Artificial Intelligence in Drug Design


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
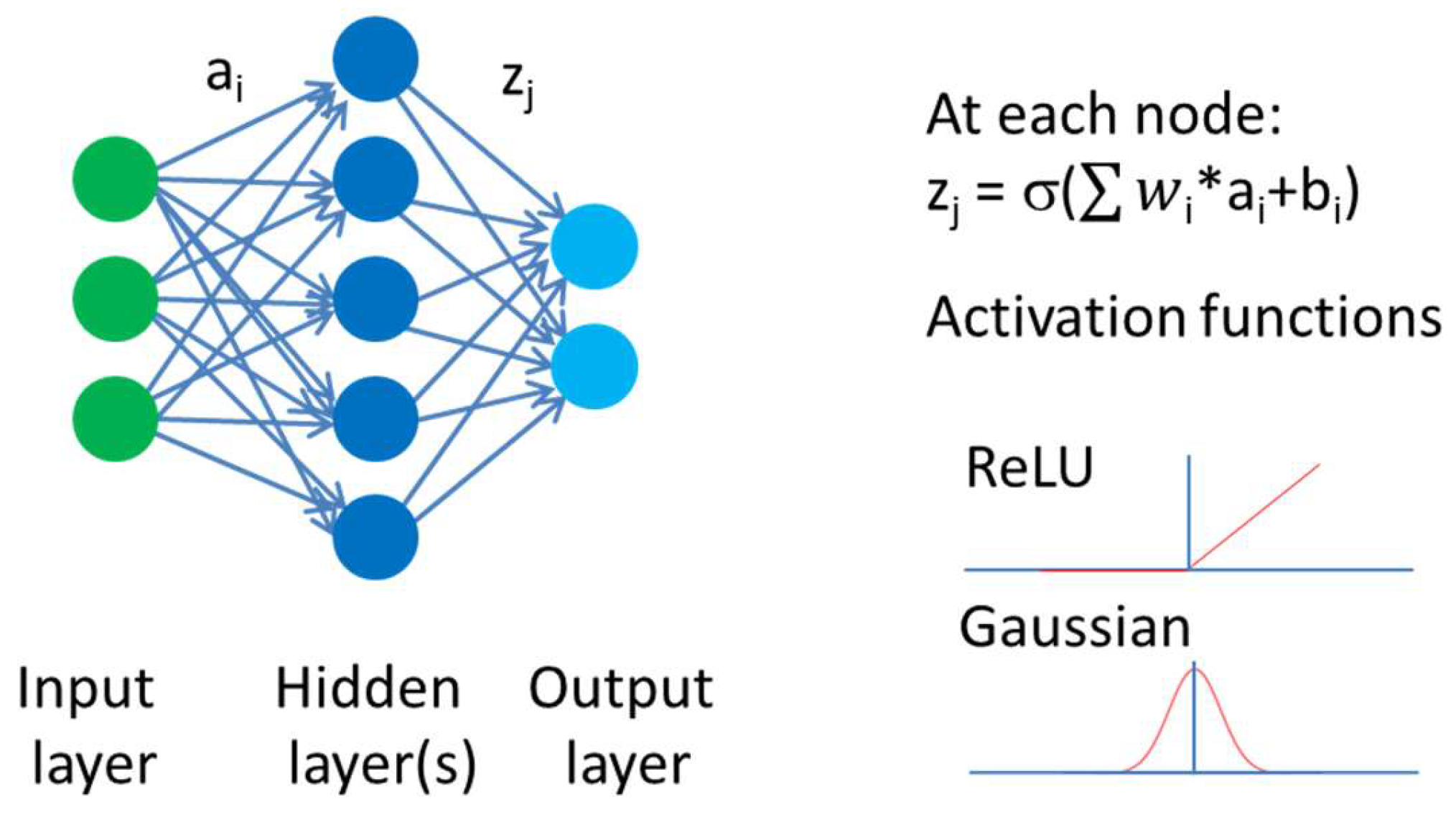
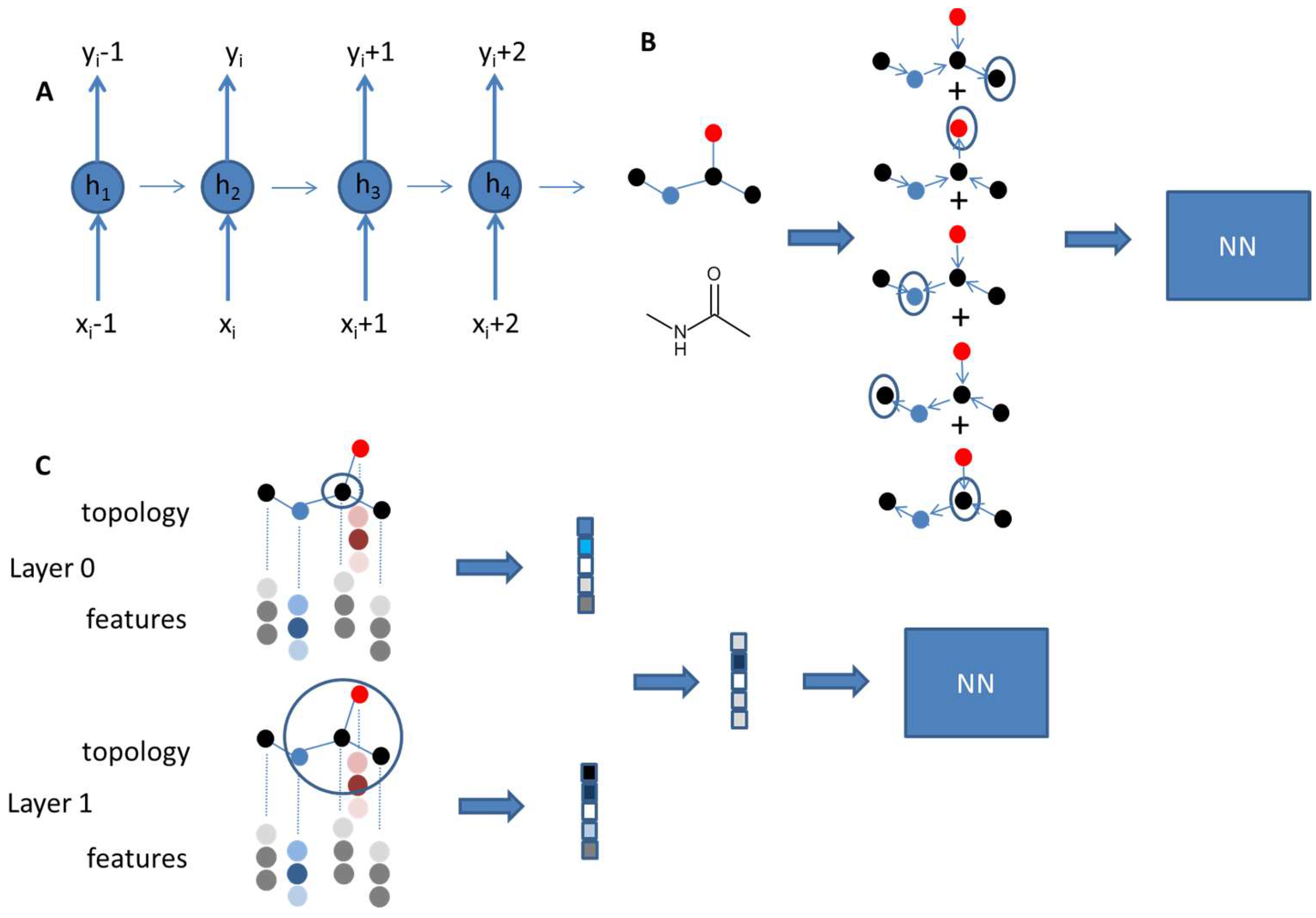
2. Artificial Intelligence in Property Prediction
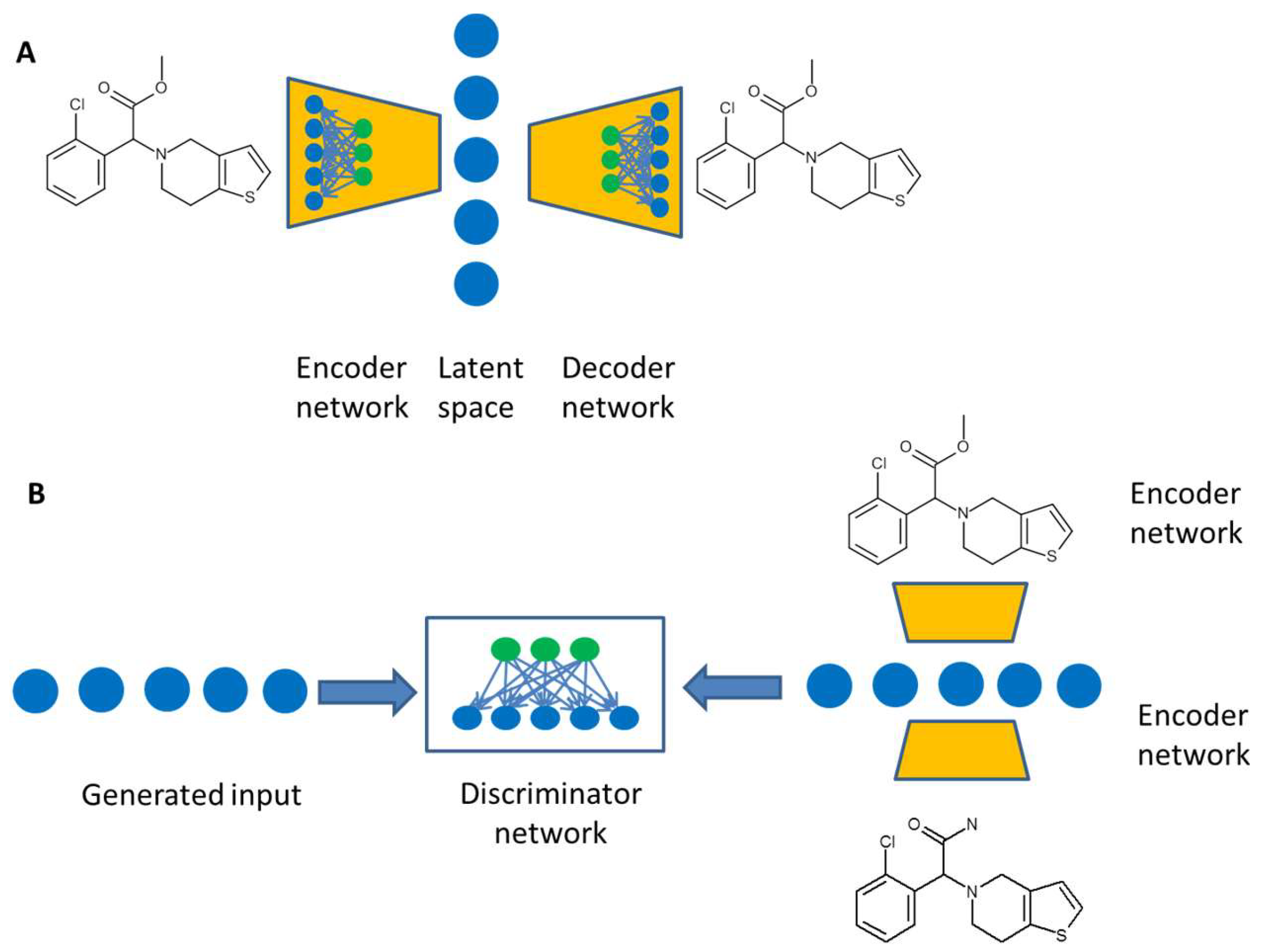
3. Artificial Intelligence for de novo Design
4. Artificial Intelligence for Synthesis Planning
5. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References and Note
- Howard, J. The business impact of deep learning. In Proceedings of the 19th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Chicago, IL, USA, 11–14 August 2013; p. 1135. [Google Scholar]
- Impact Analysis: Buisiness Impact of Deep Learning. Available online: https://www.kaleidoinsights.com/impact-analysis-businedd-impacts-of-deep-learning/ (accessed on 10 August 2018).
- Deep Learning, with Massive Amounts of Computational Power, Machines Can Now Recognize Objects and Translate Speech in Real Time. Artificial Intelligence Is Finally Getting Smart. Available online: https://www.technologyreview.com/s/513696/deep-learning/ (accessed on 10 August 2018).
- Silver, D.; Huang, A.; Maddison, C.J.; Guez, A.; Sifre, L.; van den Driessche, G.; Schrittieser, J.; Antonoglou, I.; Panneershelvam, V.; Lactot, M.; et al. Mastering the game of Go with deep neural networks and tree search. Nature 2016, 529, 484. [Google Scholar] [CrossRef] [PubMed]
- Hassabis, D. Artificial intelligence: Chess match of the century. Nature 2017, 544, 413–414. [Google Scholar] [CrossRef]
- Artificial Intelligence. Available online: https://en.wikipedia.org/wiki/Artificial_intelligence (accessed on 16 June 2018).
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep neural nets as a method for quantitative structure–activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreither, S. Deep Tox: Toxicity prediction using Deep Learning. Front. Environ. Sci. 2016, 3. [Google Scholar] [CrossRef]
- Jørgensen, P.B.; Schmidt, M.N.; Winther, O. Deep Generative Models for Molecular. Sci. Mol. Inf. 2018, 37, 1700133. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.B.; Hodas, N.O.; Vishnu, A. Deep Learning for Computational Chemistry. J. Comp. Chem. 2017, 38, 1291–1307. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Bian, Y.; Hu, Z.; Wang, L.; Xie, X.S. Deep Learning for Drug Design: An Artificial Intelligence Paradigm for Drug Discovery in the Big Data Era. AAPS J. 2018, 20, 58. [Google Scholar] [CrossRef] [PubMed]
- Gawehn, E.; Hiss, J.A.; Schneider, G. Deep Learning in Drug Discovery. Mol. Inf. 2016, 35, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Gawehn, E.; Hiss, J.A.; Brown, J.B.; Schneider, G. Advancing drug discovery via GPU-based deep learning. Expert Opin. Drug Discov. 2018, 13, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Colwell, L.J. Statistical and machine learning approaches to predicting protein–ligand interactions. Curr. Opin. Struct. Biol. 2018, 49, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tan, J.; Han, D.; Zhu, H. From machine learning to deep learning: Progress in machine intelligence for rational drug discovery. Drug Discov. Today 2017, 22, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Panteleeva, J.; Gaoa, H.; Jiab, L. Recent applications of machine learning in medicinal chemistry. Bioorg. Med. Chem. Lett. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Data analytics and deep learning in medicinal chemistry. Future Med. Chem. 2018, 10, 1541–1543. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Model. 2003, 43, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Duda, R.O.; Hart, P.E.; Stork, G.E. Pattern Classification, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2001; pp. 20–83. ISBN 0-471-05669-3. [Google Scholar]
- Rogers, D.; Brown, R.D.; Hahn, M. Using Extended-Connectivity Fingerprints with Laplacian-Modified Bayesian Analysis in High-Throughput Screening Follow-Up. J. Biomol. Screen. 2005, 10, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.; Mukherjee, P.; Sullivan, D.; Jansen, J. Profile-QSAR: A novel meta-QSAR method that combines activities across the kinase family to accurately predict affinity, selectivity, and cellular activity. J. Chem. Inf. Model. 2011, 51, 1942–1956. [Google Scholar] [CrossRef] [PubMed]
- Merget, B.; Turk, S.; Eid, S.; Rippmann, F.; Fulle, S. Profiling Prediction of Kinase Inhibitors: Toward the Virtual Assay. J. Med. Chem. 2017, 60, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Varnek, A.; Baskin, I. Machine Learning Methods for Property Prediction in Cheminformatics: Quo Vadis? J. Chem. Inf. Model. 2012, 52, 1413–1437. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Lima, A.N.; Philot, E.A.; Trossini, G.H.G.; Scott, L.P.B.; Maltarollo, V.G.; Honorio, K.M. Use of machine learning approaches for novel drug discovery. Expert Opin. Drug Discov. 2016, 2016 11, 225–239. [Google Scholar] [CrossRef]
- Ghasemi, F.; Mehridehnavi, A.; Pérez-Garrido, A.; Pérez-Sánchez, H. Neural network and deep-learning algorithms used in QSAR studies: Merits and drawbacks. Drug Discov. Today 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Pogodin, P.V.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. PASS Targets: Ligand-based multi-target computational system based on a public data and naïve Bayes approach. SAR QSAR Environ. Res. 2015, 26, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Mervin, L.H.; Afzal, A.M.; Drakakis, G.; Lewis, R.; Engkvist, O.; Bender, A. Target prediction utilising negative bioactivity data covering large chemical space. J. Cheminform. 2015, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Vidal, D.; Garcia-Serna, R.; Mestres, J. Ligand-based approaches to in silico pharmacology. Methods Mol. Biol. 2011, 672, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Steindl, T.M.; Schuster, D.; Laggner, C.; Langer, T. Parallel Screening: A Novel Concept in Pharmacophore Modelling and Virtual Screening. J. Chem. Inf. Model. 2006, 46, 2146–2157. [Google Scholar] [CrossRef] [PubMed]
- Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; Whaley, R.; Glennon, R.A.; Hert, J.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Nair, V.; Hinton, G.E. Rectified linear units improve restricted boltzmann machines. In Proceedings of the 27th International Conference on International Conference on Machine Learning, Haifa, Israel, 21–24 June 2010; pp. 807–814. [Google Scholar]
- ChEMBL. Available online: https://www.ebi.ac.uk/chembl (accessed on 15 September 2018).
- Unterthiner, T.; Mayr, A.; Klambauer, G.; Steijaert, M.; Ceulemans, H.; Wegner, J.; Hochreiter, S. Deep Learning as an Opportunity in Virtual Screening. In Proceedings of the NIPS Workshop on Deep Learning and Representation Learning, Montreal, QC, Canada, 8–13 December 2014; pp. 1058–1066. Available online: http://www.bioinf.at/publications/2014/NIPS2014a.pdf (accessed on 15 September 2018).
- Lenselink, E.B.; ten Dijke, N.; Bongers, B.; Papadatos, G.; van Vlijmen, H.W.T.; Kowalczyk, W.; IJzerman, A.P.; van Westen, G.J.P. Beyond the hype: Deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J. Cheminform. 2017, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, R.P. Time-split cross-validation as a method for estimating the goodness of prospective prediction. J. Chem. Inf. Model. 2013, 53, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Korotcov, A.; Tkachenko, V.; Russo, D.P.; Ekins, S. Comparison of Deep Learning with Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Data Sets. Mol. Pharm. 2017, 14, 4462–4475. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Dai, Z.; Chen, F.; Gao, S.; Pei, J.; Lai, L. Deep Learning for Drug-Induced Liver Injury. J. Chem. Inf. Model. 2015, 55, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Xie, Q.; Ge, W.; Qian, F.; Fang, H.; Shi, L.; Su, Z.; Perkins, R.; Tong, W. Mold2, Molecular Descriptors from 2D Structures for Chemoinformatics and Toxicoinformatics. J. Chem. Inf. Model. 2008, 48, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Lusci, A.; Pollastri, G.; Baldi, P. Deep architectures and deep learning in chemoinformatics: The prediction of aqueous solubility for drug-like molecules. J. Chem. Inf. Model. 2013, 53, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Schenkenberg, T.; Bradford, D.; Ajax, E. Line Bisection and Unilateral Visual Neglect in Patients with Neurologic Impairment. Neurology 1980, 30, 509. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Glen, R.C.; Bender, A.; Arnby, C.H.; Carlsson, L.; Boyer, S.; Smith, J. Circular fingerprints: Flexible molecular descriptors with applications from physical chemistry to ADME. IDrugs Investig. Drugs J. 2006, 9, 199–204. [Google Scholar]
- Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Gomez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R.P. Convolutional Networks on Graphs for Learning Molecular Fingerprints. In Proceedings of the 28th International Conference on Neural Information Processing Systems, Montreal, QC, Canada, 7–12 December 2015; Volume 2, pp. 2224–2232. Available online: http://arxiv.org/abs/1509.09292 (accessed on 15 September 2018).
- Li, J.; Cai, D.; He, X. Learning Graph-Level Representation for Drug Discovery. arXiv, 2017; arXiv:1709.03741v2. Available online: http://arxiv.org/abs/1709.03741 (accessed on 15 September 2018).
- Kearnes, S.; McCloskey, K.; Berndl, M.; Pande, V.; Riley, P. Molecular graph convolutions: Moving beyond fingerprints. J. Comput. Aided Mol. Des. 2016, 30, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N.O.; Baker, N. A Deep Neural Network with Minimal Chemistry Knowledge Matches the Performance of Expert-developed QSAR/QSPR Models. arXiv:1706.06689.
- Ramsundar, B.; Kearnes, S.; Riley, P.; Webster, D.; Konerding, D.; Pandey, V. Massively Multitask Networks for Drug Discovery. arXiv, 2015; arXiv:1502.02072v1. Available online: http://arxiv.org/abs/1502.02072 (accessed on 15 September 2018).
- Kearnes, S.; Goldman, B.; Pande, V. Modeling Industrial ADMET Data with Multitask Networks. arXiv, 2016; arXiv:1606.08793v3. Available online: http://arxiv.org/abs/1606.08793v3 (accessed on 15 September 2018).
- Ramsundar, B.; Liu, B.; Wu, Z.; Verras, A.; Tudor, M.; Sheridan, R.P.; Pande, V. Is Multitask Deep Learning Practical for Pharma? J. Chem. Inf. Model. 2017, 57, 2068–2076. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, J.; Liaw, A.; Sheridan, R.P.; Svetnik, V. Demystifying Multitask Deep Neural Networks for Quantitative Structure−Activity Relationships. J. Chem. Inf. Model. 2017, 57, 2490–2504. [Google Scholar] [CrossRef] [PubMed]
- Vogt, M.; Jasial, S.; Bajorath, J. Extracting Compound Profiling Matrices from Screening Data. ACS Omega 2018, 3, 4713–4723. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Isayev, O.; Roitberg, A.E. ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules. Sci. Data 2017, 4, 170193. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Steijaert, M.; Wegner, J.K.; Ceulemans, H.; Clevert, D.-A.; Hochreiter, S. Large-scale comparison of machine learning methods for drug prediction on ChEMBL. Chem. Sci. 2018, 9, 5441–5451. [Google Scholar] [CrossRef] [PubMed]
- Hartenfeller, M.; Schneider, G. Enabling future drug discovery by de novo design. WIREs Comput. Mol. Sci. 2011, 1, 742–759. [Google Scholar] [CrossRef]
- Schneider, P.; Schneider, G. De Novo Design at the Edge of Chaos. J. Med. Chem. 2016, 59, 4077–4086. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adamsk, P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. arXiv, 2016; arXiv:1610.02415v3. Available online: http://arxiv.org/abs/1610.02415 (accessed on 15 September 2018).
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminf. 2009, 1. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, T.; Olivecrona, M.; Engkvist, O.; Bajorath, J.; Chen, H. Application of Generative Autoencoder in De Novo Molecular Design. Mol. Inf. 2018, 37, 1700123. [Google Scholar] [CrossRef] [PubMed]
- Kadurin, A.; Aliper, A.; Kazennov, A.; Mamoshina, P.; Vanhaelen, Q.; Kuzma, K.; Zhavoronkov, A. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2017, 8, 10883–10890. [Google Scholar] [CrossRef] [PubMed]
- Bengio, Y. Learning Deep Architectures for AI. Found. Trends Mach. Learn. 2009, 2, 1–127. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Müller, A.T.; Huisman, B.J.H.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative Recurrent Networks for De Novo Drug Design. Mol. Inf. 2018, 37, 1700111. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.T.; Hiss, J.A.; Schneider, G. Recurrent Neural Network Model for Constructive Peptide Design. J. Chem. Inf. Model. 2018, 58, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H. Molecular de-novo design through deep reinforcement learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.; Friedrich, L.; Grisoni, F.; Schneider, G. De Novo Design of Bioactive Small Molecules by Artificial Intelligence Daniel. Mol. Inf. 2018, 37, 1700153. [Google Scholar] [CrossRef] [PubMed]
- Engkvist, O.; Norrby, P.-O.; Selmi, N.; Lam, Y.; Peng, Z.; Sherer, C.E.; Amberg, W.; Erhard, T.; Smyth, L.A. Computational prediction of chemical reactions: Current status and outlook. Drug Discov. Today 2018, 23, 1203–1218. [Google Scholar] [CrossRef] [PubMed]
- Szymkuc, S.; Gajewska, E.P.; Klucznik, T.; Molga, K.; Dittwald, P.; Startek, M.; Bajczyk, M.; Grzybowski, B.A. Computer-Assisted Synthetic Planning: The End of the Beginning. Angew. Chem. Int. Ed. 2016, 55, 5904–5937. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Baldi, P. Synthesis Explorer: A Chemical Reaction Tutorial System for Organic Synthesis Design and Mechanism Prediction. J. Chem. Educ. 2008, 85, 1699–1703. [Google Scholar] [CrossRef]
- Law, J.; Zsoldos, Z.; Simon, A.; Reid, D.; Liu, Y.; Khew, S.Y.; Johnson, A.P.; Major, S.; Wade, R.A.; Ando, H.Y. Route Designer: A Retrosynthetic Analysis Tool Utilizing Automated Retrosynthetic Rule Generation. J. Chem. Inf. Model. 2009, 49, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Kayala, M.A.; Azencott, C.A.; Chen, J.H.; Baldi, P. Learning to predict chemical reactions. J. Chem. Inf. Model. 2011, 51, 2209–2222. [Google Scholar] [CrossRef] [PubMed]
- Kayala, M.A.; Baldi, P. ReactionPredictor: Prediction of complex chemical reactions at the mechanistic level using machine learning. J. Chem. Inf. Model. 2012, 52, 2526–2540. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, P.; Fooshee, D.; Subrahmanya, N.; Baldi, P. Synergies between quantum mechanics and machine learning in reaction prediction. J. Chem. Inf. Model. 2016, 56, 2125–2128. [Google Scholar] [CrossRef] [PubMed]
- Fooshee, D.; Mood, A.; Gutman, E.; Tavakoli, M.; Urban, G.; Liu, F.; Huynh, N.; Van Vranken, D.; Baldi, P. Deep learning for chemical reaction prediction. Mol. Syst. Des. Eng. 2018, 3, 442–452. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Waller, M.P. Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction. Chem. Eur. J. 2017, 23, 5966–5971. [Google Scholar] [CrossRef] [PubMed]
- http://www.reaxys.com, Reaxys is a registered trademark of RELX Intellectual Properties SA used under license.
- Coley, W.; Barzilay, R.; Jaakkola, T.S.; Green, W.H.; Jensen, K.F. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent. Sci. 2017, 3, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Coley, C.W.; Barzilay, R.; Jaakkola, T. Predicting Organic Reaction Outcomes with Weisfeiler-Lehman Network. arXiv, 2017; arXiv:1709.04555. [Google Scholar]
- Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.; Gomes, J.; Luu Nguyen, Q.; Ho, S.; Sloane, J.; Wender, P.; Pande, V. Retrosynthetic reaction prediction using neural sequence-to-sequence models. ACS Cent. Sci. 2017, 3, 103–1113. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.; Kishimoto, A.; Buesser, B.; Diaz-Aviles, E.; Alzate, C. Chemical Reactant Recommendation Using a Network of Organic Chemistry. Available online: https://cseweb.ucsd.edu/classes/fa17/cse291-b/reading/p210-savage.pdf (accessed on 18 September 2018).
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Schreiner, P.R. Computational Chemistry: The Fate of Current Methods and Future Challenges. Angew. Chem. Int. Ed. 2018, 57, 4170–4176. [Google Scholar] [CrossRef] [PubMed]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. TensorFlow: A System for Large-Scale Machine Learning. arXiv, 2016; arXiv:1605.08695v2. [Google Scholar]
- Keras: The Python Deep Learning library. Available online: https://keras.io (accessed on 18 September 2018).
- Deepchem. Available online: https://deepchem.io (accessed on 18 September 2018).




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hessler, G.; Baringhaus, K.-H. Artificial Intelligence in Drug Design. Molecules 2018, 23, 2520. https://doi.org/10.3390/molecules23102520
Hessler G, Baringhaus K-H. Artificial Intelligence in Drug Design. Molecules. 2018; 23(10):2520. https://doi.org/10.3390/molecules23102520
Chicago/Turabian StyleHessler, Gerhard, and Karl-Heinz Baringhaus. 2018. "Artificial Intelligence in Drug Design" Molecules 23, no. 10: 2520. https://doi.org/10.3390/molecules23102520
APA StyleHessler, G., & Baringhaus, K.-H. (2018). Artificial Intelligence in Drug Design. Molecules, 23(10), 2520. https://doi.org/10.3390/molecules23102520