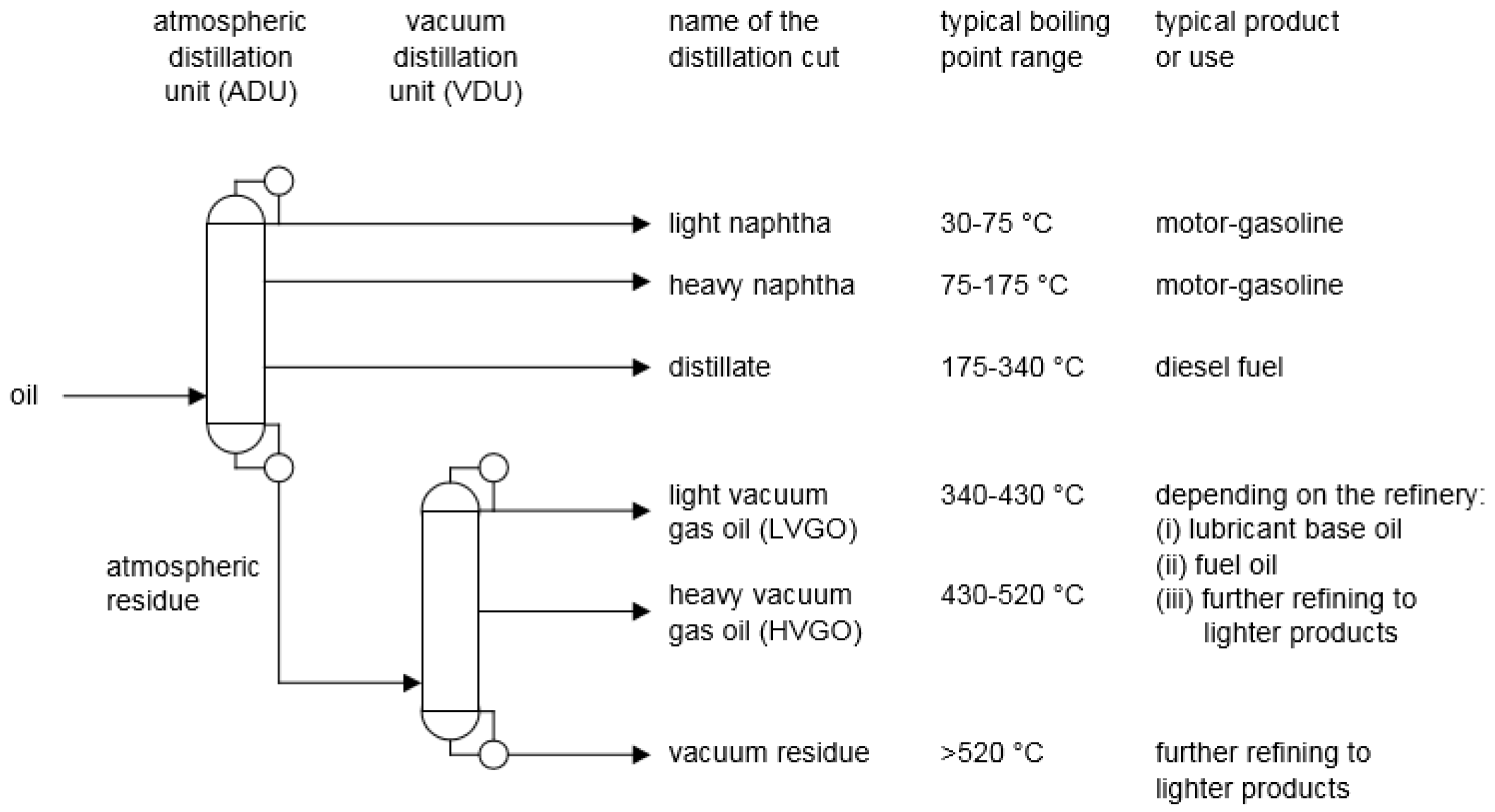
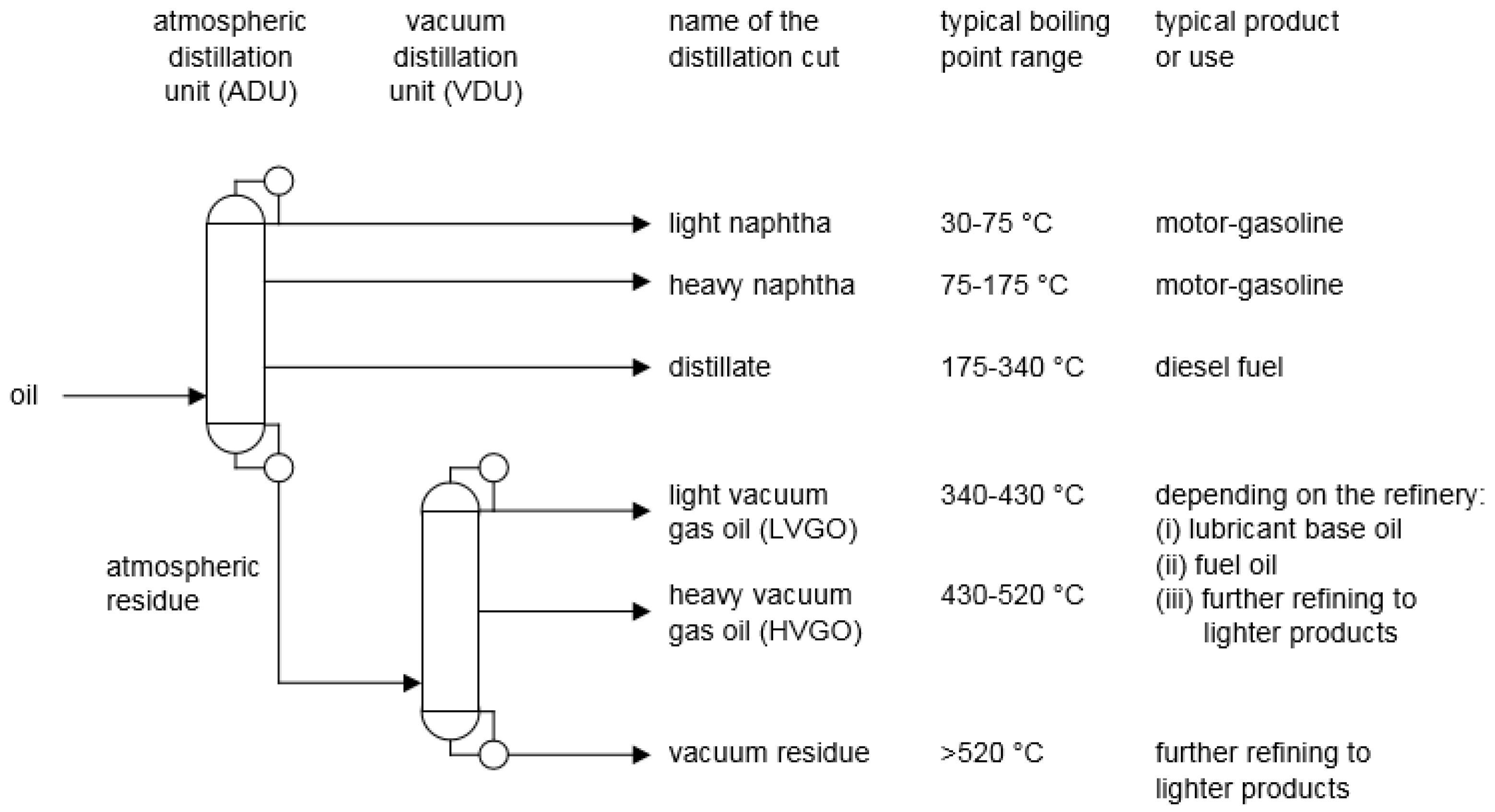
2. Refining Roadmap
- (a)
- Gaseous hydrocarbons. Broadly speaking, gaseous hydrocarbons consist of a mixture of alkanes (paraffins) and alkenes (olefins). Depending on the extent of separation in the Fischer–Tropsch refinery [11], the light hydrocarbons will be present with unconverted synthesis gas and will not be refined. For the purpose of discussion, it will be assumed that the C3-C4 hydrocarbons are recovered and will be refined. The gaseous hydrocarbons that are produced with crude oil are usually separated from the oil before the oil is transported from the oil well to the refinery, often over great distances. The gaseous hydrocarbons in a petroleum refinery are the products of refining processes and little straight run gaseous hydrocarbons enter a petroleum refinery with the crude oil.
- (b)
- Light oxygenates. The light oxygenates (typically C1-C4 oxygenates), which include methanol, are water-soluble materials. During condensation, these compounds will dissolve in water that is also condensed after Fischer–Tropsch synthesis. These compounds are unique to indirect liquefaction refineries and no equivalent fraction exists in a petroleum refinery. Methanol and/or ethanol may be imported into petroleum refinery for fuel blending and etherification.
- (c)
- Naphtha. Oil with a boiling range of 30–175 °C (typically C5-C10 hydrocarbons) is referred to as naphtha. In a Fischer–Tropsch product, the naphtha contains mainly alkanes, alkenes and alcohols, but may also contain aromatics, ketones and carboxylic acids. Petroleum naphtha obtained by distillation from crude oil consists of alkanes, cycloalkanes, aromatics, as well as some sulfur- and nitrogen-containing compounds. In petroleum, naphtha that is produced by conversion processes in the refinery, alkenes may also be present, but alkenes are seldom found in petroleum naphtha obtained by crude oil distillation.
- (d)
- Distillate. Oil with a boiling range of 175–340 °C (typically C11-C22 hydrocarbons) is referred to as distillate. Apart from boiling point, the composition is similar to that of Fischer–Tropsch naphtha, but the concentration of alkanes is much higher. For petroleum, the same is true, but the concentration of sulfur- and nitrogen-containing compounds is higher than in petroleum naphtha.
- (e)
- Atmospheric residue. Organic products with a boiling point higher than 340 °C are referred to as atmospheric residue. Depending on the type of Fischer–Tropsch technology, this product is either a wax, or an aromatic-rich oil. Atmospheric residues obtained from petroleum distillation reflect the properties of the oil and it can range from a waxy product for paraffinic crude oils to oil with high aromatic content for aromatic crude oils. The concentrations of sulfur- and nitrogen-containing compounds are also dependent on the origin of the oil and the concentration of heteroatom species is higher in the residue than in the lighter fractions. In some oils, there may also be heavy carboxylic acids.
3. Refining Gaseous Hydrocarbons
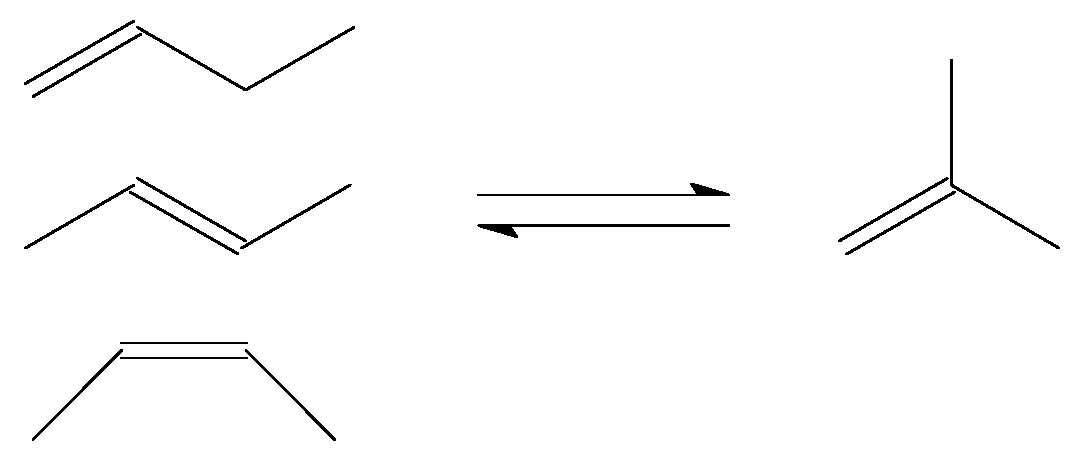
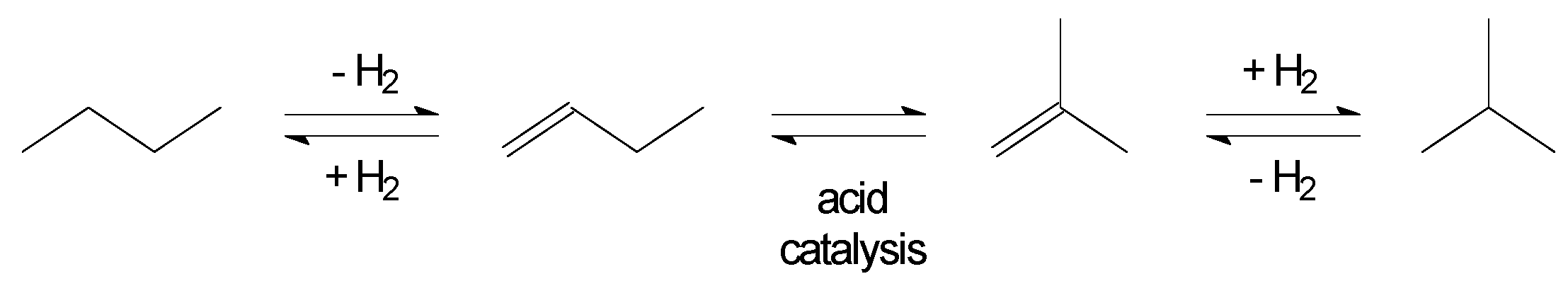
3.1. Skeletal Isomerisation of n-butenes
3.2. Hydroisomerisation of n-butane
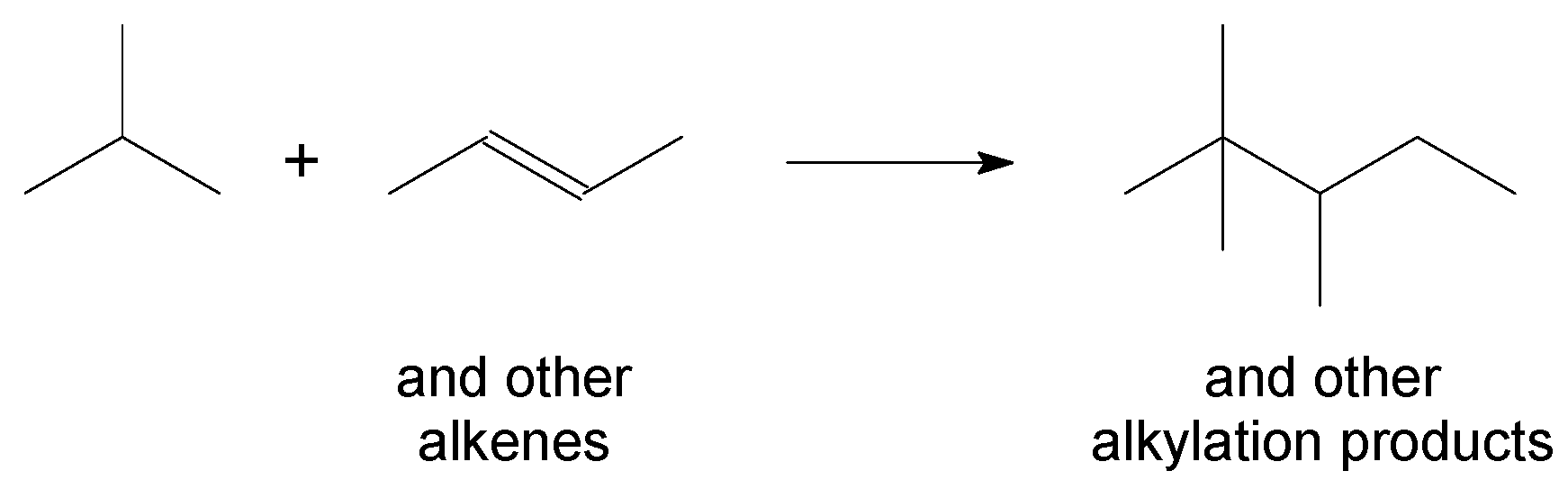
3.3. Aliphatic Alkylation
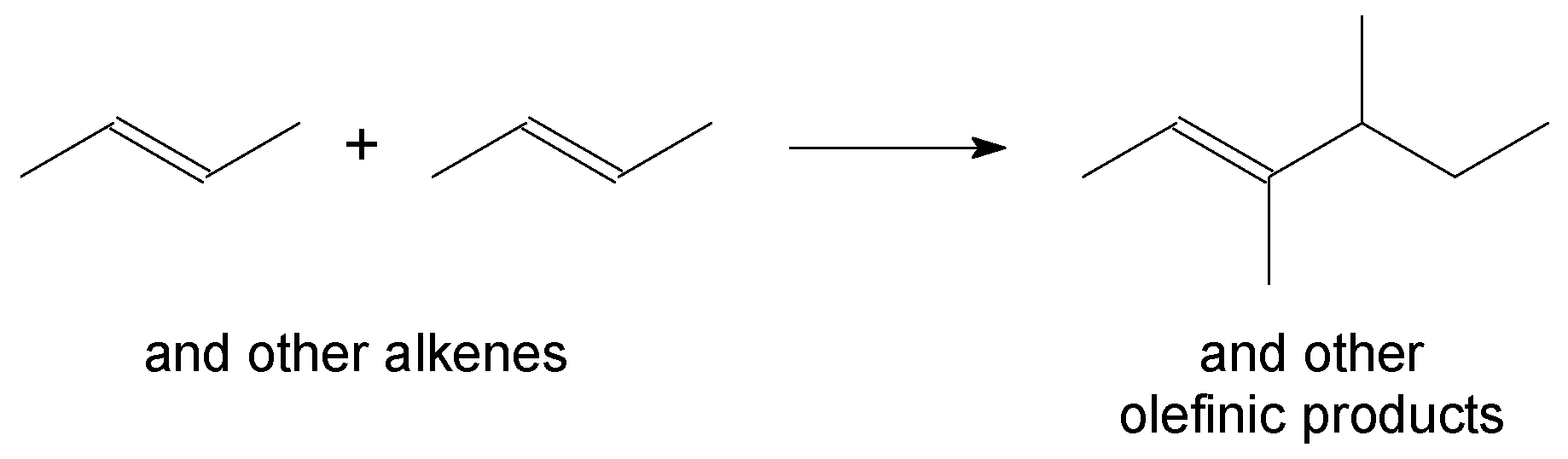
3.4. Alkene Oligomerisation
4. Light Oxygenates
4.1. Methanol to Hydrocarbons
4.2. Ethanol and Heavier Alcohols to Hydrocarbons
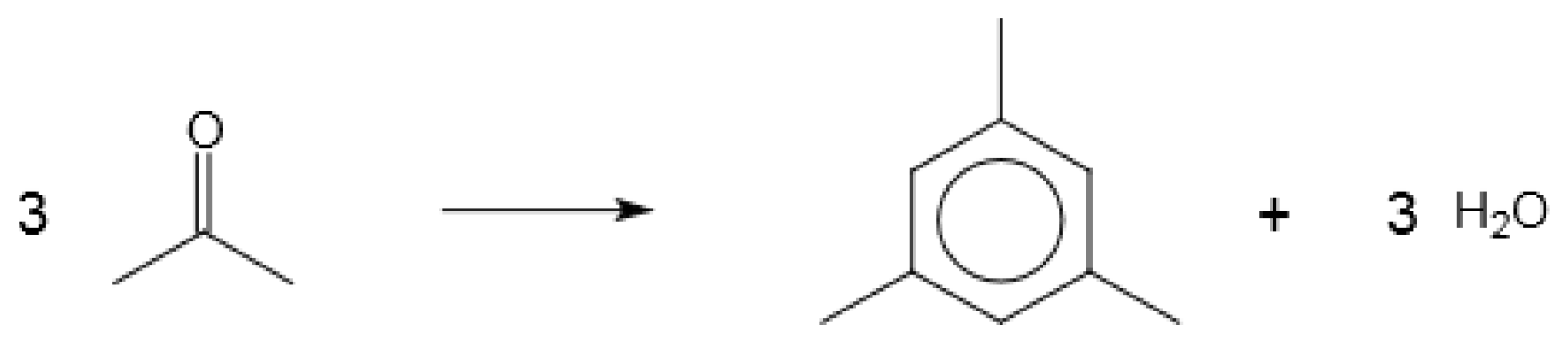
4.3. Carbonyls to Hydrocarbons
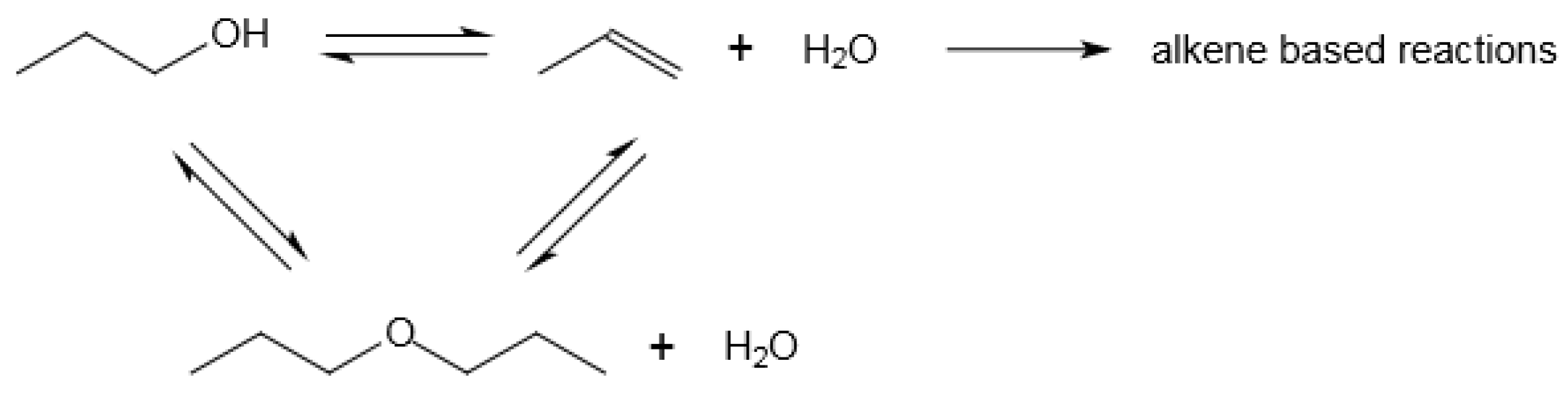
4.4. Etherification of Alkenes with Alcohols
5. Naphtha
5.1. Hydroisomerisation of Light Naphtha
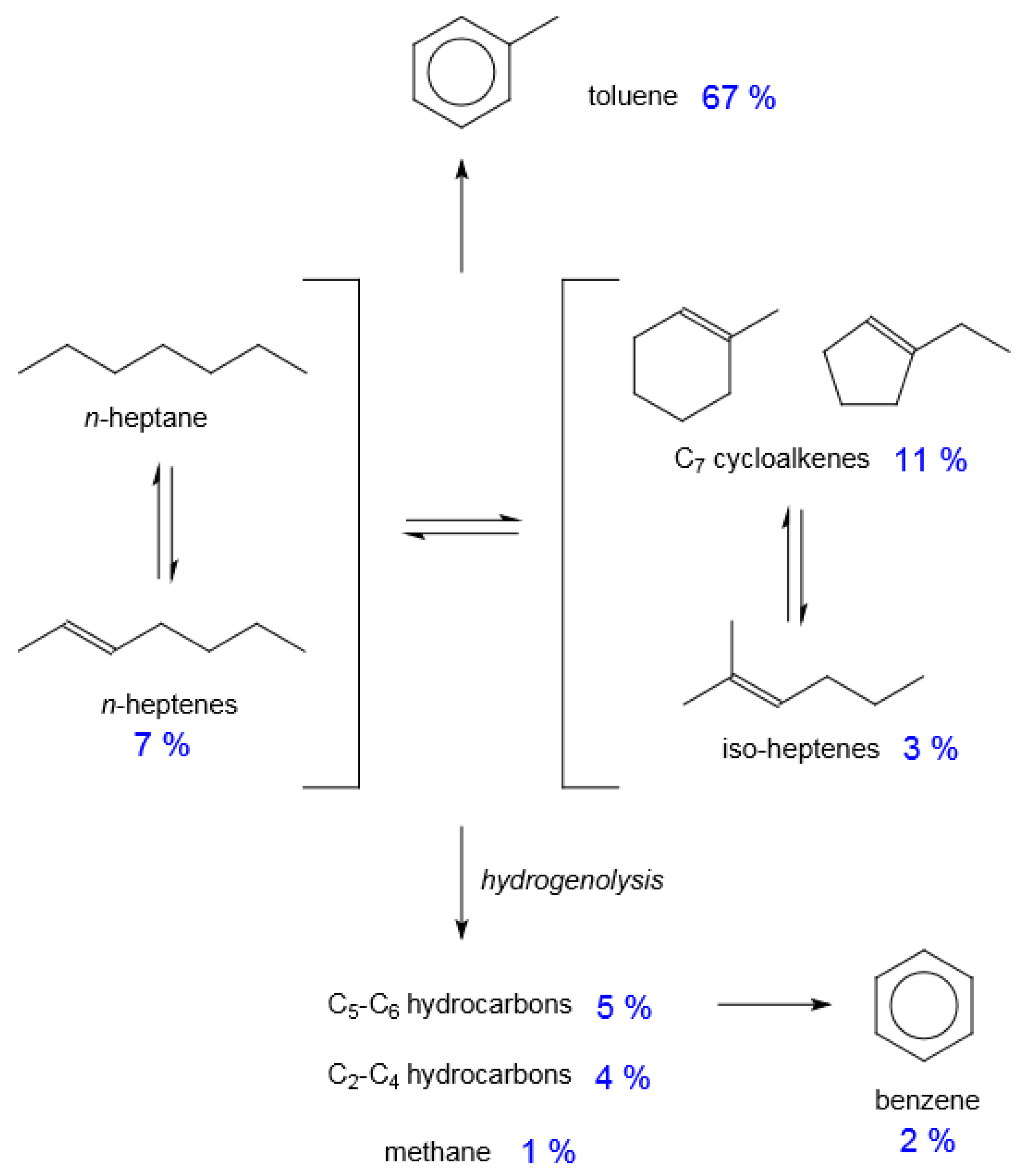
5.2. Catalytic Naphtha Reforming
6. Distillate
Hydroisomerisation of Distillate
7. Atmospheric Residue
7.1. Hydrocracking
7.2. Fluid Catalytic Cracking
8. Discussion and Conclusions
- (a)
- Catalytic naphtha reforming using Pt/K/LTL zeolite catalysts benefit from the high linear hydrocarbon content and sulfur-free nature of indirect liquefaction products. Although this type of catalyst is associated predominantly with petrochemical production, its potential benefit for fuel refinery design is clear [77], particularly for converting n-heptane, which is a challenging molecule to refine for motor-gasoline.
- (b)
- Hydroisomerisation of distillate using Pt/AEL (SAPO-11) zeolite catalysts has the potential to increase the yield of jet fuel from Fischer–Tropsch refineries. It may also become a necessary addition to refineries that want to market Fischer–Tropsch-derived diesel fuels in cold climates without relying on blending with petroleum derived distillate.
Acknowledgments
Conflicts of Interest
References
- Bertau, M.; Offermanns, H.; Plass, L.; Schmidt, F.; Wernicke, H.-J. (Eds.) Methanol: The Basic Chemical and Energy Feedstock of the Future; Asinger’s Vision Today; Springer: Heidelberg, Germany, 2014. [Google Scholar]
- Maitlis, P.M.; De Klerk, A. (Eds.) Greener Fischer-Tropsch Processes for Fuels and Feedstocks; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- De Klerk, A. Environmentally friendly refining: Fischer-Tropsch versus crude oil. Green Chem. 2007, 9, 560–565. [Google Scholar] [CrossRef]
- De Klerk, A. Fischer-Tropsch refining: Technology selection to match molecules. Green Chem. 2008, 10, 1249–1279. [Google Scholar] [CrossRef]
- Baerlocher, C.; Meier, W.M.; Olson, D.H. Atlas of Zeolite Framework Types, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Vermeiren, W.; Gilson, J.-P. Impact of zeolites on the petroleum and petrochemical industry. Top. Catal. 2009, 52, 1131–1161. [Google Scholar] [CrossRef]
- Bellussi, G.; Carati, A.; Millini, R. Industrial potential of zeolites. In Zeolites and Catalysis. Synthesis, Reactions and Applications; Čejka, J., Corma, A., Zones, S., Eds.; Wiley-VCH: Weinheim, Germany, 2010; Volume 2, pp. 449–491. [Google Scholar]
- Kustov, L.M.; Kuperman, A.; Kustov, A. Further steps of zeolites toward industrial applications: A short-range outlook. In Zeolites and Zeolite-Like Materials; Sels, B.F., Kustov, L.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 309–369. [Google Scholar]
- De Klerk, A.; Furimsky, E. Catalysis in the Refining of Fischer-Tropsch Syncrude; Royal Society of Chemistry: Cambridge, UK, 2010. [Google Scholar]
- Gary, J.H.; Handwerk, G.E.; Kaiser, M.J. Petroleum Refining. Technology and Economics, 5th ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- De Klerk, A. Fischer–Tropsch Refining; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Chuck, C.J. (Ed.) Biofuels for Aviation: Feedstocks, Technology and Implementation; Academic Press: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Van Donk, S.; Bitter, J.H.; De Jong, K.P. Deactivation of solid acid catalysts for butene skeletal isomerisation: On the beneficial and harmful effects of carbonaceous deposits. Appl. Catal. A 2001, 212, 97–116. [Google Scholar] [CrossRef]
- Wise, J.B.; Powers, D. Highly selective olefin skeletal isomerization process. ACS Symp. Ser. 1994, 552, 273–285. [Google Scholar] [CrossRef]
- Ponomareva, O.A.; Kasyanov, I.A.; Knyazeva, E.E.; Konnov, S.V.; Ivanova, I.I. Effect of the degree of zeolite recrystallization into micro-mesoporous materials on their catalytic properties in petroleum refining and petroleum chemistry processes. Pet. Chem. 2016, 56, 819–826. [Google Scholar] [CrossRef]
- Hu, H.; Ke, M.; Zhang, K.; Liu, Q.; Yu, P.; Liu, Y.; Li, C.; Liu, W. Designing ferrierite-based catalysts with improved properties for skeletal isomerization of n-butene to isobutene. RSC Adv. 2017, 7, 31535–31543. [Google Scholar] [CrossRef]
- Jo, D.; Hong, S.B.; Camblor, M.A. Monomolecular skeletal isomerization of 1-butene over selective zeolite catalysts. ACS Catal. 2015, 5, 2270–2274. [Google Scholar] [CrossRef]
- Meyers, R.A. (Ed.) Handbook of Petroleum Refining Processes, 4th ed.; McGraw-Hill: New York, NY, USA, 2016. [Google Scholar]
- Ritter, S.K. The 2016 Presidential Green Chemistry Challenge Awards: Zeolite catalyst offers a better future for gasoline. Chem. Eng. News 2016, 94, 20–21. [Google Scholar]
- Van Broekhoven, E.H.; Cabre, F.R.M.; Bogaard, P.; Klaver, G.; Vonhof, M. Process for Alkylating Hydrocarbons. U.S. Patent 5986158, 16 November 1999. [Google Scholar]
- Van Broekhoven, E.H.; Van Loevezijn, A.; Bakker, R.H.M. Alkylation Process Using a Catalyst Comprising Cerium Rich Rare Earth Containing Zeolites and a Hydrogenation Metal. U.S. Patent Appl. 20170203285, 20 July 2017. [Google Scholar]
- Nicholas, C.P. Applications of light olefin oligomerization to the production of fuels and chemicals. Appl. Catal. A 2017, 543, 82–97. [Google Scholar] [CrossRef]
- Sarazen, M.L.; Doskocil, E.; Iglesia, E. Effects of void environment and acid strength on alkene oligomerization selectivity. ACS Catal. 2016, 6, 7059–7070. [Google Scholar] [CrossRef]
- Garwood, W.E. Conversion of C2-C10 to higher olefins over synthetic zeolite ZSM-5. ACS Symp. Ser. 1983, 218, 383–396. [Google Scholar] [CrossRef]
- Knottenbelt, C. Mossgas “gas-to-liquids” diesel fuels—An environmentally friendly option. Catal. Today 2002, 71, 437–445. [Google Scholar] [CrossRef]
- Halmenschlager, C.M.; Brar, M.; Apan, I.T.; De Klerk, A. Oligomerization of Fischer-Tropsch tail gas over H-ZSM-5. Ind. Eng. Chem. Res. 2016, 55, 13020–13031. [Google Scholar] [CrossRef]
- Stöcker, M. Metanol to olefins (MTO) and methanol to gasoline (MTG). In Zeolites and Catalysis. Synthesis, Reactions and Applications; Čejka, J., Corma, A., Zones, S., Eds.; Wiley-VCH: Weinheim, Germany, 2010; Volume 2, pp. 687–711. [Google Scholar]
- Nesterenko, N.; Aguilhon, J.; Bodart, P.; Minoux, D.; Dath, J.-P. Methanol to olefins: An insight into reaction pathways and products formation. In Zeolites and Zeolite-Like Materials; Sels, B.F., Kustov, L.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 189–263. [Google Scholar]
- Chang, C.D. Hydrocarbons from methanol. Catal. Rev. Sci. Eng. 1983, 25, 1–118. [Google Scholar] [CrossRef]
- Xu, X.; Liu, Y.; Zhang, F.; Di, W.; Zhang, Y. Clean coal technologies in China based on methanol platform. Catal. Today 2017, 298, 61–68. [Google Scholar] [CrossRef]
- Haw, J.F.; Song, W.; Marcus, D.M.; Nicholas, J.B. The mechanism of methanol to hydrocarbon catalysis. Acc. Chem. Res. 2003, 36, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Bjørgen, M.; Svelle, S.; Joensen, F.; Nerlov, J.; Kolboe, S.; Bonino, F.; Palumbo, L.; Bordiga, S.; Olsbye, U. Conversion of methanol to hydrocarbons over zeolite H-ZSM-5: On the origin of the olefinic species. J. Catal. 2007, 249, 195–207. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Atatxa, A.; Aguado, R.; Bilbao, J. Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. I. Alcohols and phenols. Ind. Eng. Chem. Res. 2004, 43, 2610–2618. [Google Scholar] [CrossRef]
- Davis, B.H. Alcohol conversion selectivity as a measure of the base strength of metal oxide catalysts. Stud. Surf. Sci. Catal. 1985, 21, 309–318. [Google Scholar]
- Takahara, I.; Saito, M.; Inaba, M.; Murata, K. Dehydration of ethanol into ethylene over solid acid catalysts. Catal. Lett. 2005, 105, 249–252. [Google Scholar] [CrossRef]
- Asinger, F. Mono-Olefins. Chemistry and Technology; Pergamon Press: Oxford, UK, 1968. [Google Scholar]
- Hoang, T.Q.; Zhu, X.; Sooknoi, T.; Resasco, D.E.; Mallinson, R.G. A comparison of the reactivities of propanal and propylene on HZSM-5. J. Catal. 2010, 271, 201–208. [Google Scholar] [CrossRef]
- Salvapati, G.S.; Ramanamurthy, K.V.; Janardanarao, M. Selective catalytic self-condensation of acetone. J. Mol. Catal. 1989, 54, 9–30. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Ko, A.-N. Vapor phase reactions of acetaldehyde over type X zeolites. Appl. Catal. A 2000, 190, 149–155. [Google Scholar]
- Di Girolamo, M.; Lami, M.; Marchionna, M.; Pescarollo, E.; Tagliabue, L.; Ancillotti, F. Liquid-phase etherification/dimerization of isobutene over sulphonic acid resins. Ind. Eng. Chem. Res. 1997, 36, 4452–4458. [Google Scholar] [CrossRef]
- Linnekoski, J.A.; Krause, A.O.I.; Struckmann, L.K. Etherification and hydration of isoamylenes with ion exchange resin. Appl. Catal. A 1998, 170, 117–126. [Google Scholar] [CrossRef]
- Ali, M.A.; Brisdon, B.; Thomas, W.J. Synthesis, characterization and catalytic activity of ZSM-5 zeolites having variable silicon-to-aluminum ratios. Appl. Catal. A 2003, 252, 149–162. [Google Scholar] [CrossRef]
- Iborra, M.; Tejero, J.; Fité, C.; Cunill, F.; Izquierdo, J.F. Zeolite-catalysed liquid-phase synthesis of isopropyl tert-butyl ether by the addition of 2-propanol to isobutene. J. Catal. 2005, 231, 77–91. [Google Scholar] [CrossRef]
- Wulfers, M.J.; Jentoft, F.C. Mechanism of n-butane skeletal isomerization on H-mordenite and Pt/H-mordenite. J. Catal. 2015, 330, 507–519. [Google Scholar] [CrossRef]
- Weyda, H.; Köhler, E. Modern refining concepts—An update on naphtha-isomerization to modern gasoline manufacture. Catal. Today 2003, 81, 51–55. [Google Scholar] [CrossRef]
- Valavarasu, G.; Sairam, B. Light naphtha isomerization process: A review. Petrol. Sci. Technol. 2013, 31, 580–595. [Google Scholar] [CrossRef]
- Lamprecht, D.; De Klerk, A. Hydroisomerization of 1-pentene to iso-pentane in a single reactor. Chem. Eng. Commun. 2009, 196, 1206–1216. [Google Scholar] [CrossRef]
- Xu, D.; Wu, B.; Ren, P.; Wang, S.; Huo, C.; Zhang, B.; Guo, W.; Huang, L.; Wen, X.; Qin, Y.; et al. Controllable deposition of Pt nanoparticles into a KL zeolite by atomic layer deposition for highly efficient reforming of n-heptane to aromatics. Catal. Sci. Technol. 2017, 7, 1342–1350. [Google Scholar] [CrossRef]
- Derouane, E.G.; Vanderveken, D.J. Structural recognition and preorganization in zeolite catalysis: Direct aromatization of n-hexane on zeolite L-based catalysts. Appl. Catal. 1988, 45, L15–L22. [Google Scholar] [CrossRef]
- Ribeiro, M.F.; Guisnet, M. Aromatization of C6, C7 paraffins over Pt/LTL catalysts. In Deactivation and Regeneration of Zeolite Catalysts; Guisnet, M., Ribeiro, F.R., Eds.; Imperial College Press: London, UK, 2011; pp. 293–302. [Google Scholar]
- Azzam, K.G.; Jacobs, G.; Shafer, W.D.; Davis, B.H. Aromatization of hexane over Pt/KL catalyst: Role of intracrystalline diffusion on catalyst performance using isotope labeling. J. Catal. 2010, 270, 242–248. [Google Scholar] [CrossRef]
- Bertaux, C.; Jacobs, G.; Shafer, W.D.; Davis, B.H. Mitigation of methane selectivity on Pt/KL-zeolite aromatization catalysts by Ag promotion. Catal. Lett. 2016, 146, 763–769. [Google Scholar] [CrossRef]
- Lee, K.; Choi, M. Hierarchically micro-/mesoporous Pt/KL for alkane aromatization: Synergistic combination of high catalytic activity and suppressed hydrogenolysis. J. Catal. 2016, 340, 66–75. [Google Scholar] [CrossRef]
- Buchanan, J.S.; Santiesteban, J.G.; Haag, W.O. Mechanistic considerations in acid-catalyzed cracking of olefins. J. Catal. 1996, 158, 279–287. [Google Scholar] [CrossRef]
- Cody, I.A. Selective hydroprocessing for new lubricant standards. In Practical Advances in Petroleum Processing; Hsu, C.S., Robinson, P.R., Eds.; Springer: Heidelberg, Germany, 2006; Volume 2, pp. 79–104. [Google Scholar]
- Maesen, T.L.M.; Schenk, M.; Vlugt, T.J.H.; De Jonge, J.P.; Smit, B. The shape selectivity of paraffin hydroconversion on TON-, MTT-, and AEL-type sieves. J. Catal. 1999, 188, 403–412. [Google Scholar] [CrossRef]
- Soualah, A.; Lemberton, J.L.; Pinard, L.; Chater, M.; Magnoux, P.; Moljord, K. Hydroisomerization of long-chain n-alkanes on bifunctional Pt/zeolite catalysts: Effect of the zeolite structure on the product selectivity and on the reaction mechanism. Appl. Catal. A 2008, 336, 23–28. [Google Scholar] [CrossRef]
- Miller, S.J. Studies on wax isomerization for lubes and fuels. Stud. Surf. Sci. Catal. 1994, 84, 2319–2326. [Google Scholar]
- Scherzer, J.; Gruia, A.J. Hydrocracking Science and Technology; CRC Press: Boca Raton, FL, USA, 1996. [Google Scholar]
- Calemma, V.; Gambaro, C. Effect of feed distribution on hydrocracking of Fischer-Tropsch wax. ACS Symp. Ser. 2011, 1084, 239–253. [Google Scholar] [CrossRef]
- Rigutto, M. Cracking and hydrocracking. In Zeolites and Catalysis. Synthesis, Reactions and Applications; Čejka, J., Corma, A., Zones, S., Eds.; Wiley-VCH: Weinheim, Germany, 2010; Volume 2, pp. 547–584. [Google Scholar]
- Wei, J. Nonlinear phenomena in zeolite diffusion and reaction. Ind. Eng. Chem. Res. 1994, 33, 2467–2472. [Google Scholar] [CrossRef]
- Maesen, T.L.M.; Beerdsen, E.; Calero, S.; Dubbeldam, D.; Smit, B. Understanding cage effects in the n-alkane conversion on zeolites. J. Catal. 2006, 237, 278–290. [Google Scholar] [CrossRef]
- Hanaoka, T.; Miyazawa, T.; Shimura, K.; Hirata, S. Jet fuel synthesis from Fischer-Tropsch product under mild hydrocracking conditions using Pt-loaded catalysts. Chem. Eng. J. 2015, 263, 178–185. [Google Scholar] [CrossRef]
- Wojciechowski, B.W. Dichotomies in catalytic cracking. Ind. Eng. Chem. Res. 1997, 36, 3323–3335. [Google Scholar] [CrossRef]
- Sousa-Aguiar, E.F. Y Zeolites as a major component of FCC catalysts: Main challenges in the modification thereof. In Zeolites and Zeolite-Like Materials; Sels, B.F., Kustov, L.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 265–282. [Google Scholar]
- Lloyd, L. Handbook of Industrial Catalysts; Springer: Heidelberg, Germany, 2011. [Google Scholar]
- Akah, A. Application of rare earths in fluid catalytic cracking: A review. J. Rare Earths 2017, 35, 941–956. [Google Scholar] [CrossRef]
- Liu, X.; Liu, S.; Liu, Y. A potential substitute for CeY zeolite used in fluid catalytic cracking process. Microporous Mesoporous Mater. 2016, 226, 162–168. [Google Scholar] [CrossRef]
- Mathieu, Y.; Corma, A.; Echard, M.; Bories, M. Single and combined effects of Bottom Cracking (BCA) and Propylene Booster (PBA) separate particles additives addition to a Fluid Catalytic Cracking (FCC) catalyst on the FCC product distribution and quality. Appl. Catal. A 2012, 439–440, 57–73. [Google Scholar] [CrossRef]
- Magee, J.; Dolbear, G. Petroleum Catalysis in Nontechnical Language; PennWell: Tulsa, OK, USA, 1998. [Google Scholar]
- Kubička, D.; Černý, R. Upgrading of Fischer-Tropsch waxes by fluid catalytic cracking. Ind. Eng. Chem. Res. 2012, 51, 8849–8857. [Google Scholar] [CrossRef]
- Abbot, J.; Wojciechowski, B.W. Catalytic cracking on HY and HZSM-5 of a Fischer-Tropsch product. Ind. Eng. Chem. Prod. Res. Dev. 1985, 24, 501–507. [Google Scholar] [CrossRef]
- Schwartz, M.M. The Selective Catalytic Cracking of Fischer-Tropsch Liquids to High Value Transportation Fuels; DOE Contract No. DE-AC22-91PC90057, Final Report; U.S. Department Energy Pittsburgh Energy Technology Center: Pittsburgh, PA, USA, 1995.
- Dupain, X.; Krul, R.A.; Schaverien, C.J.; Makkee, M.; Moulijn, J.A. Production of clean transportation fuels and lower olefins from Fischer-Tropsch synthesis waxes under fluid catalytic cracking conditions. The potential of highly paraffinic feedstocks for FCC. Appl. Catal. B 2006, 63, 277–295. [Google Scholar] [CrossRef]
- Komvokis, V.G.; Karakoulia, S.; Iliopoulou, E.F.; Papapetrou, M.C.; Vasalos, I.A.; Lappas, A.A.; Triantafyllidys, K.S. Upgrading of Fischer-Tropsch synthesis bio-waxes via catalytic cracking: Effect of acidity, porosity and metal modification of zeolitic and mesoporous aluminosilicate catalysts. Catal. Today 2012, 196, 42–55. [Google Scholar] [CrossRef]
- De Klerk, A. Fischer-Tropsch fuels refinery design. Energy Environ. Sci. 2011, 4, 1177–1205. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
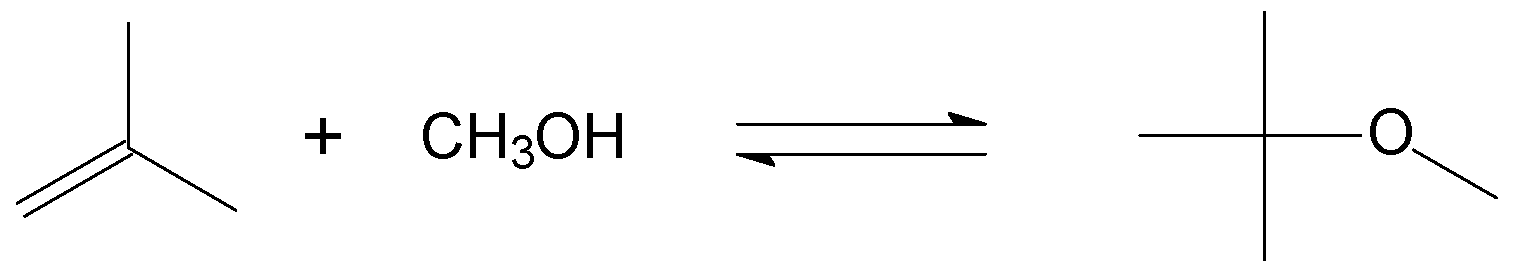{kind=link}
{kind=link}
{kind=link}
| Framework Type | Type Used in Refining | Ring Size/atomsunit | Channel Size/nm × nm |
|---|---|---|---|
| AEL | SAPO-11 | 10 | 0.65 × 0.40 |
| CHA | SAPO-34 | 8 | 0.38 × 0.38 |
| FAU | Y-zeolite | 12 | 0.74 × 0.74 |
| FER | Ferrierite | 8 and 10 | 0.35 × 0.48 and 0.42 × 0.54 |
| LTL | L-zeolite | 12 | 0.71 ×0.71 |
| MFI | ZSM-5 | 10 and 10 | 0.51 × 0.55 and 0.53 × 0.56 |
| MFS | ZSM-57 | 8 and 10 | 0.33 × 0.48 and 0.51 × 0.54 |
| MOR | Mordenite | 8, 8 and 12 | 0.26 × 0.57, 0.34 × 0.48 and 0.65 × 0.70 |
| MWW | MCM-22 | 10 and 10 | 0.40 × 0.55 and 0.41 × 0.51 |
| TON | ZSM-22 | 10 | 0.46 × 0.57 |
| Product | Product Distribution/wt % | ||
|---|---|---|---|
| Methanol Synthesis | Low Temperature Fischer-Tropsch Synthesis a | High Temperature Fischer-Tropsch Synthesis | |
| gaseous hydrocarbons | |||
| methane | 1 | 4 | 13 |
| ethane/ethene | - | 2 | 10 |
| C3-C4 hydrocarbons | - | 8 | 24 |
| light oxygenates | |||
| C1-C4 oxygenates | 99 | 4 | 10 |
| oil products | |||
| light naphtha | - | 4 | 15 |
| heavy naphtha | - | 8 | 18 |
| distillate | - | 20 | 7 |
| atmospheric residue | - | 50 b | 3 c |
| Description | Pt/Cl/Al2O3 | Pt/SO42−/ZrO2 | Pt/MOR |
|---|---|---|---|
| Operating temperature (°C) | 130–150 | 180–210 | 250–280 |
| Water tolerance (µg/g) | 0 | 20 | 200 |
| Sulfur tolerance (µg/g) | 0 | 20 | 200 |
| C7 hydrocarbon tolerance (%) | 2 | 2 | 5 |
| Technology | Zeolites | Comments |
|---|---|---|
| Gaseous hydrocarbons | ||
| skeletal isomerisation of n-butenes | FER | performance limited by rate of coking |
| hydroisomerisation of n-butane | - | catalyst selection related to feed purity |
| aliphatic alkylation | FAU | new industrial process; metal promoted |
| alkene oligomerisation | MFI, MWW, TON, MFS | product target drives catalyst selection |
| Light oxygenates | ||
| methanol to hydrocarbons | MFI, CHA | MFI for fuels, CHA for light alkenes |
| C2+ alcohols to hydrocarbons | - | |
| carbonyls to aromatic hydrocarbons | - | |
| etherification of alkenes with alcohols | - | |
| Naphtha | ||
| hydroisomerisation of light naphtha | MOR | more tolerant to feed impurities |
| catalytic naphtha reforming | LTL | high selectivity for C6/C7 n-alkanes |
| Distillate | ||
| hydroisomerisation of distillate | AEL | less hydrocracking at high conversion |
| Atmospheric residue | ||
| hydrocracking | AEL, MFI | amorphous supports used for wax |
| fluid catalytic cracking | FAU, MFI | FAU standard, MFI to boost propene |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klerk, A.D. Zeolites as Catalysts for Fuels Refining after Indirect Liquefaction Processes. Molecules 2018, 23, 115. https://doi.org/10.3390/molecules23010115
Klerk AD. Zeolites as Catalysts for Fuels Refining after Indirect Liquefaction Processes. Molecules. 2018; 23(1):115. https://doi.org/10.3390/molecules23010115
Chicago/Turabian StyleKlerk, Arno De. 2018. "Zeolites as Catalysts for Fuels Refining after Indirect Liquefaction Processes" Molecules 23, no. 1: 115. https://doi.org/10.3390/molecules23010115
APA StyleKlerk, A. D. (2018). Zeolites as Catalysts for Fuels Refining after Indirect Liquefaction Processes. Molecules, 23(1), 115. https://doi.org/10.3390/molecules23010115