For high resolution mass spectrometry, a Waters GCT Premier instrument was used with electron impact ionization (70 eV) at an ion source temperature of 200 °C.
General Information on Syntheses
Compounds were synthesized as described below. Solvents were of analytical quality, if not stated otherwise. The purity of all synthesized compounds was verified using NMR and analytical HPLC. Analytical thin layer chromatography (TLC) was performed using aluminium foil coated with silica 60 F
254 (Merck, Darmstadt, Germany). Preparative thin layer chromatography (PTLC) was performed using glass plates coated with silica 60 F
254 (Merck). Detection was done using UV/254 nm and spraying with molybdophosphoric acid and subsequent heating. Compound mixtures were separated using column chromatography (CC) on silica gel 60 (63–200 µm, Merck) using cyclohexane/ethyl acetate mixtures. Further purification was performed using preparative HPLC (Varian Prepstar with a Dynamax Rainin detector; column SepServ, Berlin, Germany, 250 × 21 mm, RP-18, 7 μm, flow rate 15 mL). Honokiol (purity > 98%) was purchased from APIChem Technology Co. (Hangzhou, China). Proton NMR spectra of the newly described compounds are given under “
Supplementary Materials”.
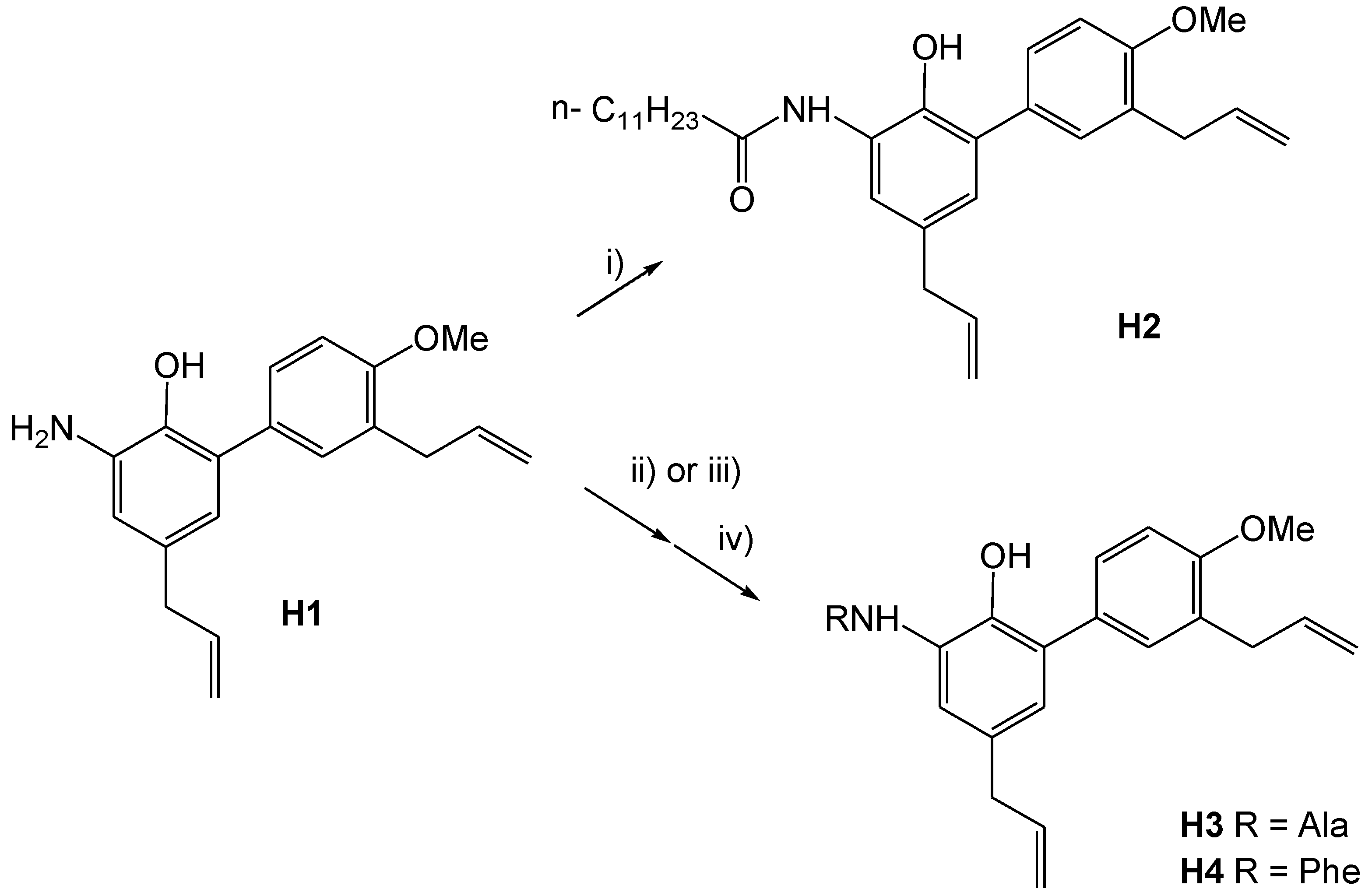
3-Dodecanoylamino-4′-O-methylhonokiol (H2)
Dodecanoyl chloride (0.34 mmol, 80 μL) was added with intense stirring at room temperature (RT) to a solution of 100 mg (0.34 mmol) of H1 and pyridine (0.41 mmol, 33 μL) in abs. Et2O (4 mL). The reaction mixture was stirred at room temperature overnight and filtered. The precipitate was extracted with Et2O (4 mL). NaHCO3 (1 M, 10 mL) was added to the combined filtrate and washings. The organic phase was separated, and the aqueous phase was extracted with Et2O (3 × 10 mL). The combined organic phases were washed with brine (10 mL), dried over Na2SO4, concentrated under reduced pressure, and purified using CC (silica, cyclohexane/AcOEt 5:1) resulting in 107 mg (59%) of a light brown solid (H2).
Compound
H2: IR (ATR,
νmax, cm
−1): 3304, 2956, 2919, 2847, 2485, 1639, 1606, 1539, 1499, 1467, 1411, 1243, 906, 725, 577;
1H-NMR (CDCl
3) δ 7.57 (bs, 1H, NH), 7.40 (d,
J = 1.5 Hz, 1H, H-4), 7.34 (dd,
J = 8.4, 2.3 Hz, 1H, H-6′), 7.27 (d,
J = 2.1 Hz, 1H, H-2′), 7.17 (s, 1H, OH), 6.94 (d,
J = 8.4 Hz, 1H, H-5′), 6.89 (d,
J = 1.9 Hz, 1H, H-6), 6.01 (ddt,
J = 16.8, 10.2, 6.6 Hz, 1H, H-2‴), 5.96 (ddt,
J = 16.8, 10.0, 6.7 Hz, 1H, H-2″), 5.02–5.13 (m, 4H, H-3‴, H-3″), 3.87 (s, 3H, OCH
3), 3.43 (d,
J = 6.6 Hz, 2H, H-1‴), 3.33 (d,
J = 6.7 Hz, 2H, H-1″), 2.42 (t,
J = 7.5 Hz, 2H, Acyl-H
α), 1.74 (quint,
J = 7.3 Hz, 2H, Acyl-H
β), 1.21–1.42 (m, 16H, Acyl-H
γ to H
ω-1), 0.88 (t,
J = 6.8 Hz, 3H, Acyl-H
ω),
13C-NMR (CDCl
3) δ 172.7 (CO), 156.8 (C-4′), 142.3 (C-2), 137.5 (C-2″), 136.7 (C-2‴), 132.2 (C-5), 130.7 (C-2′), 130.7 (C-1), 129.6 (C-1′), 129.0 (C-3′), 128.1 (C-6′), 126.7 (C-6), 126.2 (C-3), 120.3 (C-4), 115.8 (C-3″), 115.6 (C-3‴), 110.5 (C-5′), 55.5 (OCH
3), 39.5 (C-1″), 37.5 (Acyl-C
α), 34.3 (C-1‴), 31.9 (Acyl-C
ω-2), {2 × 29.6, 29.4, 2 × 29.3, 29.2 (Acyl-C
γ-to C
ω-3)}, 25.7 (Acyl-C
β), 22.7 (Acyl-C
ω-1), 14.1 (Acyl-C
ω), ESI
+ calcd for C
31H
43NO
3: [M]
+ 477.324; found ESI-MS
m/
z (rel. int.): 478.63 [M + H]
+ (100). From the same batch, the compound has been tested in parallel towards CB
1/CB
2 receptor agonistic activity by Bertini et al. [
22]; however, spectroscopic data and synthesis are only given here.
3-(N-l-Alanyl)-4′-O-methylhonokiol (H3)
l-N-(9-Fluorenylmethoxycarbonyl)alanylchloride, Fmoc-l-Ala-Cl: Fmoc-l-Ala-OH · H2O was first dehydrated at 50 °C over P2O5 in vacuo for 12 h. Under dry conditions 400 mg (1.28 mmol) l-N-(9-Fluorenylmethoxycarbonyl)alanin was suspended in 5 mL dichloromethane, thionyl chloride (freshly distilled, 1.23 mL, 17.7 mmol) was added, and the mixture was sonicated at RT. After 7 min, the reaction mixture was a homogeneous solution and it was sonicated for an additional 15 min. Dichloromethane and the excess of thionyl chloride were removed in vacuo to yield 404 mg (96%) of Fmoc-l-Ala-Cl as a grey solid, which was used without further purification for a second step.
l-3-(N-(9-Fluorenylmethoxycarbonyl)alanyl)-4′-O-methylhonokiol (H3a): Zn (dust) was treated with HCl (2 M, aqueous solution), washed subsequently with water, EtOH and diethylether, dried in vacuo, and stored under argon. Under argon, H1 (259 mg, 0.841 mmol), Fmoc-l-Ala-Cl (279 mg, 0.846 mmol), and Zn dust (55 mg, 0.841 mmol) were suspended in abs. THF (50 mL) and stirred at RT overnight. Over a period of two hours, the reaction color turned from brownish to yellowish. Reaction completion was proven using TLC analysis (cyclohexane/AcOEt = 5:3). Zn dust was filtered off and the filtrate was evaporated in vacuo. The residue was dissolved in dichloromethane (15 mL), washed with brine (2 × 5 mL), and dried over Na2SO4. Evaporation of the solvent in vacuo and CC (silica, cyclohexane/AcOEt = 5:1) yielded in H3a (428 mg; 87%).
3-(N-l-Alanyl)-4′-O-methylhonokiol (H3): H3a (401 mg, 0.682 mmol) was dissolved in diethyl ether (3 mL) and piperidine (1 mL, 1 mmol) was added. The mixture was stirred at RT for 1 h. The reaction mixture was evaporated under reduced pressure and dichloromethane (5 mL) was added to the residue. The resulting precipitate was filtered off and the filtrate was concentrated under reduced pressure to dryness. The residue (377 mg) was purified using CC (silica, dichloromethane/MeOH, gradient: 0–10% v/v methanol) to yield 220 mg (88%) of H3 as an orange oil.
Compound H3: IR (ATR, νmax, cm−1): 3272, 3075, 2974, 2929, 2834, 1638, 1606, 1590, 1530, 1500, 1460, 1440, 1244, 1029, 993, 910, 813, 594; 1H-NMR (CDCl3 + D2O) δ 7.35 (dd, J = 8.1, 1.8 Hz, 1H, H-6′), 7.29 (d, J = 2.2 Hz, 1H, H-2′), 7.18 (s, 1H, H-4), 6.92 (s, 1H, H-6), 6.91 (d, J = 8.4 Hz, 1H, H-5′), 6.01 (ddt, J = 16.8, 9.8, 6.6 Hz, 1H, H-2‴), 5.95 (ddt, J = 16.9, 9.9, 6.6 Hz, 1H, H-2″), 5.01–5.13 (m, 4H, H-3″, H-3‴), 3.85 (s, 3H, O–CH3), 3.73 (q, J = 6.2 Hz, 1H, CH3CH(NH2)CO), 3.42 (d, J = 6.6 Hz, 2H, H-1‴), 3.32 (d, J = 6.2 Hz, 2H, H-1″), 2.03 (s, 2H, NH2), 1.45 (d, J = 6.6 Hz, 3H, CH3CH(NH2)CO); 13C-NMR (CDCl3 + D2O) δ 174.6 (CO), 156.7 (C-4′), 143.2 (C-2), 137.5 (C-2″), 136.8 (C-2‴), 131.9 (C-5), 131.0 (C-1), 130.8 (C-2′), 130.1 (C-1′), 128.6 (C-3′), 128.2 (C-6′), 127.5 (C-6), 125.6 (C-3), 120.6 (C-4), 115.7 (C-3″), 115.5 (C-3‴), 110.3 (C-5′), 55.5 (O–CH3), 50.5 (CH3CH(NH2)CO), 39.4 (C-1″), 34.3 (C-1‴), 21.1 (CH3CH(NH2)CO); ESI+ calcd for C22H26N2O3: [M]+ 366.194; found ESI-MS m/z (rel. int.): 367.18 [M + H]+ (100).
3-(N-l-Phenylalanyl)-4′-O-methylhonokiol (H4)
l-N-(9-Fluorenylmethoxycarbonyl)phenylalanylchloride, Fmoc-l-Phe-Cl: Under dry conditions 300 mg (0.774 mmol) l-N-(9-Fluorenylmethoxycarbonyl)phenylalanin was suspended in dry dichloromethane (4 mL), thionyl chloride (freshly distilled, 0.776 mL, 10.7 mmol) was added, and the mixture was sonicated at RT for 45 min. The sonication bath was heated up to 40 °C over that period of time. Dichloromethane and the excess of thionyl chloride were removed in vacuo to yield 294 mg (94%) of Fmoc-l-Phe-Cl as a grey solid, which was used without further purification in the next step.
3-(N-l-Phenylalanyl)-4′-O-methylhonokiol (H4): Zn dust was treated with aqueous HCl (2 M), washed subsequently with water, ethanol, and diethylether, dried in vacuo, and stored under argon. Under argon, H1 (75 mg, 0.25 mmol), Fmoc-l-Phe-Cl (103 mg, 0.254 mmol), and Zn dust (17 mg, 0.25 mmol) were suspended in dry THF (10 mL) and stirred for 4 h. Reaction completion was proven using TLC analysis (cyclohexane/AcOEt = 5:3). Piperidine (0.5 mL, 5 mmol) was added and the reaction mixture was stirred for 20 min at RT, the solvent was evaporated under reduced pressure and the residue was diluted with dichloromethane (5 mL), filtered, and evaporated under reduced pressure. The residue (170 mg) was purified using CC (silica, cyclohexane/AcOEt, gradient: 0–100% v/v AcOEt) to yield 50 mg (45%) of H4 as an orange oil.
Compound H4: IR (ATR, νmax, cm−1): 3270, 3073, 2974, 2834, 1638, 1605, 1530, 1500, 1453, 1439 s, 1244, 1028, 910; 1H-NMR (CDCl3) δ 9.69 (bs, 1H, CONH), 7.39 (dd, J = 8.4, ≈2 Hz, 1H, H-6′), 7.30–7.36 (m, 3H, Ph-Hmeta Ph-Hpara), 7.22–7.30 (m, 3H, H-2′, Ph-Hortho), 7.10 (s, 1H, H-4), 6.95 (s, 1H, H-6), 6.92 (d, J = 8.4 Hz, 1H, H-5′), 6.01 (ddt, J = 16.9, 9.9, 6.6 Hz, 1H, H-2‴), 5.97 (ddt, J = 16.9, 10.2, 6.6 Hz, 1H, H-2″), 5.02–5.14 (m, 4H, H-3″, H-3‴), 3.86 (s, 3H, OCH3), 3.82 (m, 1H, PhCH2CH(NH2)CO), 3.43 (d, J = 6.6 Hz, 2H, H-1‴), 3.34 (m, 1H, PhCH2CH(NH2)CO), 3.33 (d, J = 6.9 Hz, 2H, H-1″), 2.84 (dd, J = 13.6, 9.2 Hz, 1H, PhCH2CH(NH2)CO); 13C-NMR (CDCl3) δ 173.7 (CO), 156.6 (C-4′), 143.5 (C-2), 139.1 (Ph-Cipso), 137.5 (C-2″), 136.8 (C-2‴), 131.8 (C-5), 131.2 (C-1), 130.8 (C-2′), 130.2 (C-1′), 129.3 (Ph-Cortho), 128.8 (Ph-Cmeta), 128.6 (C-3′), 128.2 (C-6′), 127.6 (C-6), 127.0 (Ph-Cpara), 125.7 (C-3), 120.5 (C-4), 115.7 (C-3″), 115.5 (C-3‴), 110.3 (C-5′), 56.3 (PhCH2CH(NH2)CO), 55.5 (OCH3), 40.5 (PhCH2CH(NH2)CO), 39.3 (C-1″), 34.3 (C-1‴); ESI+ calcd for C28H30N2O3: [M]+ 442.226; found ESI-MS m/z (rel. int.): 442.20 [M + H]+ (100).
3-Amino-4′-O-ethylhonokiol (H5)
4′-
O-Ethyl-3-nitro-honokiol (
H5a): Aqueous nitric acid (65%, 0.474 mL, 6.79 mmol) was added under intense stirring within ≈5 s to a solution of
H10 (200 mg, 0.679 mmol; synthesis see Schuehly et al. [
12] supplemental; cpd
16b) in AcOEt (5 mL) at RT. The reaction mixture was stirred for 60 s and quenched with NaHCO
3 (1 M, 10 mL). The organic phase was separated, and the aqueous phase was extracted with AcOEt (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over Na
2SO
4, concentrated under reduced pressure, and purified using CC (silica, cyclohexane/AcOEt 99:1) to yield 120 mg (50%) of 4′-
O-ethyl-3-nitro-honokiol (
H5a) as a dark yellow oil.
Compound H5a: IR (ATR, νmax, cm−1): 3177, 3078, 2978, 2903, 1638, 1607, 1537, 1502, 1314, 1244, 1045, 912, 672, 555 cm−1; 1H-NMR (CDCl3) δ 11.03 (s, 1H, OH), 7.90 (d, J = 2.2 Hz, 1H, H-4), 7.45 (d, J = 2.2 Hz, 1H, H-6), 7.38 (dd, J = 8.4, 2.2, 1H, H-6′), 7.33 (d, J = 2.2 Hz, 1H, H-2′), 6.92 (d, J = 8.4 Hz, 1H, H-5′), 6.03 (ddt, J = 16.9, 10.3, 6.6 Hz, 1H, H-2‴), 5.96 (ddt, J = 16.5, 10.3, 6.6 Hz, 1H, H-2″), 5.03–5.17 (m, 4H, H-3″ and H-3‴), 4.10 (q, J = 7.2 Hz, 2H, OCH2CH3), 3.45 (d, J = 6.4 Hz, 2H, H-1‴), 3.40 (d, J = 6.6 Hz, 2H, H-1″), 1.46 (t, J = 6.9 Hz, 3H, OCH2CH3); 13C-NMR (CDCl3) δ 156.6 (C-4′), 151.3 (C-2), 138.9 (C-6), 136.8 (C-2‴), 136.0 (C-2″), 133.8 (C-3), 132.9 (C-1), 131.7 (C-5), 130.7 (C-2′), 128.5 (C-3′), 128.2 (C-6′), 127.4 (C-1′), 122.8 (C-4), 117.1 (C-3‴), 115.6 (C-3″), 111.0 (C-5′), 63.7 (OCH2CH3), 38.9 (C-1″), 34.5 (C-1‴), 14.9 (OCH2CH3); ESI– calcd for C20H21NO4: [M]− 339.147; found ESI-MS m/z (rel. int.): 338.21 ([M − H]− (100).
3-Amino-4′-O-ethylhonokiol (H5): SnCl2×2H2O (1.62 g, 7.16 mmol) was added to a solution of H5a (270 mg, 0.796 mmol) in abs. MeOH (10 mL). The reaction mixture was stirred for 48 h at RT, concentrated under reduced pressure and diluted with AcOEt (15 mL). The spumy precipitate resulting from the addition of aqueous NaHCO3 (1 M, 20 mL) was filtered off with Celite® (Sigma-Aldrich, Buchs, Switzerland) and rinsed with AcOEt (30 mL). The organic layer was separated from the combined filtrate and washings, dried over Na2SO4, and concentrated under reduced pressure. The residue (127 mg) was purified using PTLC (silica, cyclohexane/AcOEt = 5:1) to yield 65 mg (26%) of H5 as a brown oil.
Compound H5: IR (ATR, νmax, cm−1): 3544, 3370, 3075, 2977, 2900, 1637, 1607, 1503, 1486,1474, 1242, 1215, 1122, 1044, 908, 808; 1H-NMR (CDCl3) δ 7.26 (d ≈ 9 Hz, 1H, H-6′), 7.25 (s, 1H, H-2′), 6.93 (d, J = 8.1 Hz, 1H, H-5′), 6.61 (s, 1H, H-4), 6.51 (s, 1H, H-6), 6.00 (ddt, J = 16.9, 9.9, 6.6 Hz, 1H, H-2‴), 5.96 (ddt, J = 17.0, 9.9, 6.9 Hz, 1H, H-2″), 5.02–5.14 (m, 4H, H-3″, H-3‴), 4.19 (bs, 2H, NH2), 4.09 (q, J = 7.0 Hz, 2H, OCH2CH3), 3.43 (d, J = 6.6 Hz, 2H, H-1‴), 3.28 (d, J = 6.9 Hz, 2H, H-1″), 1.46 (t, J = 7.0 Hz, 3H, OCH2CH3); 13C-NMR (CDCl3) δ 156.4 (C-4′), 139.0 (C-2), 137.9 (C-2″), 136.6 (C-2‴), 133.6 (C-3), 132.4 (C-5), 130.3 (C-2′), 129.9 (C-3′), 129.0 (C-1′), 127.9 (C-1), 127.7 (C-6′), 120.5 (C-6), 115.8 (C-3‴), 115.7 (C-4), 115.4 (C-3″), 111.9 (C-5′), 63.7 (OCH2CH3), 39.6 (C-1″), 34.5 (C-1‴), 14.9 (OCH2CH3); ESI+ calcd C20H23NO2: [M]+ 309.173; found ESI-MS m/z (rel. int.): 310.11 [M + H]+ (100).
3-Amino-2-O-ethyl-4′-O-methylhonokiol (H6)
In a microwave process vial, KOH (132 mg, 2.36 mmol) was added to a solution of 4′-
O-methyl-3-nitro-honokiol (synthesis see [
10]) (192 mg, 0.590 mmol) in abs. MeOH (2 mL) and the mixture was stirred at RT for 10 min. Diethyl sulfate (155 μL, 2.36 mmol) was added and the reaction mixture was irradiated in a microwave oven at 90 °C for 60 min. After cooling to RT, the reaction mixture was neutralized with aqueous HCl (2 M). MeOH was evaporated under reduced pressure (40 °C, 100 mbar). The residue was extracted with dichloromethane (3 × 2 mL), the organic phase washed with water (3 × 5 mL), dried over Na
2SO
4, and concentrated under reduced pressure, resulting in 198 mg of crude 2-
O-ethyl-4′-
O-methyl-3-nitro-honokiol, which was used without further purification.
SnCl2×2H2O (1.09 g, 4.84 mmol) was added to a solution of crude 2-O-ethyl-4′-O-methyl-3-nitro-honokiol (190 mg, 0.54 mmol) in abs. EtOH (10 mL). The reaction mixture was stirred at RT for 48 h. The spumy precipitate resulting from the addition of aqueous NaHCO3 (1 M, 20 mL) was filtered using Celite®, the residue was extracted with EtOH (50 mL). Combined organic layers were evaporated under reduced pressure (100 mbar). The residue was extracted with dichloromethane (3 × 10 mL), which was washed with brine (3 × 5 mL), dried over Na2SO4, and concentrated under reduced pressure to yield 107 mg of a crude product, which after purification using PTLC (silica, cyclohexane/AcOEt = 5:1), yielded 26 mg (14%) of H6.
Compound H6: IR (ATR, νmax, cm−1): 3417, 3310, 3076, 2975, 2928, 2836, 1674, 1638, 1607, 1522, 1502, 1435, 1245, 1026, 910, 812, 600, 523; 1H-NMR (CDCl3) δ 7.47 (d, J = 2.2 Hz, 1H, H-2′), 7.46 (dd, J = 8.1, 2.2 Hz, 1H, H-6′), 6.92 (d, J = 8.4 Hz, 1H, H-5′), 6.60 (s, 2H, H-4, H-6), 6.07 (ddt, J = 16.6, 10.2, 6.2 Hz, 1H, H-2‴), 6.00 (ddt, J = 16.9, 9.9, 6.9 Hz, 1H, H-2″), 5.04–5.17 (m, 4H, H-3″, H-3‴), 3.96 (bs, 2H, NH2), 3.89 (s, 3H, OCH3), 3.57 (q, J = 6.9 Hz, 2H, OCH2CH3), 3.46 (d, J = 6.6 Hz, 2H, H-1‴), 3.33 (d, J = 6.9 Hz, 2H, H-1″), 1.16 (t, J = 7.1 Hz, 3H, OCH2CH3); 13C-NMR (CDCl3) δ 156.4 (C-4′), 142.1 (C-2), 140.0 (C-3), 137.6 (C-2″),137.0 (C-2‴), 136.0 (C-5), 134.4 (C-1), 131.0 (C-1′), 130.5 (C-2′), 128.0 (C-3′), 127.6 (C-6′), 120.5 (C-6), 115.5 (C-3″), 115.2 (C-3‴), 114.7 (C-4), 109.9 (C-5′), 67.6 (C–OCH2CH3), 55.4 (OCH3), 39.9 (C-1″), 34.3 (C-1‴), 15.7 (OCH2CH3); ESI+ calcd for C21H25NO2: [M]+ 323.189; found ESI-MS m/z (rel. int.): 324.23 [M + H]+ (100).
3-Acetylamino-2-O-ethyl-4′-O-methylhonokiol (H7)
In a 10 mL round-bottom flask, H6 (34 mg, 0.105 mmol) was mixed with water (0.3 mL), and acetic anhydride (0.21 mmol, 20 μL) was added. The flask was rotated at 80 °C for about 10 min in a water bath. After cooling to RT, the reaction mixture was quenched with aqueous NaHCO3 (1 M, 2 mL) and extracted with dichloromethane (3 × 2 mL). The combined extracts were washed with aqueous NaHCO3 (1 M, 2 mL), water (2 mL), dried over Na2SO4, and concentrated under reduced pressure. The crude product (27 mg) was purified using PTLC (cyclohexane/AcOEt = 5:3), yielding 21 mg (55%) of H7 as a brownish oil.
Compound H7: IR (ATR, νmax, cm−1): 3469, 3368, 3075, 2974, 2928, 2903, 2834, 1638, 1606, 1504, 1438, 1243, 1209, 1030, 909, 810, 596; 1H-NMR (CDCl3) δ 8.17 (s, 1H, H-4), 7.96 (bs, 1H, NH), 7.38 (s, 1H, H-2′), 7.37 (dd, J ≈ 8, 2.2 Hz, 1H, H-6′), 6.90 (d, J = 8.1 Hz, 1H, H-5′), 6.86 (d, J = 1.1 Hz, 1H, H-6), 6.03 (ddt, J = 16.8, 9.9, 6.9 Hz, 1H, H-2‴), 5.98 (ddt, J = 16.9, 9.9, 6.6 Hz, 1H, H-2″), 5.02–5.15 (m, 4H, H-3″, H-3‴), 3.87 (s, 3H, OCH3), 3.52 (q, J = 7.1 Hz, 2H, OCH2CH3), 3.42 (d, J = 6.6 Hz, 2H, H-1‴), 3.38 (d, J = 7.0 Hz, 2H, H-1″), 2.21 (s, 3H, CH3CO), 1.14 (t, J = 7.0 Hz, 3H, OCH2CH3); 13C-NMR (CDCl3) δ 168.09 (CO), 156.7 (C-4′), 143.1 (C-2), 137.3 (C-2″), 136.9 (C-2‴), 136.2 (C-5), 133.5 (C-1), 131.9 (C-3), 130.4 (C-1′), 130.3 (C-2′), 128.4 (C-3′), 127.6 (C-6′), 125.3 (C-6), 118.7 (C-4), 115.8 (C-3″), 115.4 (C-3‴), 110.2 (C-5′), 68.7 (O–CH2–CH3), 55.4 (OCH3), 40.0 (C-1″), 34.3 (C-1‴), 24.9 (CH3–C=O), 15.6 (OCH2CH3); ESI+ calcd for C23H27NO3: [M]+ 365.199; found ESI-MS m/z (rel. int.): 366.29 [M + H]+ (100).
5′-(N-Acetylamino)-2-O-methyl-honokiol (H9)
In a 10 mL round-bottom flask,
H12 (for synthesis, see Reference [
11]) (37 mg, 0.125 mmol) was suspended with water (1 mL), and acetic anhydride (0.21 mmol, 20 μL) was added. The flask was allowed to rotate for 10 min at 80 °C in a water bath. After cooling to RT, the reaction mixture was quenched with aqueous NaHCO
3 (1 M, 1 mL) and extracted with dichloromethane (3 × 1.5 mL). The combined extracts were washed with aqueous NaHCO
3 (1 M, 1.5 mL) and water (1.5 mL), dried over Na
2SO
4, and concentrated under reduced pressure. The crude product (29 mg) was purified using PTLC (silica, cyclohexane/AcOEt 5:3), yielding 16 mg (39%) of
H9, a light orange solid.
Compound H9: IR (ATR, νmax, cm−1): 3276, 3075, 3001, 2917, 2848, 1750, 1637, 1595, 1540, 1480, 1239, 1239, 1139, 1025, 910, 873, 809; 1H-NMR (CDCl3) δ 7.62 (s, br 1H, NH), 7.18 (s, 1H, H-2′), 7.10 (d ≈ 8 Hz, 1H, H-4), 7.07 (s, 2H, H-6, H-6′), 6.89 (d, J = 8 Hz, 1H, H-3), 6.06 (ddt, J = 16.9, 10.3, 6.6 Hz, 1H, H-2‴), 5.97 (ddt, J ≈ 17, 9.9, 6.9 Hz, 1H, H-2″), 5.03–5.16 (m, 4H, H-3″, H-3‴), 3.77 (s, 3H, OCH3), 3.50 (d, J = 5.9 Hz, 2H, H-1‴), 3.35 (d, J = 6.6 Hz, 2H, H-1″), 2.22 (s, 3H, CO-CH3); 13C-NMR (CDCl3) δ 170.5 (CO), 154.7 (C-2), 146.0 (C-4′), 137.7 (C-2″), 136.8 (C-2‴), 132.3 (C-5), 130.9 (C-6), 130.4 (C-5′), 130.2 (C-3′), 129.6 (C-1), 128.8 (C-2′), 128.2 (C-4), 125.2 (C-1′), 121.4 (C-6′), 115.7 (C-3‴), 115.6 (C-3″), 111.4 (C-3), 55.8 (OCH3), 39.4 (C-1″), 35.9 (C-1‴), 23.7 (CO–CH3); ESI+ calcd for C21H23NO3: [M]+ 337.168; found ESI-MS m/z (rel. int.): 338.08 [M + H]+ (100).
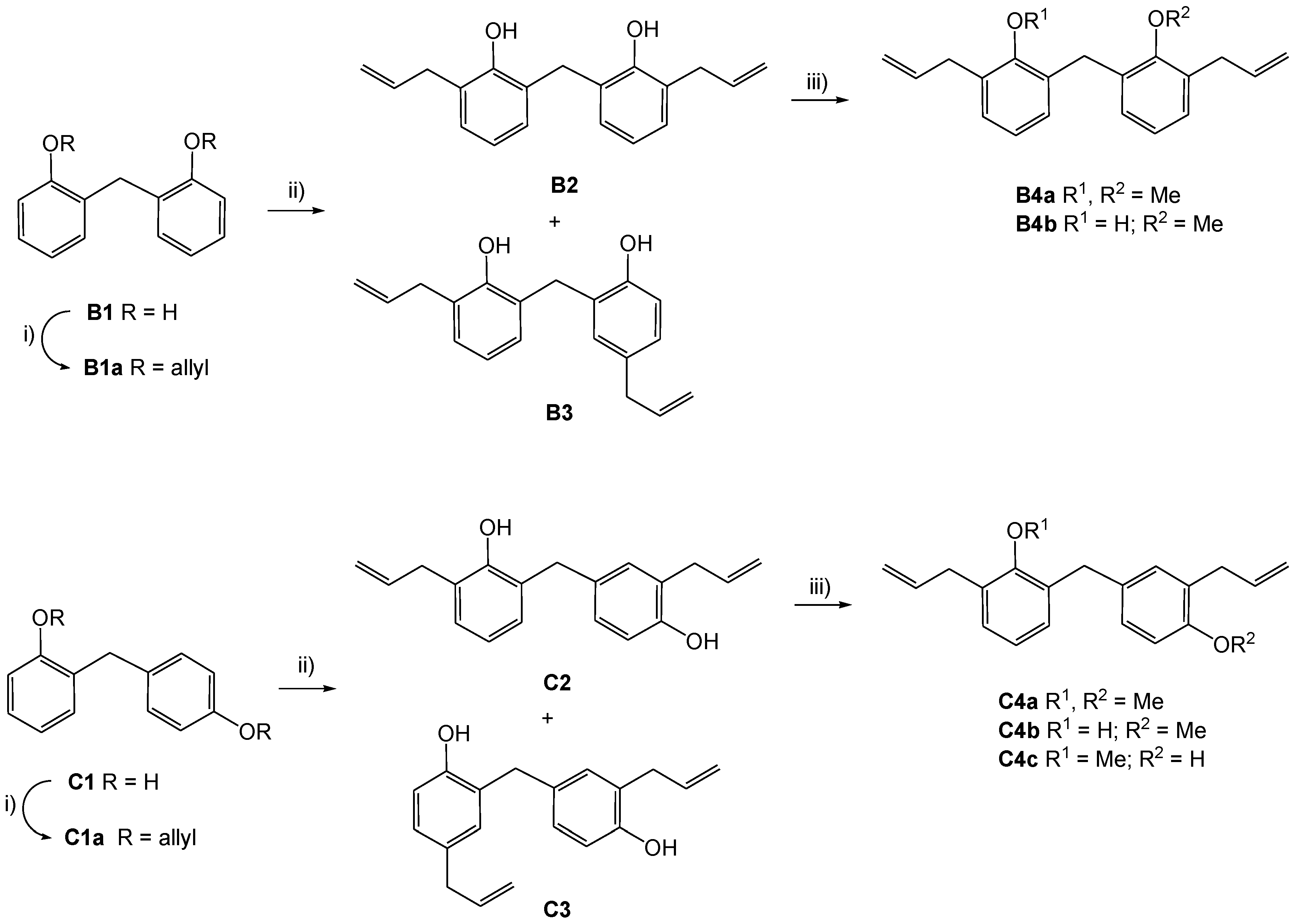
Bis(2-Allyloxyphenyl)-methane (B1a)
B1 (400 mg, 2.0 mmol) was O-allylated with allylbromide (968 mg, 0.70 mL, 8.0 mmol) and K2CO3 (2.21 g, 16 mmol) in acetone (10 mL). The reaction mixture was stirred for 5 h with reflux, and a further 12 h at RT and filtered. The residue was extracted with acetone (50 mL) and the combined organic solutions were evaporated under reduced pressure yielding B1a (559 mg, 73%).
Compound B1a: IR (ATR, νmax, cm−1): 3025, 2920, 2905, 1595, 1584, 1491, 1450, 1421, 1248, 1116, 1103, 1022, 924, 751 cm−1; 1H-NMR (CDCl3) δ 7.19 (td, J = 7.6, 1.5 Hz, 2H, H-4), 7.14 (dd, J = 7.5, 1.2 Hz, 2H, H-6), 6.90 (t, J ≈ 8 Hz, 2H, H-5), 6.89 (d, 2H, J = 7.5 Hz, H-3), 6.02–6.13 (m, 2H, CH2–CH=CH2), 5.42 (dquint, J = 17.2, 4.3 Hz, 2H, CH2–CH=CH2), 5.27 (dquint, J = 10.5, 1.4 Hz, 2H, CH2–CH=CH2), 4.58 (dq, J = 5.0, 1.5 Hz, 2H, CH2–CH=CH2), 4.10 (s, 2H, Ar–CH2–Ar); 13C-NMR (CDCl3) δ 156.5 (C-2), 133.6 (CH=CH2), 130.5 (C-6), 129.6 (C-1), 126.9 (C-4), 120.5 (C-5), 116.8 (CH=CH2), 111.6 (C-3), 30.8 (O–CH2), 29.8 (Ar–CH2–Ar); EI+ calcd for C19H20O2: [M]+ 280.1463; found EI-MS m/z (rel. int.): 280.1465 [M]+ (100). Compound B1a has been mentioned in several patents; however, chemical data were not given therein.
2-Allyl-6-(3-allyl-2-hydroxybenzyl)-phenol (B2) and 4-Allyl-2-(3-allyl-2-hydroxybenzyl)-phenol (B3)
Claisen rearrangement of
B1a (561 mg, 2 mmol) in
N,
N-diethylaniline (4 mL) following the procedure of Chattopadhyay et al. [
21] resulted in
B2 (410 mg; 73%) and
B3 (89 mg; 15%) as white wax like solids.
Compound B2: IR (KBr, νmax, cm−1): 3407 (br, OH), 3073, 3035, 2975, 2937, 1639, 1590, 1461, 1448, 1372, 1294, 1234, 1210, 1192, 1086, 999, 922, 912, 754 cm−1; 1H-NMR (CDCl3) δ 7.20 (dd, J = 7.7, 1.5 Hz, 2H, H-6), 7.02 (d, J = 7.7 Hz,2H, H-4), 6.88 (t, J = 7.7 Hz, 2H, H-5), 6.43 (s, br, 2H, OH), 6.06 (ddt, J = 16.8, 10.3, 6.2 Hz, 2H, CH2–CH=CH2), 5.18–5.24 (m, 4H, CH2–CH=CH2), 3.97 (s, 2H, Ar–CH2–Ar), 3.45 (d, J = 6.6 Hz, 4H, CH2–CH=CH2); 13C-NMR (CDCl3) δ 151.4 (C-2), 136.6 (CH2–CH=CH2), 129.1 (C-6), 128.7 (C-4), 127.1 (C-1), 125.8 (C-3), 121.1 (C-5), 116.6 (CH2–CH=CH2), 35.4 (CH2–CH=CH2), 30.8 (Ar–CH2–Ar); EI+ calcd for C19H20O2: [M]+ 280.1463; found EI-MS m/z (rel. int.): 280.1469 [M]+ (58). Compound B2 has been mentioned in several patents; however, chemical data were not given therein.
Compound B3: IR (KBr, νmax, cm−1): 3387 (OH), 3077, 2853, 1638, 1611, 1592, 1506, 1465, 1434, 1256, 1232, 1215, 1107, 996, 914, 759 cm−1. 1H-NMR (CDCl3) δ 7.17 (dd, J = 7.8, 1.5 Hz, 1H, H-6), 7.06 (d, J = 2.2 Hz,1H, H-6′), 6.98 (dd, J = 7.3, 1.5 Hz, 1H, H-4), 6.91 (dd, J = 8.4, 2.2 Hz, 1H, H-4′), 6.84 (t, J = 7.5 Hz, 1H, H-5), 6.74 (d, J = 8.0 Hz, 1H, H-3′), 6.31 (s, vb, 2H, OH), 6.02 (ddt, J = 16.8, 10.3, 6.2 Hz, 1H, 2″-H), 5.92 (ddt, J = 16.9, 10.3, 6.6 Hz, 1H, H-2″), 5.19 (dq, J ≈ 16, 1.7 Hz, 1H, H-3″), 5.18 (dq, J ≈ 9, 1.5 Hz, 1H, H-3″), 5.03 (dq, J ≈ 17, 1.8 Hz, 1H, H-3‴), 5.04 (dq, J ≈ 9, 1.3 Hz, 1H, H-3‴), 3.89 (s, 2H, Ar–CH2–Ar), 3.41 (d, J = 6.2 Hz, 2H, H-1″), 3.28 (d, J = 7.0 Hz, 2H, H-1‴); 13C-NMR (CDCl3) δ 151.3 (C-2), 151.2 (C-2′), 137.8 (C-2‴), 136.5 (C-2″), 132.8 (C-5′), 130.8 (C-6′), 129.1 (C-6), 128.8 (C-4), 128.0 (C-4′), 127.1 (C-1), 126.6 (C-1′), 125.6 (C-3), 121.2 (C-5), 116.9 (C-3″), 116.0 (C-3′), 115.4 (C-3‴), 39.4 (C-1‴), 35.6 (C-1″), 30.8 (Ar–CH2–Ar); EI+ calcd for C19H20O2: [M]+ 280.1463; found EI-MS m/z (rel. int.): 280.1464 [M]+ (100).
2,4′-Diallyloxy-diphenylmethane (C1a)
C1 (3.77 g, 18.8 mmol) was allylated with allylbromide (8.97 g, 6.5 mL, 74.1 mmol) and K2CO3 (20.8 g, 0.15 mol) in acetone (100 mL) as described above yielding the diallyloxy-diphenylmethane (4.12 g, 78%).
Compound C1a: IR (ATR, νmax, cm−1): 3077, 3022, 2915, 2860, 1610, 1599, 1584, 1508, 1490, 1451, 1237, 1221, 1020, 996, 921, 749 cm−1; 1H-NMR (CDCl3) δ 7.22 (td, J ≈ 8, 1.3 Hz, 1H, H-4), 7.21 (d, J ≈ 8 Hz, 2H, H-2′ and H-6′), 7.14 (d, J = 7.7 Hz, 1H, H-6), 6.94 (t, J = 7.3 Hz, 1H, H-5), 6.90 (d, 1H, J ≈ 8 Hz, H-3), 6.89 (d, 2H, J = 8.4 Hz, H-3′ and H-5′), 6.03–6.17 (m, 2H, H-2″ and H-2‴), 5.45 (dq, J = 17.2, 1.5 Hz, 1H, H-3‴), 5.44 (dq, J = 17.3, 1.5 Hz, 1H, H-3″), 5.32 (dq, J = 10.5, 1.5 Hz, 1H, H-3‴), 5.31 (dq, J = 10.5, 1.5 Hz, 1H, H-3″), 4.58 (dt, J = 5.1, 1.5, 2H, H-1″), 4.55 (dt, J = 5.1, 1.5, 2H, H-1‴), 4.02 (s, 2H, Ar–CH2–Ar); 13C-NMR (CDCl3) δ 156.7(C-4′), 156.2 (C-2), 2 × 133.5 (2 × CH=CH2), 133.3 (C-1′*), 130.4 (C-1*), 130.2 (C-6), 129.8 (C-2′ and C-6′), 127.2 (C-C-4), 120.6 (C-5), 117.4 (C-3‴), 116.9 (C-3″), 114.5 (C-3′ and C-5′), 111.7 (C-3), 68.8 and 68.7 (2 × O–CH2), 35.1 (Ar–CH2–Ar); EI+ calcd for C19H20O2: [M]+ 280.1463; found EI-MS m/z (rel. int.): 280.1 [M]+ (45), 133.1 [C6H4–O–allyl]+ (100).
2-Allyl-4-(3-allyl-2-hydroxybenzyl)-phenol (C2) and 2-Allyl-4-(5-allyl-2-hydroxybenzyl)-phenol (C3)
Claisen rearrangement of
C1a (14.5 g, 51.7 mmol) in
N,
N-diethylaniline (115 mL) following the procedure of Chattopadhyay et al. [
21] resulted in
C2 (10.6 g; 73%) and
C3 (1.2 g; 8.3%)
Compound C2: IR (ATR, νmax, cm−1): 3507 (br, OH), 3076, 3009, 2977, 2906, 1637, 1609, 1591, 1501, 1461, 1432, 1328, 1255, 1182, 996, 911, 751 cm−1; 1H-NMR (CDCl3) δ 6.95–7.06 (m, 4H, H-4,H-6, H-2′, H-6′), 6.86 (t, J = 7.5 Hz, 1H, H-5), 6.74 (d, J = 7.7 Hz, 1H, H-5′), 5.94–6.09 (m, 2H, H-2″, H-2‴), 5.12–5.20 (m, 4H, H-3″, H-3‴), 4.99 (s, b, 2H, OH), 3.92 (s, 2H, Ar–CH2–Ar), 3.41 (d, J = 6.2 Hz, 2H, H-1″), 3.38 (d, J = 6.2 Hz, 2H, H-1‴); 13C-NMR (CDCl3) δ 152.6 (C-4′), 152.4 (C-2), 136.5 (C-2″), 136.4 (C-2‴), 132.1 (C-1′), 130.7 (C-2′), 129.1 (C-6), 128.6 (C-4), 128.0 (C-6′), 127.6 (C-1), 125.6 (C-3, C-3′), 120.6 (C-5), 116.4 (C-3″, C-3‴), 116.0 (C-5′), 35.7 (Ar–CH2–Ar), 35.3 (C-1″), 35.1′ (C-1‴); EI+ calcd for C19H20O2: [M]+ 280.1463; found EI-MS m/z (rel. int.): 280.1465 [M]+ (100).
Compound C3: IR (ATR, νmax, cm−1): 3277 (br, OH), 3078, 3013, 2977, 2902, 1638, 1608, 1502, 1434, 1341, 1247, 1207, 1094, 992, 793, 634, 617 cm−1; 1H-NMR (CDCl3) δ 6.90–7.04 (m, 4H, H-4, H-6, H-2′, H-6′), 6.68–6.78 (m 2H, H-3, H-5′), 5.88–6.07 (m, 2H, H-2″, H-2‴), 5.10–5.20 (m, 2H, H-3‴), 4.99–5.10 (m, 2H, H-3″), 4.87 (s, 1H, OH), 4.57 (s, 1H, OH), 3.89 (s, 2H, Ar–CH2–Ar), 3.38 (d, J = 6.2 Hz, 2H, 1‴-H), 3.30 (d, J = 6.6 Hz, 2H, H-1′); 13C-NMR (CDCl3) δ 152.6 (C-4′), 152.1 (C-2), 137.9 (C-2″), 136.4 (C-2‴), 132.2 (C-5), 132.0 (C-1′), 131.0 (C-6), 130.2 (C-2′), 127.9 (C-6′), 127.7 (C-4), 127.1 (C-1), 125.6 (C-3′), 116.3 (C-3‴), 116.0, 115.8 (C-3, C-5′), 115.3 (C-3″), 39.3 (C-1″), 35.7 (Ar–CH2–Ar), 35.1 (C-1‴ EI+ calcd for C19H20O2: [M]+ 280.1463; found EI-MS m/z (rel. int.): 280.1 [M]+ (100), 147.1 [CH2–C6H4–O–allyl]+ (100).
General Procedure for the Methylation of Dihydroxy-Diphenylmethanes
The corresponding diol (1 eq) was dissolved in aqueous KOH (10%, 2.5 eq) and stirred for 10 min. Me2SO4 (1.2 eq) was added and the resulting mixture was stirred for 20 min at RT and for 1 h at 95 °C. After cooling to RT, the mixture was acidified with aqueous HCl (2M) and extracted with chloroform (3 ×). The organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was chromatographed over silica with cyclohexane/AcOEt = 5:1.
Bis(3-allyl-2-methoxyphenyl)methane (B4a) and 6-allyl-2-(3-allyl-2-methoxybenzyl)-phenol (B4b)
Methylation of 420 mg (1.5 mmol) B2 resulted in 39 mg of starting material, 40 mg B4a (8.6%) and 249 mg B4b (57%).
Compound B4a: IR (ATR, νmax, cm−1): 3001, 2978, 2941, 2827, 1463, 1427, 1291, 1164, 1080, 1009, 911, 766 cm−1; 1H-NMR (CDCl3) δ 7.09 (dd, 2H, J = 7.6 and 1.3 Hz, H-4), 7.00 (t, 2H, J = 7.5 Hz, H-5), 6.93 (dd, 2H, J = 7.4 and 1.3 Hz, H-6), 5.97–6.09 (m, 2H, -CH=CH2), 5.06–5.21 (m, 4H, -CH=CH2), 4.10 (s, 2H, Ar–CH2–Ar), 3.72 (s, 6H, OCH3), 3.46 (d, 4H, J 6.6 Hz, CH2-CH=CH2); 13C-NMR (CDCl3) δ 154.4 (C-2), 137.3 (-CH=CH2), 134.0 (C-1), 132.9 (C-3), 128.9 (C-6), 128.5 (C-4), 124.1 (C-5), 115.8 (-CH=CH2), 34.0 (-CH2-CH=CH2), 29.6 (Ar–CH2–Ar). EI+ calcd for C21H24O2: [M]+ 308.1776; found EI-MS m/z (rel. int.): 308.1776 [M]+ (100).
Compound B4b: IR (ATR, νmax, cm−1): 3320 (br, OH), 3001, 2978, 2975, 2944, 2908, 1641, 1632, 1591, 1462, 1446, 1430, 1220, 1086, 998, 910, 780, 604 cm−1; 1H-NMR (CDCl3) δ 7.35 (s, 1H, OH), 7.18 (dd, J = 7.1, 1.8 Hz, 1H, H-6′), 7.13 (d, J = 7.1 Hz, 1H, H-6), 7.08 (dd, J ≈ 7, ≈ 2 Hz, 1H, H-4′), 7.06 (t, J = 7.5 Hz, 1H, H-5′), 7.00 (d, J = 7.7 Hz, 1H, H-4), 6.81 (t, J = 7.5 Hz, 1H, H-5), 5.92–6.08 (m, 2H, H-2′ and H-2‴), 5.03–5.15 (m, 4H, H-3″ and H-3‴), 3.92 (2 × s, 5H, Ar–CH2–Ar and OCH3), 3.47 (d, J = 6.3 Hz, 2H, H-1‴), 3.40 (d, J = 6.6 Hz, 2H, H-1″); 13C-NMR (CDCl3) δ 154.2 (C-2′), 151.2 (C-2), 137.0 (C-2″), 136.8 (C-2‴), 2 × 133.0, (C-1′ and C-3′), 129.1 (C-4′), 128.9 (C-6′), 128.6 (C-4), 128.4 (C-6), 127.2 (C-3), 126.6 (C-1), 125.3 (C-5′), 120.1 (C-5), 116.2 (C-3‴), 115.4 (C-3″), 62.3 (OCH3), 34.7 (C-1″), 33.8 (C-1‴), 31.4 (Ar–CH2–Ar); EI+ calcd for C21H24O2: [M]+ 294.1620; found EI-MS m/z (rel. int.): 294.1619 [M]+ (100).
3,3′-Diallyl-2,4′-dimethoxy-diphenyl-methan (C4a), 6-allyl-2-(3-allyl-4-methoxybenzyl)-phenol (C4b) and 2-allyl-4-(3-allyl-2-methoxybenzyl)-phenol (C4c)
Methylation of 2.10 g (7.5 mmol) C2 resulted in 143 mg of starting material, 318 mg C4a (14%), 665 mg C4b (30%) and 442 mg C4c (20%).
Compound C4a: IR (ATR, νmax, cm−1): 3001, 2977, 2939, 2833, 1638, 1500, 1463, 1429, 1248, 1125, 1080, 1033, 1010, 910, 768 cm−1; 1H-NMR (CDCl3) δ 7.07 (dd, J = 7.3, 1.5 Hz, 1H, H-4), 6.98–7.02 (m, 3H, H-2′, H-5 and H-6′), 6.96 (dd, J = 7.6, 1.3 Hz, 1H, H-6), 6.78 (d, J = 8.4 Hz, 1H, H-5′), 5.92–6.07 (m, 2H, H-2″ and H-2‴), 5.06–5.13 (m, 2H, H-3″), 4.99–5.06 (m, 2H, H-3‴), 3.96 (s, 2H, Ar–CH2–Ar), 3.80 (s, 3H, C4′-OCH3), 3.68 (s, 3H, C2-OCH3), 3.46 (d, J = 6.3 Hz, 2H, H-1″), 3.36 (d, J = 6.6 Hz, 2H, H-1‴); 13C-NMR (CDCl3) δ 156.3 (C-2), 155.6 (C-2′), 137.3 (C-2″), 137.1 (C-2‴), 134.7 (C-1), 133.0 (C-3), 132.8 (C-1′), 130.2 (C-2′), 129.0 (C-6), 128.5 (C-4), 128.4 (C-3′), 127.5 (C-6′), 120.5 (C-5), 115.8 (C-3″), 115.3 (C-3‴), 110.3 (C-5′), 61.2 (C2-OCH3), 55.4 (C4′-OCH3), 34.9 (Ar–CH2–Ar), 34.3 (C-1‴), 34.0 (C-1″); MS (ESI+) m/z (%): calculated for C21H24O2: [M]+ 308.178; found: EI-MS m/z (rel. int.): 308.4 [M]+ (100).
D4b: IR (ATR, νmax, cm−1): 3526 (br, OH), 3003, 2976, 2905, 2834, 1637, 1608, 1592, 1499, 1461, 1247, 1182, 1032, 996, 910, 752 cm−1; 1H-NMR (CDCl3) δ 6.97–7.02 (m, 4H, H-4, H-6, H-2′ and H-6′), 6.82 (t, J = 7.5 Hz, 1H, H-5), 6.76 (d, J = 8.8 Hz, 1H, H-5′), 5.90–6.05 (m, 2H, H-2″and H-2‴), 5.11–5.17 (m, 2H, H-3″), 4.98–5.05 (m, 2H, H-3‴), 4.94 (s, 1H, OH), 3.91 (s, 2H, Ar–CH2–Ar), 3.78 (s, 3H, OCH3), 3.38 (d, J = 6.2 Hz, 2H, H-1″), 3.34 (d, J = 6.5 Hz, 2H, H-1‴); 13C-NMR (CDCl3) δ 152.9 (C-4′), 152.4 (C-2), 136.9 (C-2‴), 136.5 (C-2″), 131.4 (C-1′), 130.2 (C-2′), 129.1 (C-6′), 128.8 (C-3′), 128. 6 (C-4), 127.6 (C-1), 127.2 (C-6), 125.5 (C-3), 120.5 (C-5), 116.4 (C-3″), 115.3 (C-3‴), 55.4 (OCH3), 35.8 (Ar–CH2–Ar), 35.3 (C-1″), 34.3 (C-1‴); MS (EI) m/z (%): calculated for C20H22O2: [M]+ 294.162; found: EI-MS m/z (rel. int.): 294.4 [M]+ (80), 253.2 (65).
D4c: IR (ATR, νmax, cm−1): 3393 (br, OH), 3005, 2977, 2940, 2911, 2830, 1638, 1610, 1591, 1505, 1464, 1428, 1251, 1198, 1080, 996, 910, 768 cm−1; 1H-NMR (CDCl3) δ 7.09 (dd, J = 7.3, 1.7 Hz, 1H, H-4), 7.01 (t, J = 7.3 Hz, 1H, H-5), 6.93–6.99 (m, 3H, H-2′, H-6 and H-6′), 6.73 (d, J = 7.7 Hz, 1H, H-5′), 5.94–6.08 (m, 2H, H-2″ and H-2‴), 5.06–5.20 (m, 4H, H-3″ and H-3‴), 4.95 (s, 1H, OH), 3.96 (s, 2H, Ar–CH2–Ar), 3.68 (s, 3H, OCH3), 3.46 (d, J = 6.6 Hz, 2H, H-1″), 3.38 (d, J = 6.5 Hz, 2H, H-1‴), 13C-NMR (CDCl3) δ 156.3 (C-2), 152.3 (C-4′), 137.3 (C-2″), 136.5 (C-2‴), 134.6 (C-1), 133.3 (C-1′), 133.0 (C-3), 130.1 (C-2′), 129.0 (C-6), 128.6 (C-4), 128.2 (C-6′), 125.1 (C-3′), 124.1 (C-5), 116.3 (C-3″), 115.8 (C-3‴), 115.7 (C-5′), 61.2 (OCH3), 35.2 (C-1‴), 34.9 (Ar–CH2–Ar), 33.9 (C-1″); MS (EI) m/z (%): calculated for C20H22O2: [M]+ 294.162; found: EI-MS m/z (rel. int.): 294.4 [M]+ (75), 253.2 (40).

 ,
, 




{kind=link}
{kind=link}
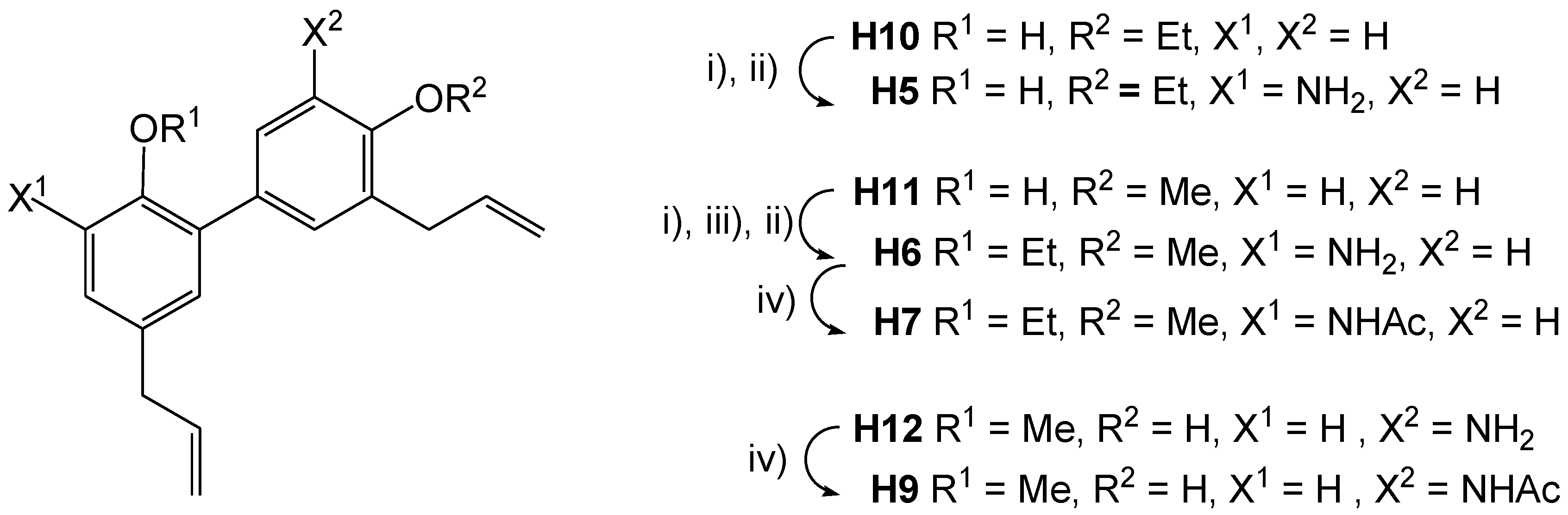{kind=link}
{kind=link}




