
Unravelling the Distribution of Secondary Metabolites in Olea europaea L.: Exhaustive Characterization of Eight Olive-Tree Derived Matrices by Complementary Platforms (LC-ESI/APCI-MS and GC-APCI-MS)

 ,
, 
Abstract
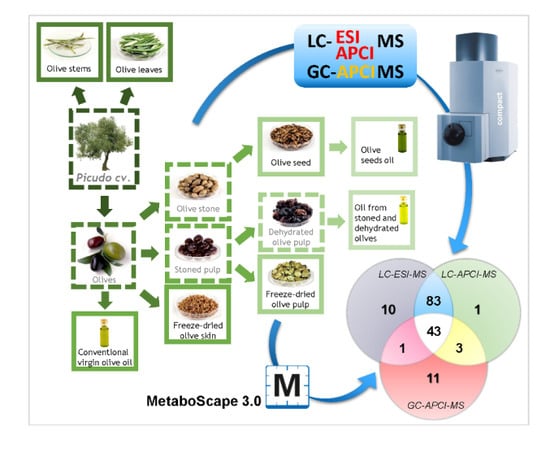
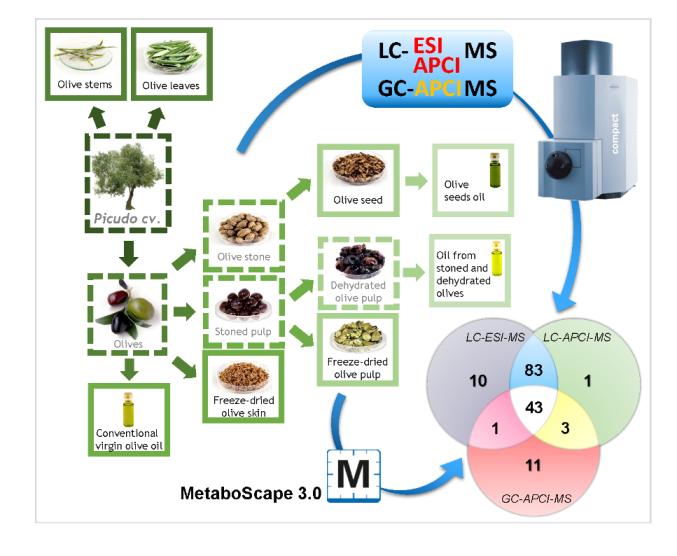{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Comprehensive Qualitative Determination of the Matrices under Study
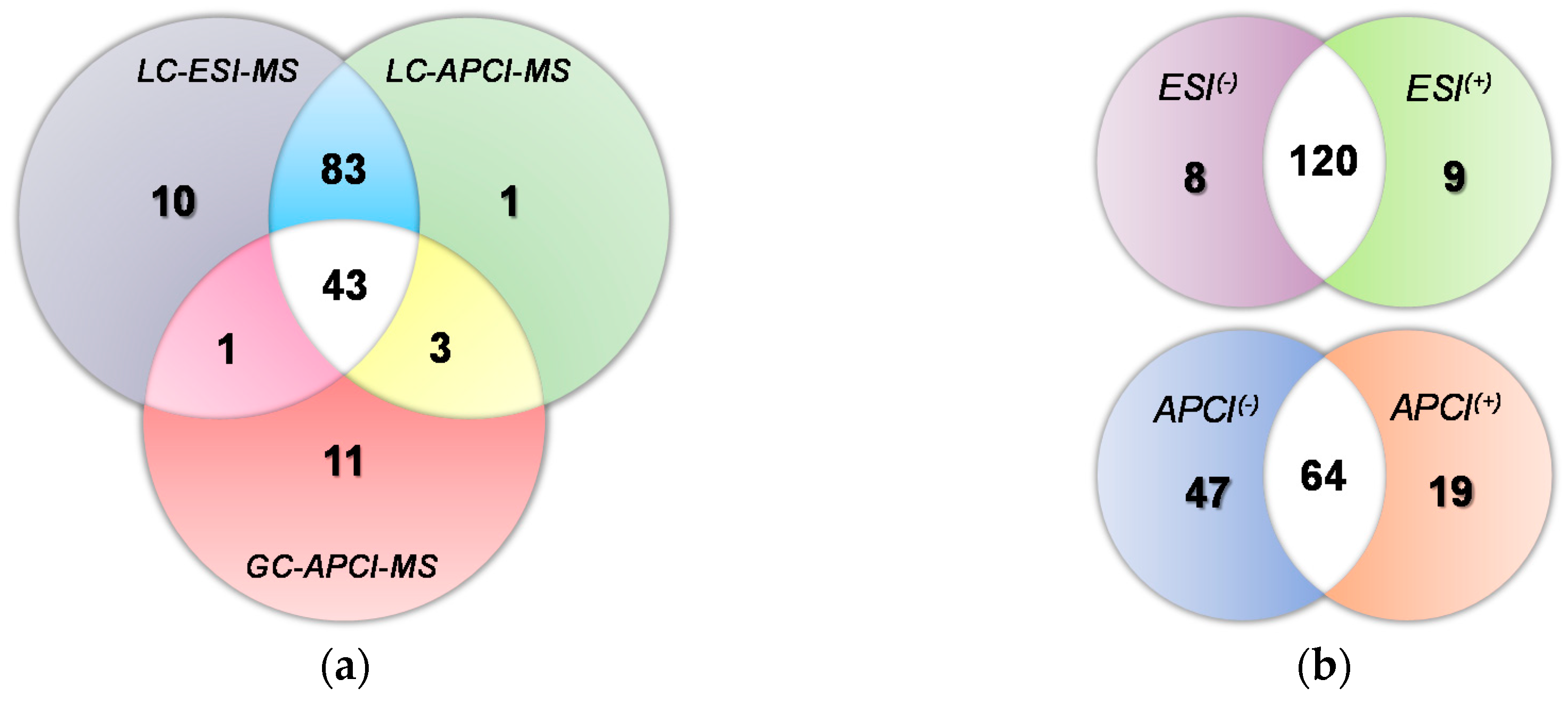
2.2. Comparison of the Potential of the Evaluated Analytical Platforms
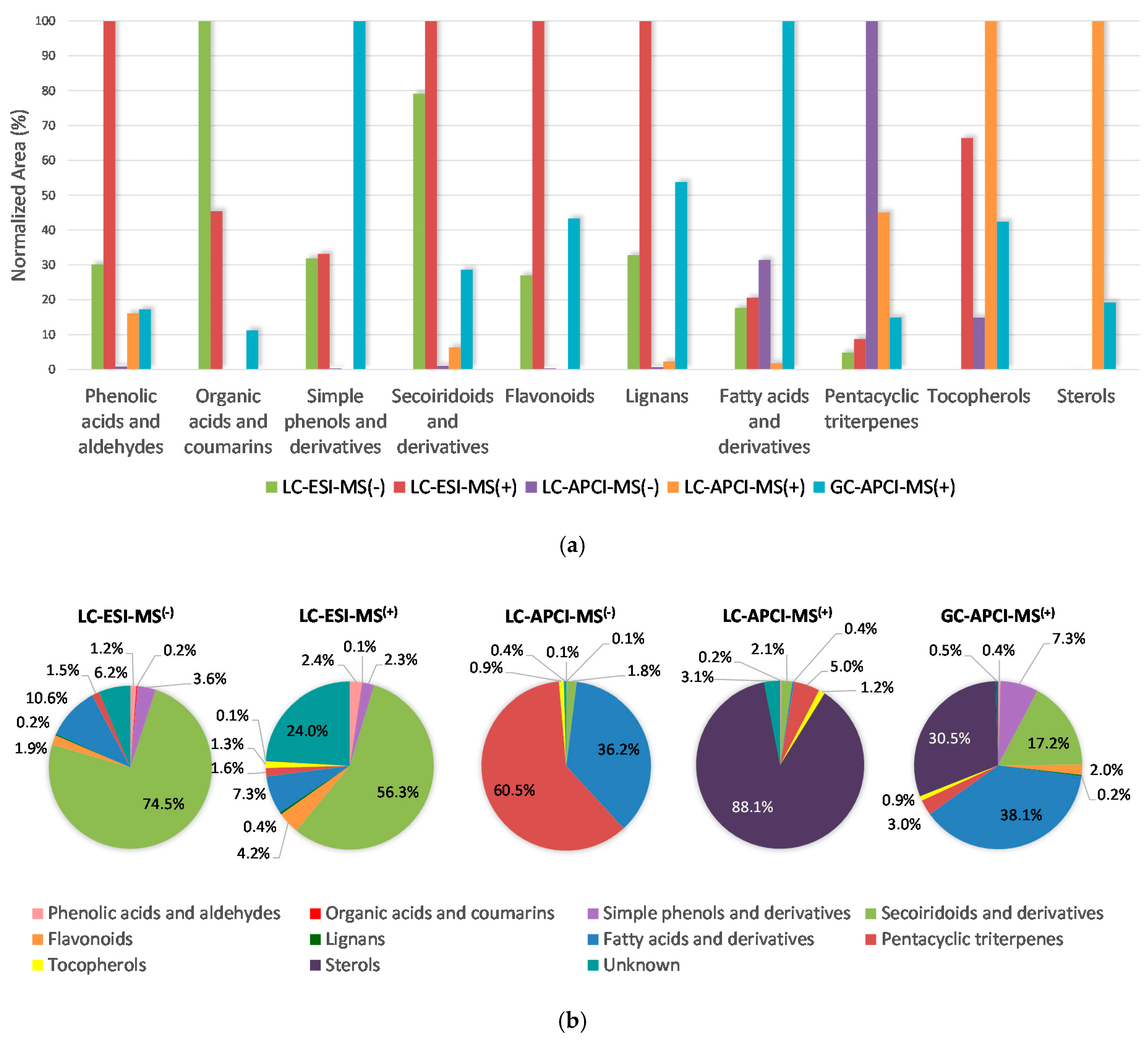
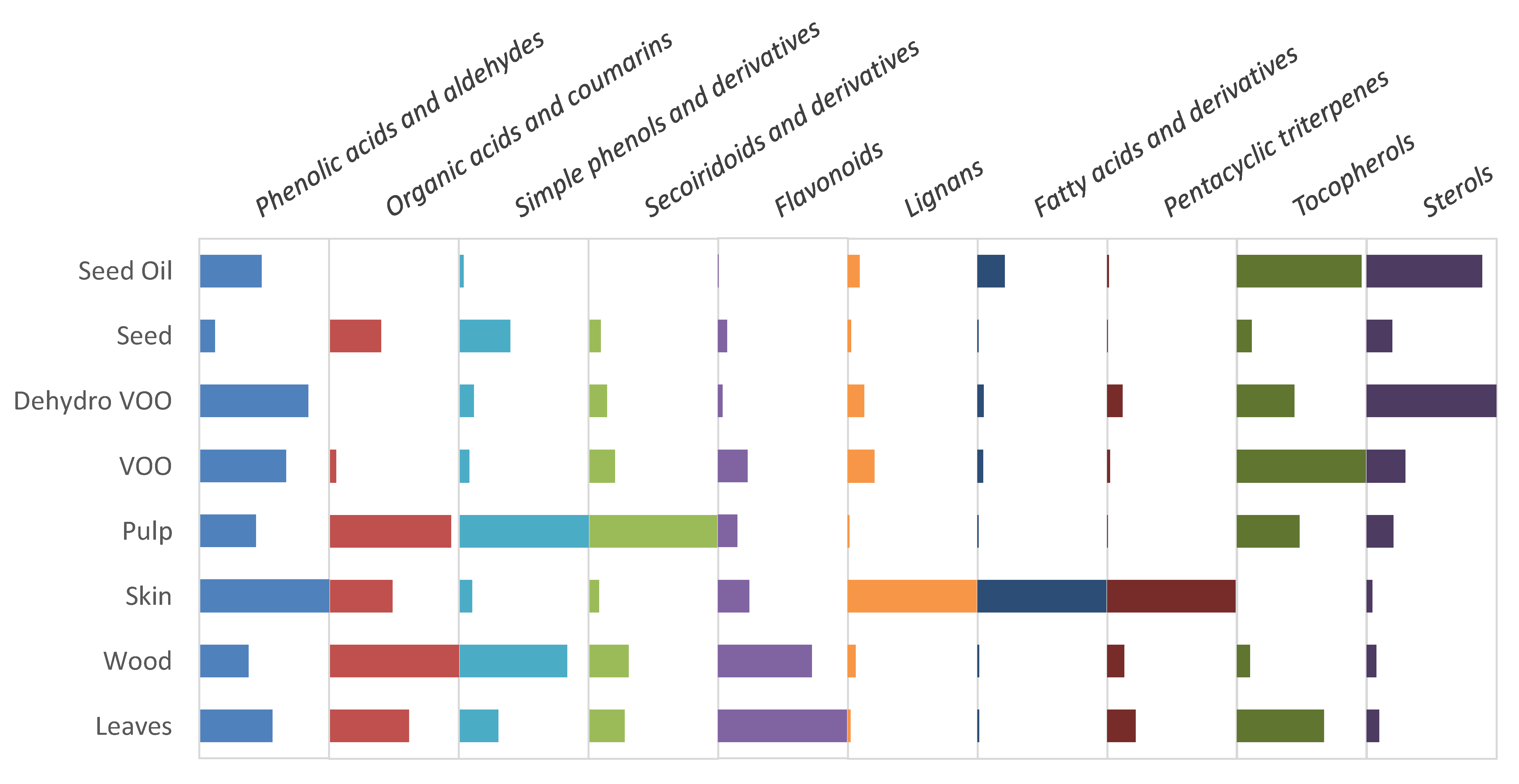
2.3. Establishing the Relative Prevalence of Each Determined Chemical Class in the Samples under Evaluation
3. Materials and Methods
3.1. Chemicals and Standards
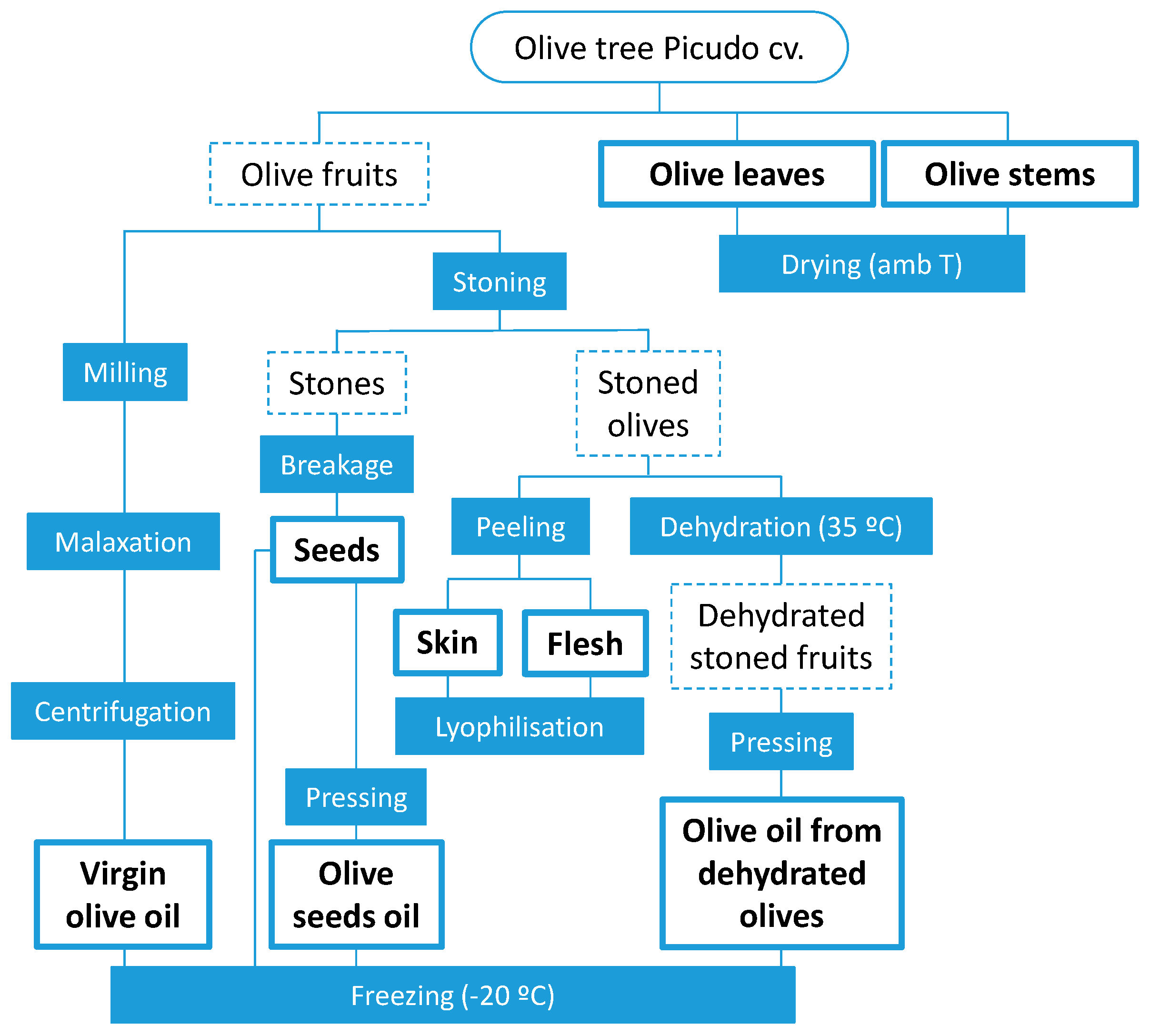
3.2. Samples and Sample Treatment
3.3. GC-MS and LC-MS Methodologies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaniewski, D.; Van Campo, E.; Boiy, T.; Terral, J.F.; Khadari, B.; Besnard, G. Primary domestication and early uses of the emblematic olive tree: Palaeobotanical, historical and molecular evidence from the Middle East. Biol. Rev. 2012, 87, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Vossen, P. Olive Oil: History, Production, and Characteristics of the World’s Classic Oils. HortScience 2007, 42, 1093–1100. [Google Scholar]
- Hashmi, M.A.; Khan, A.; Hanif, M.; Farooq, U.; Perveen, S. Traditional uses, phytochemistry, and pharmacology of Olea europaea (olive). Evid.-Based Complement. Altern. Med. 2015, 2015. [Google Scholar] [CrossRef]
- Ghanbari, R.; Anwar, F.; Alkharfy, K.M.; Gilani, A.H.; Saari, N. Valuable nutrients and functional bioactives in different parts of olive (Olea europaea L.)—A review. Int. J. Mol. Sci. 2012, 13, 3291–3340. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Quesada, C.; López-Biedma, A.; Warleta, F.; Campos, M.; Beltrán, G.; Gaforio, J.J. Bioactive properties of the main triterpenes found in olives, virgin olive oil, and leaves of Olea europaea. J. Agric. Food Chem. 2013, 61, 12173–12182. [Google Scholar] [CrossRef] [PubMed]
- Özcan, M.M.; Matthäus, B. A review: Benefit and bioactive properties of olive (Olea europaea L.) leaves. Eur. Food Res. Technol. 2017, 243, 89–99. [Google Scholar] [CrossRef]
- Fernandez del Rio, L.; Gutierrez-Casado, E.; Varela-Lopez, A.; Villalba, J.M. Olive oil and the hallmarks of aging. Molecules 2016, 21, 163. [Google Scholar] [CrossRef] [PubMed]
- Kanakis, P.; Termentzi, A.; Michel, T.; Gikas, E.; Halabalaki, M.; Skaltsounis, A.L. From olive drupes to olive Oil. An HPLC-orbitrap-based qualitative and quantitative exploration of olive key metabolites. Planta Med. 2013, 79, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Termentzi, A.; Halabalaki, M.; Skaltsounis, A.L. From Drupes to Olive Oil: An Exploration of Olive Key Metabolites. In Olive and Olive Oil Bioactive Constituents; AOCS Press: Urbana, IL, USA, 2015; ISBN 9781630670429. [Google Scholar]
- Talhaoui, N.; Gómez-Caravaca, A.M.; León, L.; De La Rosa, R.; Fernández-Gutiérrez, A.; Segura-Carretero, A. From olive fruits to olive Oil: Phenolic compound transfer in six different olive cultivars grown under the same agronomical conditions. Int. J. Mol. Sci. 2016, 17, 337. [Google Scholar] [CrossRef] [PubMed]
- Covas, M.I.; Fitó, M.; De La Torre, R. Minor Bioactive Olive Oil Components and Health: Key Data for Their Role in Providing Health Benefits in Humans. In Olive and Olive Oil Bioactive Constituents; AOCS Press: Urbana, IL, USA, 2015; ISBN 9781630670429. [Google Scholar]
- Piroddi, M.; Albini, A.; Fabiani, R.; Giovannelli, L.; Luceri, C.; Natella, F.; Rosignoli, P.; Rossi, T.; Taticchi, A.; Servili, M.; et al. Nutrigenomics of extra-virgin olive oil: A review. BioFactors 2016, 43, 17–41. [Google Scholar] [CrossRef] [PubMed]
- Frankel, E.; Bakhouche, A.; Lozano-Sánchez, J.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Literature review on production process to obtain extra virgin olive oil enriched in bioactive compounds. Potential use of byproducts as alternative sources of polyphenols. J. Agric. Food Chem. 2013, 61, 5179–5188. [Google Scholar] [CrossRef] [PubMed]
- Romero, C.; Medina, E.; Mateo, M.A.; Brenes, M. New by-products rich in bioactive substances from the olive oil mill processing. J. Sci. Food Agric. 2018, 98, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Dermeche, S.; Nadour, M.; Larroche, C.; Moulti-Mati, F.; Michaud, P. Olive mill wastes: Biochemical characterizations and valorization strategies. Process. Biochem. 2013, 48, 1532–1552. [Google Scholar] [CrossRef]
- Borja, B.R.; Raposo, F.; Rincón, B. Treatment technologies of liquid and solid wastes from two-phase olive oil mills. Grasas Aceites 2006, 57, 32–46. [Google Scholar] [CrossRef]
- Ciriminna, R.; Meneguzzo, F.; Fidalgo, A.; Ilharco, L.M.; Pagliaro, M. Extraction, benefits and valorization of olive polyphenols. Eur. J. Lipid Sci. Technol. 2015, 503–511. [Google Scholar] [CrossRef]
- Zbakh, H.; El Abbassi, A. Potential use of olive mill wastewater in the preparation of functional beverages: A review. J. Funct. Foods 2012, 4, 53–65. [Google Scholar] [CrossRef]
- Şahin, S.; Bilgin, M. Olive tree (Olea europaea L.) leaf as a waste by-product of table olive and olive oil industry: A review. J. Sci. Food Agric. 2017, 98, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Rahmanian, N.; Jafari, S.M.; Wani, T.A. Bioactive profile, dehydration, extraction and application of the bioactive components of olive leaves. Trends Food Sci. Technol. 2015, 42, 150–172. [Google Scholar] [CrossRef]
- Rodríguez, G.; Lama, A.; Rodríguez, R.; Jiménez, A.; Guillén, R.; Fernández-Bolaños, J. Olive stone an attractive source of bioactive and valuable compounds. Bioresour. Technol. 2008, 99, 5261–5269. [Google Scholar] [CrossRef] [PubMed]
- Roselló-Soto, E.; Koubaa, M.; Moubarik, A.; Lopes, R.P.; Saraiva, J.A.; Boussetta, N.; Grimi, N.; Barba, F.J. Emerging opportunities for the effective valorization of wastes and by-products generated during olive oil production process: Non-conventional methods for the recovery of high-added value compounds. Trends Food Sci. Technol. 2015, 45, 296–310. [Google Scholar] [CrossRef]
- Alves, E.; Rey, F.; Costa, E.; Moreira, A.S.P.; Pato, L.; Pato, L.; Domingues, M.R.M.; Domingues, P. Olive (Olea europaea L. cv. galega vulgar) seed oil: A first insight into the major lipid composition of a promising agro-industrial by-product at two ripeness stages. Eur. J. Lipid Sci. Technol. 2018, 120. [Google Scholar] [CrossRef]
- Guermazi, Z.; Gharsallaoui, M.; Perri, E.; Gabsi, S.; Benincasa, C. Integrated approach for the eco design of a new process through the life cycle analysis of olive oil: Total use of olive by-products. Eur. J. Lipid Sci. Technol. 2017, 119. [Google Scholar] [CrossRef]
- Calixto, F.S.; Díaz Rubio, M.E. Method for Obtaining Olive Oil and at Least One Multifunctional Ingredient from Olives. Patent WO 2013030426 A1, 7 March 2013. [Google Scholar]
- Luque De Castro, M.D. Towards a comprehensive exploitation of agrofood residues: Olive tree-Olive oil as example. C. R. Chim. 2014, 17, 252–260. [Google Scholar] [CrossRef]
- Piccinonna, S.; Ragone, R.; Stocchero, M.; Del Coco, L.; De Pascali, S.A.; Schena, F.P.; Fanizzi, F.P. Robustness of NMR-based metabolomics to generate comparable data sets for olive oil cultivar classification. An inter-laboratory study on Apulian olive oils. Food Chem. 2016, 199, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Servili, M.; Baldioli, M.; Selvaggini, R.; Macchioni, A.; Montedoro, G.F. Phenolic compounds of olive fruit: One- and two-dimensional nuclear magnetic resonance characterization of nuzhenide and its distribution in the constitutive parts of fruit. J. Agric. Food Chem. 1999, 47, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R.; Vaidyanathan, S.; Bianchi, G.; Kell, D.B. Metabolic profiling using direct infusion electrospray ionisation mass spectrometry for the characterisation of olive oils. Analyst 2002, 127, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Guodong, R.; Xiaoxia, L.; Weiwei, Z.; Wenjun, W.; Jianguo, Z. Metabolomics reveals variation and correlation among different tissues of olive (Olea europaea L.). Biol. Open 2017, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Khlif, I.; Kanakis, P.; Termentzi, A.; Allouche, N.; Halabalaki, M.; Skaltsounis, A.L. UHPLC-DAD-FLD and UHPLC-HRMS/MS based metabolic profiling and characterization of different Olea europaea organs of Koroneiki and Chetoui varieties. Phytochem. Lett. 2015, 11, 424–439. [Google Scholar] [CrossRef]
- Tóth, G.; Alberti, Á.; Sólyomváry, A.; Barabás, C.; Boldizsár, I.; Noszál, B. Phenolic profiling of various olive bark-types and leaves: HPLC–ESI/MS study. Ind. Crops Prod. 2015, 67, 432–438. [Google Scholar] [CrossRef]
- Ammar, S.; Contreras, M.d.M.; Gargouri, B.; Segura-Carretero, A.; Bouaziz, M. RP-HPLC-DAD-ESI-QTOF-MS based metabolic profiling of the potential Olea europaea by-product “wood” and its comparison with leaf counterpart. Phytochem. Anal. 2017, 28, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Obied, H.K.; Bedgood, D.R.; Prenzler, P.D.; Robards, K. Chemical screening of olive biophenol extracts by hyphenated liquid chromatography. Anal. Chim. Acta 2007, 603, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Servili, M.; Sordini, B.; Esposto, S.; Taticchi, A.; Urbani, S.; Sebastiani, L. Metabolomics of Olive Fruit: A Focus on the Secondary Metabolites. In The Olive Tree Genome; Springer: Cham, Switherland, 2016; pp. 123–139. ISBN 978-3-319-48886-8. [Google Scholar]
- Bajoub, A.; Pacchiarotta, T.; Hurtado-Fernández, E.; Olmo-García, L.; García-Villalba, R.; Fernández-Gutiérrez, A.; Mayboroda, O.A.O.A.; Carrasco-Pancorbo, A. Comparing two metabolic profiling approaches (liquid chromatography and gas chromatography coupled to mass spectrometry) for extra-virgin olive oil phenolic compounds analysis: A botanical classification perspective. J. Chromatogr. A 2015, 1428, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Olmo-García, L.; Polari, J.J.; Li, X.; Bajoub, A.; Fernández-Gutiérrez, A.; Wang, S.C.; Carrasco-Pancorbo, A. Deep insight into the minor fraction of virgin olive oil by using LC-MS and GC-MS multi-class methodologies. Food Chem. 2018, 261, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, Y.; Xu, L.; Bai, Y.; Liu, H. Online coupling techniques in ambient mass spectrometry. Analyst 2016, 141, 5913–5921. [Google Scholar] [CrossRef] [PubMed]
- Lanina, S.A.; Toledo, P.; Sampels, S.; Kamal-Eldin, A.; Jastrebova, J.A. Comparison of reversed-phase liquid chromatography-mass spectrometry with electrospray and atmospheric pressure chemical ionization for analysis of dietary tocopherols. J. Chromatogr. A 2007, 1157, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Cañabate-Díaz, B.; Segura Carretero, A.; Fernández-Gutiérrez, A.; Belmonte Vega, A.; Garrido Frenich, A.; Martínez Vidal, J.L.; Duran Martos, J. Separation and determination of sterols in olive oil by HPLC-MS. Food Chem. 2007, 102, 593–598. [Google Scholar] [CrossRef]
- Caruso, D.; Colombo, R.; Patelli, R.; Giavarini, F.; Galli, G. Rapid evaluation of phenolic component profile and analysis of oleuropein aglyeon in olive oil by atmospheric pressure chemical ionization-mass spectrometry (APCI-MS). J. Agric. Food Chem. 2000, 48, 1182–1185. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Fernández, E.; Pacchiarotta, T.; Longueira-Suárez, E.; Mayboroda, O.A.; Fernández-Gutiérrez, A.; Carrasco-Pancorbo, A. Evaluation of gas chromatography-atmospheric pressure chemical ionization-mass spectrometry as an alternative to gas chromatography-electron ionization-mass spectrometry: Avocado fruit as example. J. Chromatogr. A 2013, 1313, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Saimaru, H.; Orihara, Y. Biosynthesis of acteoside in cultured cells of Olea europaea. J. Nat. Med. 2010, 64, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Obied, H.K.; Prenzler, P.D.; Ryan, D.; Servili, M.; Taticchi, A.; Esposto, S.; Robards, K. Biosynthesis and biotransformations of phenol-conjugated oleosidic secoiridoids from Olea europaea L. Nat. Prod. Rep. 2008, 25, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Rufino-Palomares, E.E.; Perez-Jimenez, A.; Reyes-Zurita, F.J.; Garcia- Salguero, L.; Mokhtari, K.; Herrera-Merchan, A.; Medina, P.P.; Peragon, J.; Lupianez, J.A. Anti-cancer and Anti-angiogenic Properties of Various Natural Pentacyclic Tri-terpenoids and Some of their Chemical Derivatives. Curr. Org. Chem. 2015, 19, 919–947. [Google Scholar] [CrossRef]
- Lerma-García, M.J.; Concha-Herrera, V.; Herrero-Martínez, J.M.; Simó-Alfonso, E.F. Classification of extra virgin olive oils produced at La Comunitat Valenciana according to their genetic variety using sterol profiles established by high-performance liquid chromatography with mass spectrometry detection. J. Agric. Food Chem. 2009, 57, 10512–10517. [Google Scholar] [CrossRef] [PubMed]
- Zarrouk, W.; Carrasco-Pancorbo, A.; Zarrouk, M.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Multi-component analysis (sterols, tocopherols and triterpenic dialcohols) of the unsaponifiable fraction of vegetable oils by liquid chromatography-atmospheric pressure chemical ionization-ion trap mass spectrometry. Talanta 2009, 80, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Dong, L.; Hurst, W.J.; Van Breemen, R.B. Quantitative analysis of phytosterols in edible oils using APCI liquid chromatography-tandem mass spectrometry. Lipids 2013, 48, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.P.; Rady, A.H.; Castañeda, S. The formation of oxo- and hydroxy-fatty acids in heated fats and oils. J. Am. Oil Chem. Soc. 1994, 71, 441–444. [Google Scholar] [CrossRef]
- Jiménez-Sánchez, C.; Olivares-Vicente, M.; Rodríguez-Pérez, C.; Herranz-López, M.; Lozano-Sánchez, J.; Segura-Carretero, A.; Fernández-Gutiérrez, A.; Encinar, J.A.; Micol, V. AMPK modulatory activity of olive-tree leaves phenolic compounds: Bioassay-guided isolation on adipocyte model and in silico approach. PLoS ONE 2017, 12, e0173074. [Google Scholar] [CrossRef] [PubMed]
- Maestro-Durán, R.; León Cabello, R.; Ruiz-Gutiérrez, V.; Fiestas, P.; Vázquez-Roncero, A. Glucósidos fenólicos amargos de las semillas de olivo (Olea europaea). Grasas Aceites 1994, 45, 332–335. [Google Scholar] [CrossRef]
- Servili, M.; Baldioli, M.; Selvaggini, R.; Miniati, E.; Macchioni, A.; Montedoro, G. High-performance liquid chromatography evaluation of phenols in olive fruit, virgin olive oil, vegetation waters, and pomace and 1D- and 2D-nuclear magnetic resonance characterization. J. Am. Oil Chem. Soc. 1999, 76, 873–882. [Google Scholar] [CrossRef]
- Olmo-García, L.; Bajoub, A.; Fernández-Gutiérrez, A.; Carrasco-Pancorbo, A. Evaluating the potential of LC coupled to three alternative detection systems (ESI-IT, APCI-TOF and DAD) for the targeted determination of triterpenic acids and dialcohols in olive tissues. Talanta 2016, 150, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Olmo-García, L.; Wendt, K.; Kessler, N.; Bajoub, A.; Fernández-Gutiérrez, A.; Baessmann, C.; Carrasco-Pancorbo, A. Exploring the capability of LC-MS and GC-MS multi-class methods to discriminate olive oils from different geographical indications and to identify potential origin markers. Eur. J. Lipid Sci. Technol. 2018. Under review. [Google Scholar]
- Wolf, S.; Schmidt, S.; Müller-Hannemann, M.; Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinform. 2010, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olmo-García, L.; Kessler, N.; Neuweger, H.; Wendt, K.; Olmo-Peinado, J.M.; Fernández-Gutiérrez, A.; Baessmann, C.; Carrasco-Pancorbo, A. Unravelling the Distribution of Secondary Metabolites in Olea europaea L.: Exhaustive Characterization of Eight Olive-Tree Derived Matrices by Complementary Platforms (LC-ESI/APCI-MS and GC-APCI-MS). Molecules 2018, 23, 2419. https://doi.org/10.3390/molecules23102419
Olmo-García L, Kessler N, Neuweger H, Wendt K, Olmo-Peinado JM, Fernández-Gutiérrez A, Baessmann C, Carrasco-Pancorbo A. Unravelling the Distribution of Secondary Metabolites in Olea europaea L.: Exhaustive Characterization of Eight Olive-Tree Derived Matrices by Complementary Platforms (LC-ESI/APCI-MS and GC-APCI-MS). Molecules. 2018; 23(10):2419. https://doi.org/10.3390/molecules23102419
Chicago/Turabian StyleOlmo-García, Lucía, Nikolas Kessler, Heiko Neuweger, Karin Wendt, José María Olmo-Peinado, Alberto Fernández-Gutiérrez, Carsten Baessmann, and Alegría Carrasco-Pancorbo. 2018. "Unravelling the Distribution of Secondary Metabolites in Olea europaea L.: Exhaustive Characterization of Eight Olive-Tree Derived Matrices by Complementary Platforms (LC-ESI/APCI-MS and GC-APCI-MS)" Molecules 23, no. 10: 2419. https://doi.org/10.3390/molecules23102419
APA StyleOlmo-García, L., Kessler, N., Neuweger, H., Wendt, K., Olmo-Peinado, J. M., Fernández-Gutiérrez, A., Baessmann, C., & Carrasco-Pancorbo, A. (2018). Unravelling the Distribution of Secondary Metabolites in Olea europaea L.: Exhaustive Characterization of Eight Olive-Tree Derived Matrices by Complementary Platforms (LC-ESI/APCI-MS and GC-APCI-MS). Molecules, 23(10), 2419. https://doi.org/10.3390/molecules23102419