
Poly-Gamma-Glutamic Acid (γ-PGA)-Based Encapsulation of Adenovirus to Evade Neutralizing Antibodies


 , ,
, ,  and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
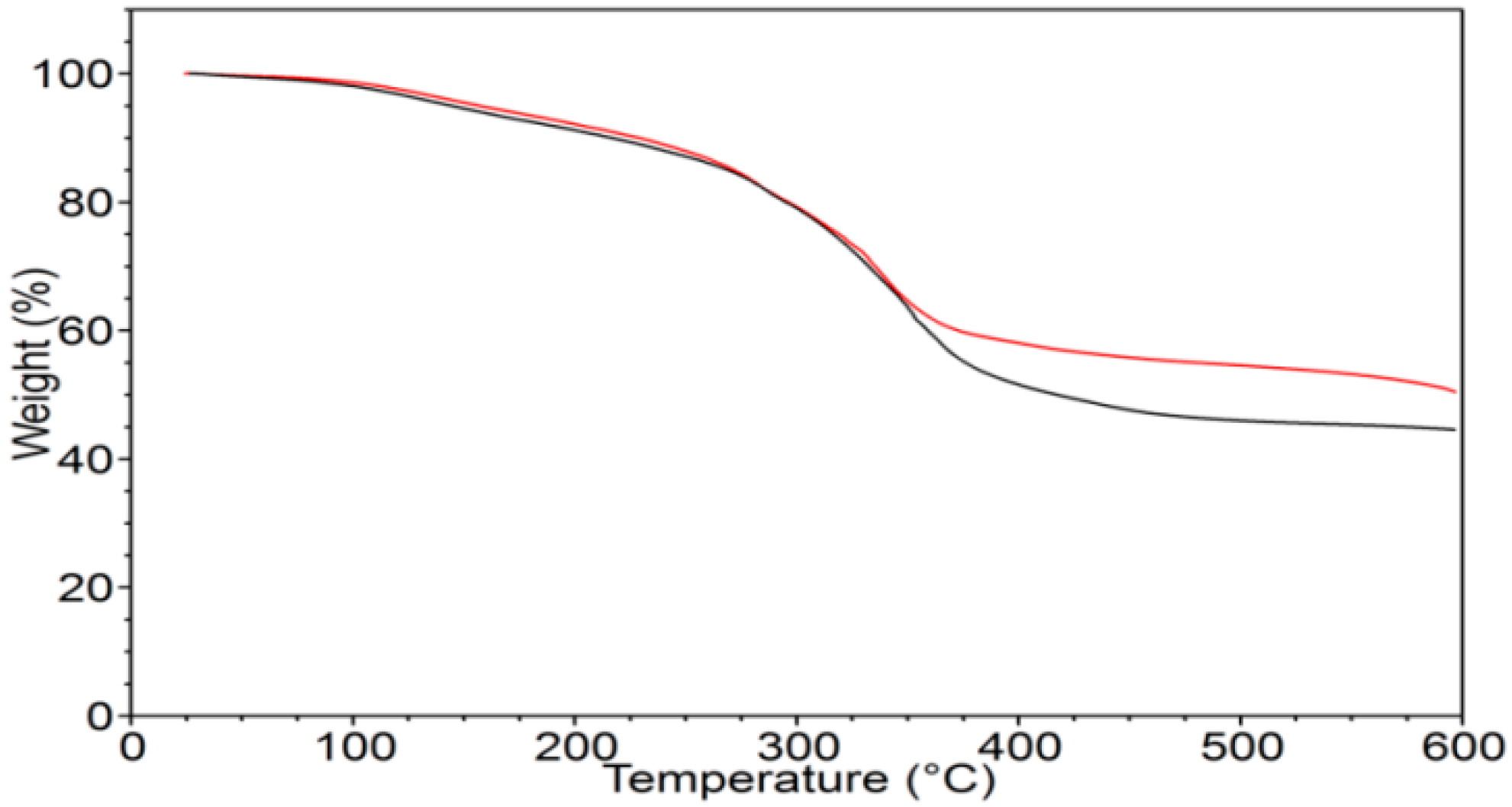
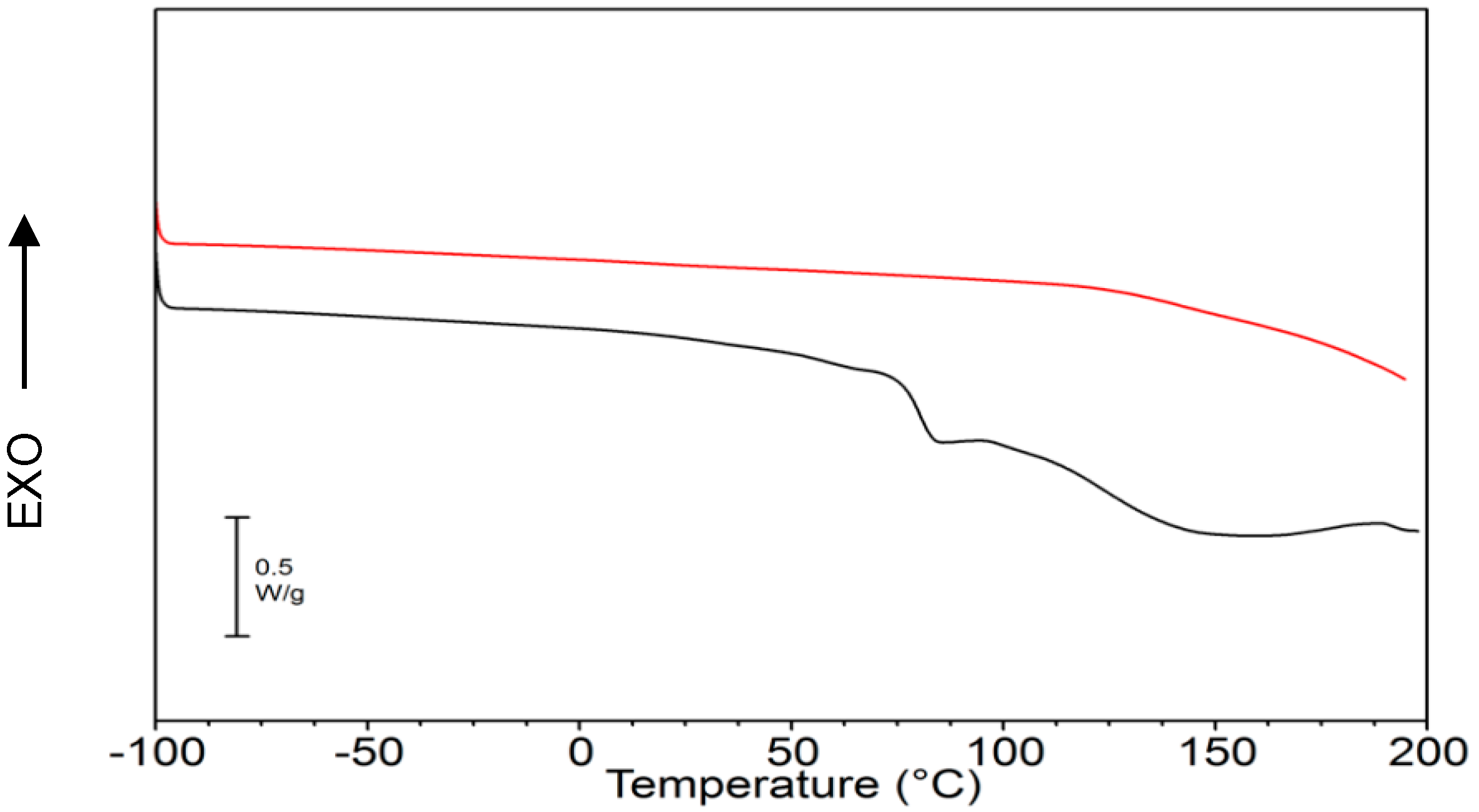
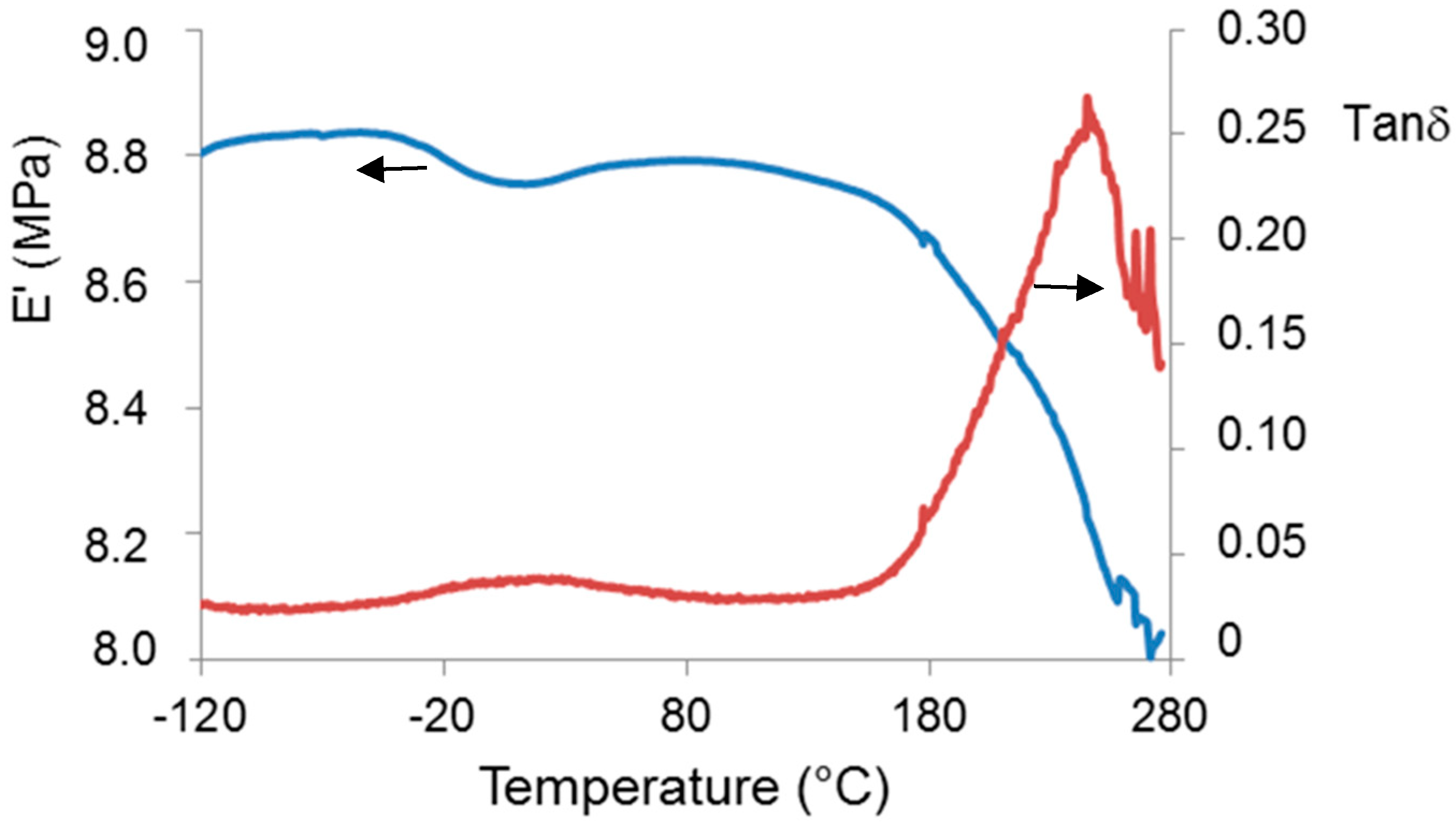
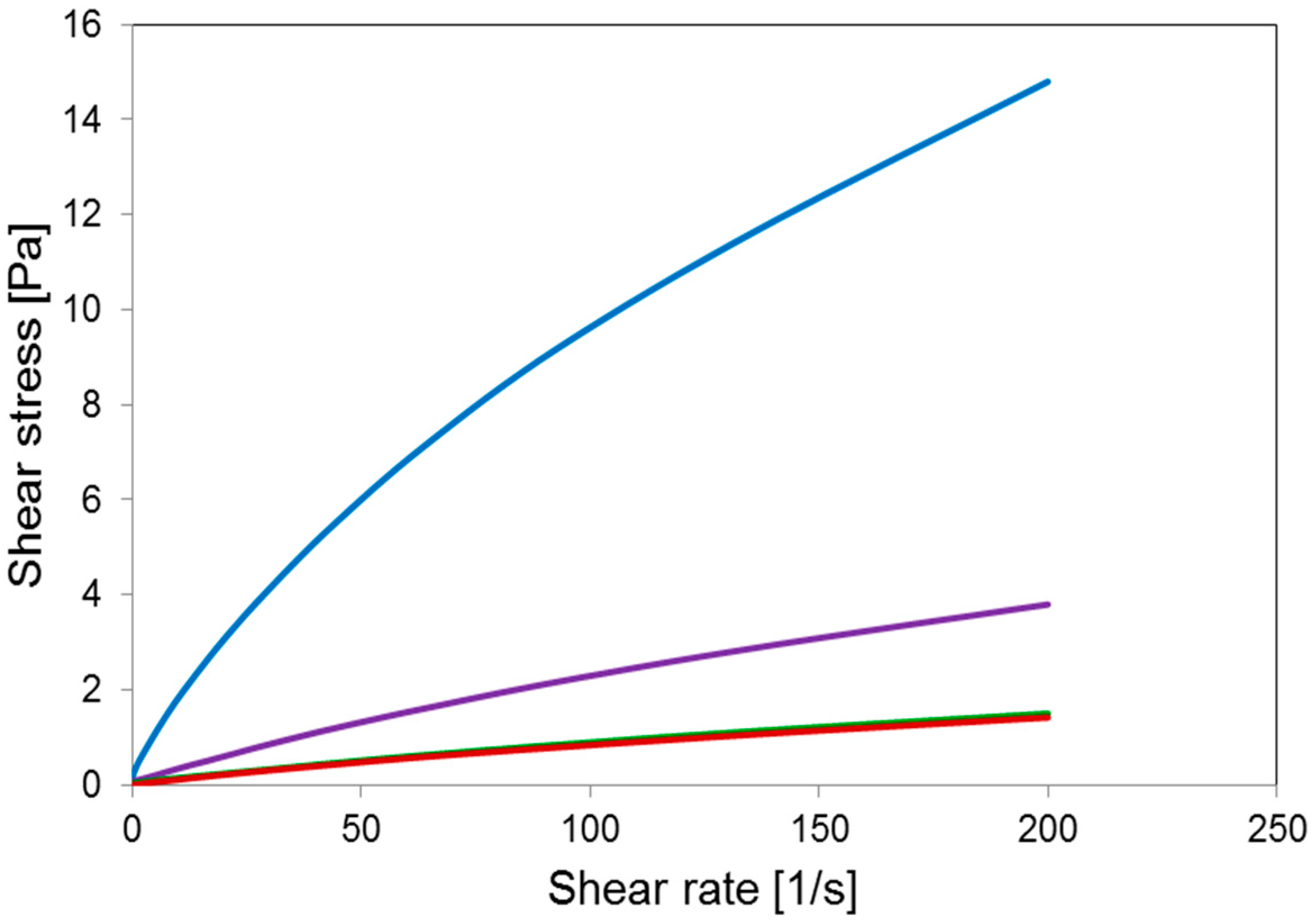
2.1. γ-PGA Identification and Characterization
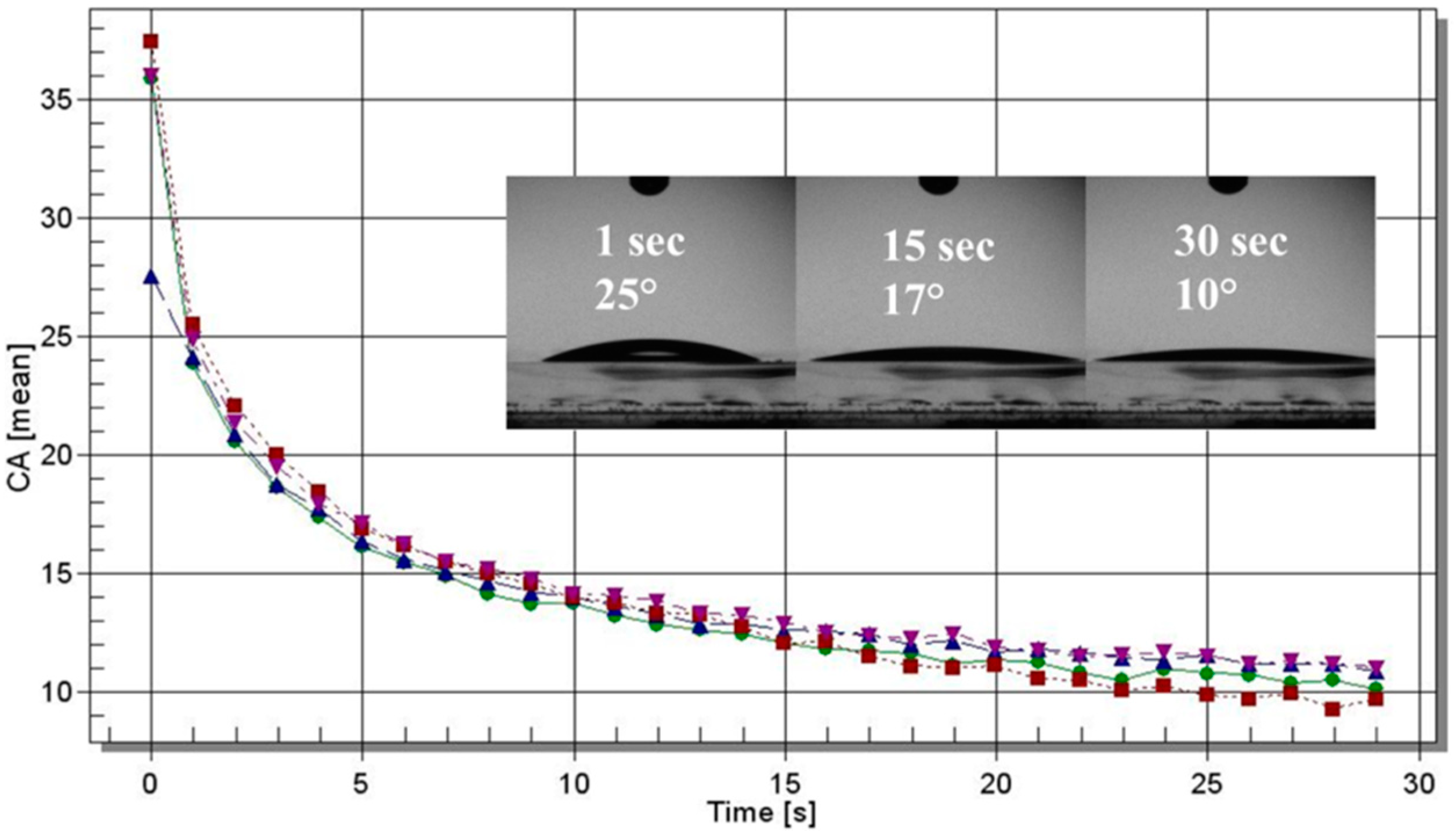
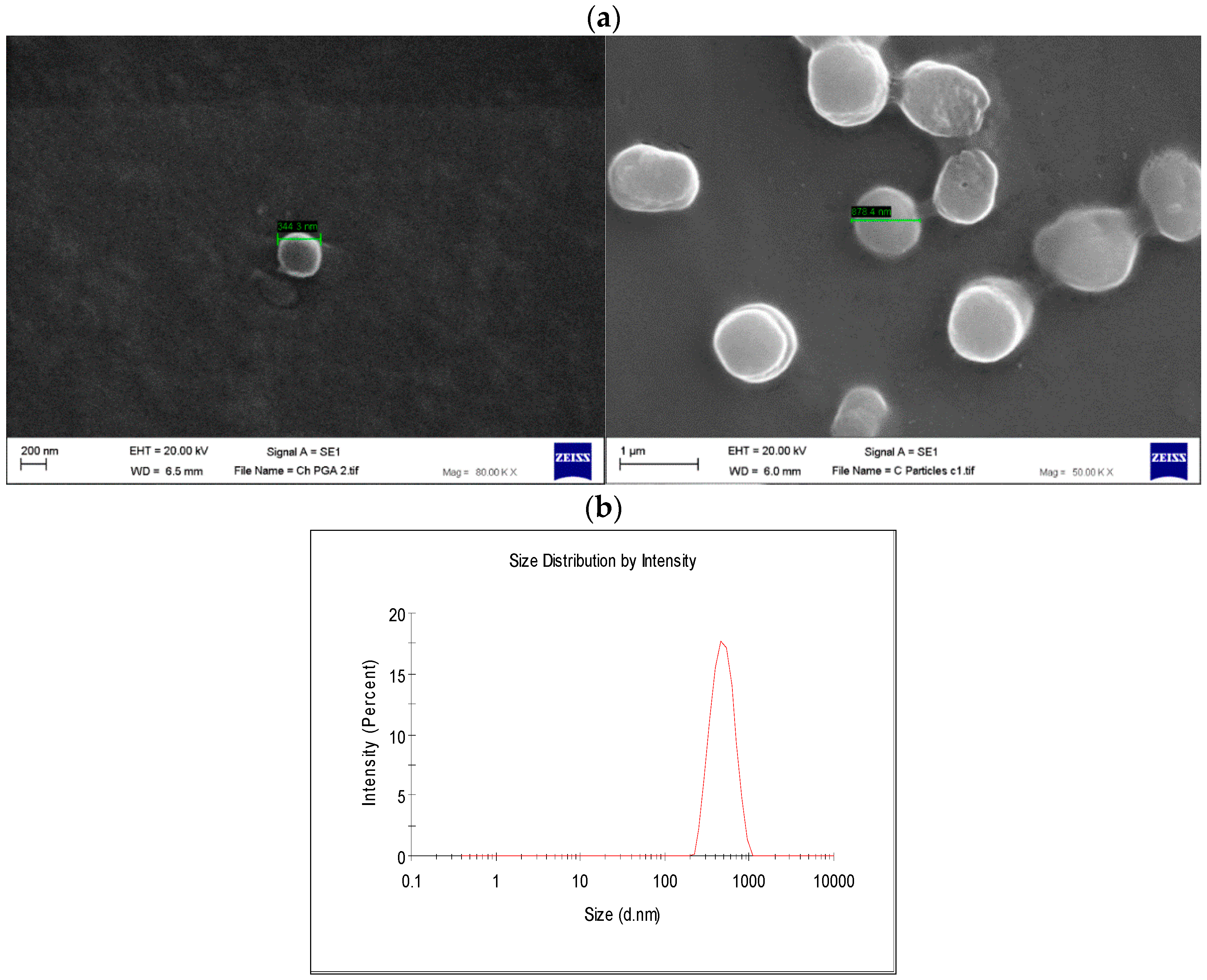
2.2. Nanoparticle Characterization
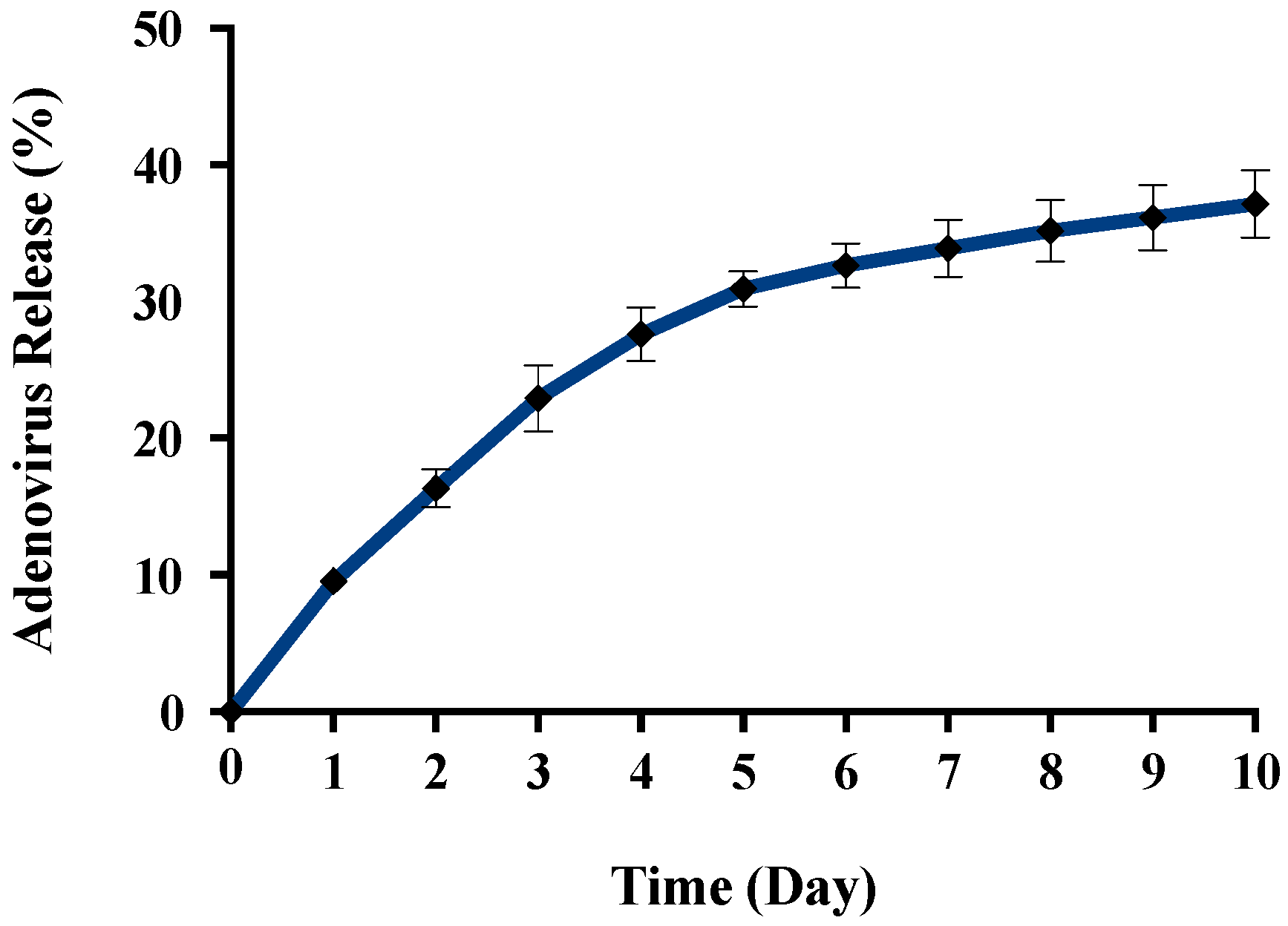
2.3. Adenovirus Encapsulation and Release Profile
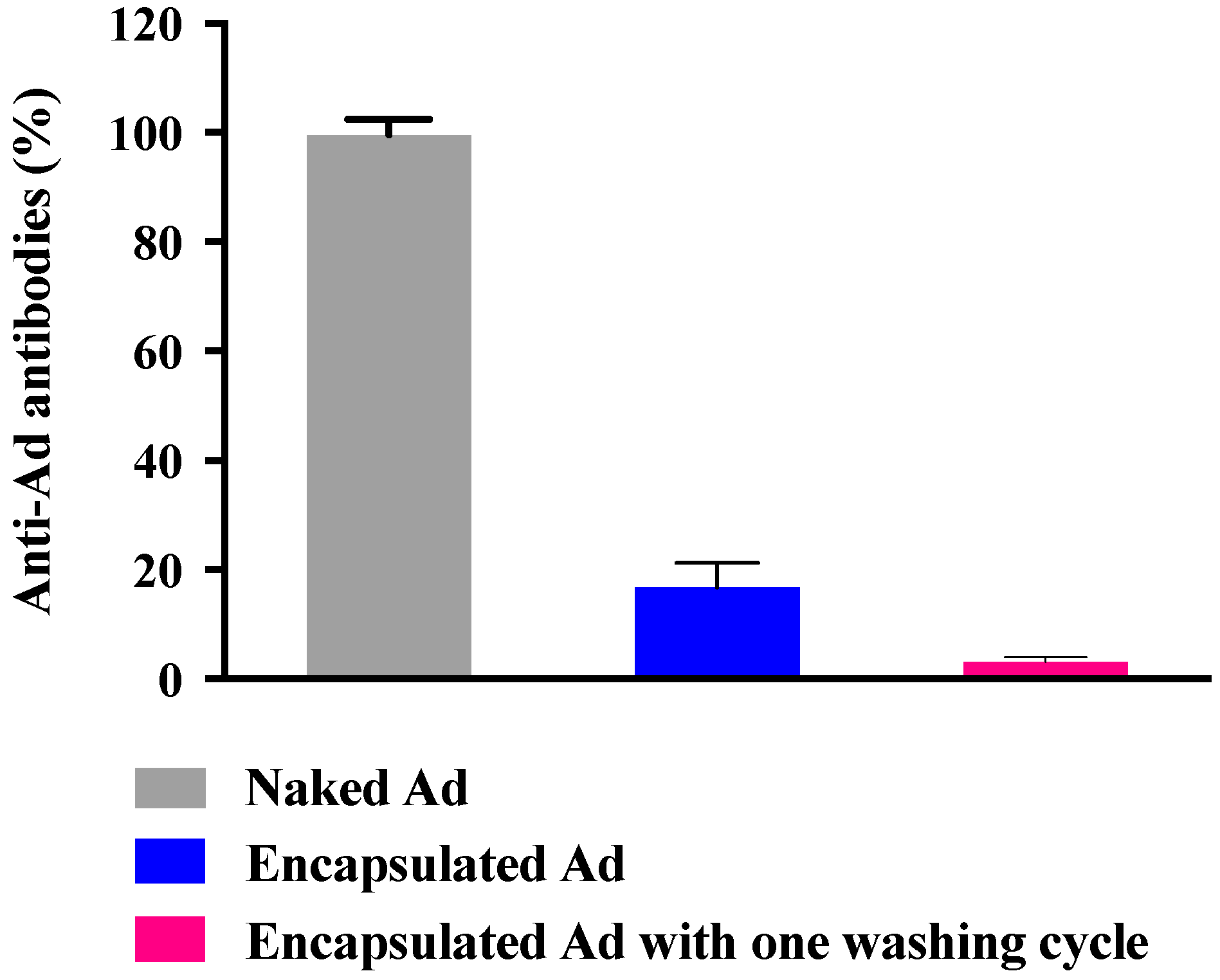
2.4. Adenovirus Immunogenicity
2.5. Fluorescent Labeled NPs
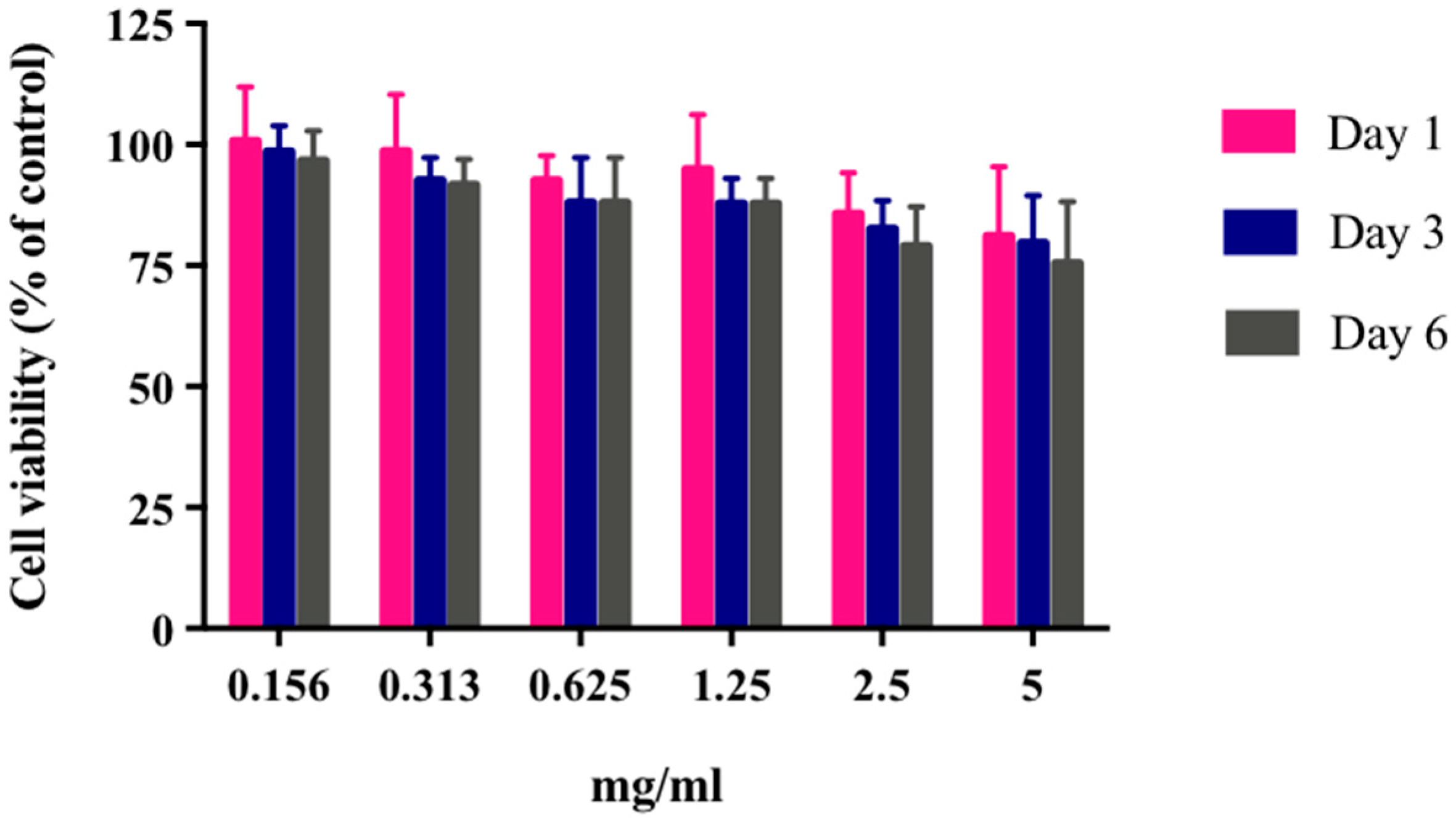
2.6. Cytotoxicity Assay
3. Discussion
4. Materials and Methods
4.1. γ-PGA Production, Purification, and Characterization
4.2. Adenovirus Preparation
4.3. Adenovirus Encapsulation
4.4. In Vitro Virus Release from Chitosan-γ-PGA NPs
4.5. Fluorescent Labeled NPs
4.6. Adenovirus Immunogenicity
4.7. Cytotoxicity Assay
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global Cancer Statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and Adolescent Cancer Statistics, 2014. CA Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.; Burmester, J.K. Gene Therapy for Cancer Treatment: Past, Present and Future. Clin. Med. Res. 2006, 4, 218–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dorp, W.; van Beek, R.D.; Laven, J.S.; Pieters, R.; De Muinck Keizer-Schrama, S.M.; van den Heuvel-Eibrink, M.M. Long-term Endocrine Side Effects of Childhood Hodgkin′s Lymphoma Treatment: A Review. Hum. Reprod. Update 2012, 18, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.; Carle, G.; Faneca, H.; de Lima, M.C.; Pierrefite-Carle, V. Suicide Gene Therapy in Cancer: Where Do We Stand Now? Cancer Lett. 2012, 324, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Maya, S.; Sarmento, B.; Lakshmanan, V.K.; Menon, D.; Jayakumar, R. Actively Targeted Cetuximab Conjugated Gamma-Poly (Glutamic Acid)-Docetaxel Nanomedicines for Epidermal Growth Factor Receptor Over Expressing Colon Cancer Cells. J. Biomed. Nanotechnol. 2014, 10, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Hirst, D.G.; O’Sullivan, J.M. Gold Nanoparticles as Novel Agents for Cancer Therapy. Br. J. Radiol. 2012, 85, 101–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannon-Peppas, L.; Blanchette, J.O. Nanoparticle and Targeted Systems for Cancer Therapy. Adv. Drug Deliv. Rev. 2004, 56, 1649–1659. [Google Scholar] [CrossRef] [PubMed]
- Felgner, S.; Kocijancic, D.; Frahm, M.; Weiss, S. Bacteria in Cancer Therapy: Renaissance of an Old Concept. Int. J. Microbiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Patyar, S.; Joshi, R.; Byrav, D.S.; Prakash, A.; Medhi, B.; Das, B.K. Bacteria in Cancer Therapy: A Novel Experimental Strategy. J. Biomed. Sci. 2010, 17, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanneman, M.; Dranoff, G. Combining Immunotherapy and Targeted Therapies in Cancer Treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Fan, X.; Wang, X.; Ye, E.; Loh, X.J.; Li, Z.; Wu, Y. Hierarchically Self-Assembled Supramolecular Host–Guest Delivery System for Drug Resistant Cancer Therapy. Biomacromolecules 2018, 19, 1926–1938. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Z.; Loh, X.J.; Chen, K.; Li, Z.; Wu, Y.L. Targeted and Sustained Corelease of Chemotherapeutics and Gene by Injectable Supramolecular Hydrogel for Drug-Resistant Cancer Therapy. Macromol. Rapid Commun. 2018, e1800117. [Google Scholar] [CrossRef] [PubMed]
- Liikanen, I.; Tähtinen, S.; Guse, K.; Gutmann, T.; Savola, P.; Oksanen, M.; Kanerva, A.; Hemminki, A. Oncolytic Adenovirus Expressing Monoclonal Antibody Trastuzumab for Treatment of HER2-Positive Cancer. Mol. Cancer Ther. 2016, 15, 2259–2269. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic Virus Therapy: A New Era of Cancer Treatment at Dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic Viruses and Their Application to Cancer Immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cockle, J.V.; Picton, S.V.; Melcher, A. Future Clinical Potential of Oncolytic Virotherapy for Pediatric CNS Tumors. CNS Oncol. 2013, 2, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Huang, Q.; Qiu, F.; Sui, M. Non-viral Delivery Systems for the Application in p53 Cancer Gene Therapy. Curr. Med. Chem. 2015, 22, 4118–4136. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Dhar, D.; Wold, W.S.M. Oncolytic (Replication-Competent) Adenoviruses as Anticancer Agents. Expert Opin. Biol. Ther. 2010, 10, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.J.; Kang, E.; Choi, J.W.; Kim, S.W.; Yun, C.O. Therapeutic Targeting of Chitosan–PEG–Folate-Complexed Oncolytic Adenovirus for Active and Systemic Cancer Gene Therapy. J. Control. Rel. 2013, 169, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Croyle, M.A.; Chirmule, N.; Zhang, Y.; Wilson, J.M. “Stealth” Adenoviruses Blunt Cell-Mediated and Humoral Immune Responses against the Virus and Allow for Significant Gene Expression upon Readministration in the Lung. J. Virol. 2001, 75, 4792–4801. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.L.; Van, Y.T.; Yeh, L.C.; Lin, H.G.; Chang, Y.N. Production of a Biopolymer Flocculant from Bacillus licheniformis and its Flocculation Properties. Bioresour. Technol. 2001, 78, 267–272. [Google Scholar] [CrossRef]
- Bhat, A.R.; Irorere, V.U.; Bartlett, T.; Hill, D.; Kedia, G.; Morris, M.R.; Charalampopoulos, D.; Radecka, I. Bacillus subtilis natto: A Non-Toxic Source of Poly-γ-Glutamic Acid that Could be Used as a Cryoprotectant for Probiotic Bacteria. AMB Express 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Khalil, I.R.; Burns, A.T.H.; Radecka, I.; Kowalczuk, M.; Khalaf, T.; Adamus, G.; Johnston, B.; Khechara, M.P. Bacterial-Derived Polymer Poly-γ-Glutamic Acid (γ-PGA)-Based Micro/Nanoparticles as a Delivery System for Antimicrobials and Other Biomedical Applications. Int. J. Mol. Sci. 2017, 18, 313. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Feng, J.; Sirisansaneeyakul, S.; Song, C.; Chisti, Y. Genetic and metabolic engineering for microbial production of poly-γ-glutamic acid. Biotechnol. Adv. 2018, 36, 1424–1433. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.H.; Park, C.; Kim, C.J.; Poo, H.; Soda, K.; Ashiuchi, M. Natural and Edible Biopolymer Poly-Gamma-Glutamic Acid: Synthesis, Production, And Applications. Chem. Rec. 2005, 5, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Ashiuchi, M. Microbial Production and Chemical Transformation of Poly-γ-Glutamate. Microb. Biotechnol. 2013, 6, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, I.; Singhal, R. Poly (Glutamic Acid)-An Emerging Biopolymer of Commercial Interest. Bioresour. Technol. 2011, 102, 5551–5561. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Soma, L.A.; Fromm, J.R. Targeted Therapy for Hodgkin Lymphoma and Systemic Anaplastic Large Cell Lymphoma: Focus on Brentuximab Vedotin. Oncotargets Ther. 2014, 7, 45–56. [Google Scholar] [CrossRef]
- Jang, J.; Cho, M.; Chun, J.H.; Cho, M.H.; Park, J.; Oh, H.B.; Yoo, C.K.; Rhie, G.E. The Poly-γ-d-Glutamic Acid Capsule of Bacillus anthracis Enhances Lethal Toxin Activity. Infect. Immun. 2011, 79, 3846–3854. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Chun, J.H.; Ha, H.J.; Park, J.; Kim, B.S.; Oh, H.B.; Rhie, G.E. Poly-Gamma-D-Glutamic Acid and Protective Antigen Conjugate Vaccines Induce Functional Antibodies Against the Protective Antigen and Capsule of Bacillus anthracis in Guinea-Pigs and Rabbits. FEMS Immunol. Med. Microbiol. 2009, 57, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Ramgopal, Y.; Mondal, D.; Venkatraman, S.S.; Godbey, W.T. Sustained Release of Complexed and Naked DNA from Polymer Films. J. Biomed. Mater. Res. B Appl. Biomater. 2008, 85, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Bangari, D.S.; Mittal, S.K. Current Strategies and Future Directions for Eluding Adenoviral Vector Immunity. Curr. Gene Ther. 2006, 6, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.; Herzog, R.W. Progress and Prospects: Immune Responses to Viral Vectors. Gene Ther. 2010, 17, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Vogels, R.; Zuijdgeest, D.; Van Rijnsoever, R.; Hartkoorn, E.; Damen, I.; De Bethune, M.P.; Kostense, S.; Penders, G.; Helmus, N.; Koudstaal, W.; et al. Replication-Deficient Human Adenovirus Type 35 Vectors for Gene Transfer and Vaccination: Efficient Human Cell Infection and Bypass of Pre-existing Adenovirus Immunity. J. Virol. 2003, 77, 8263–8271. [Google Scholar] [CrossRef] [PubMed]
- Schnell, M.A.; Zhang, Y.; Tazelaar, J.; Gao, G.P.; Yu, Q.C.; Qian, R.; Chen, S.J.; Varnavski, A.N.; LeClair, C.; Raper, S.E.; et al. Activation of Innate Immunity in Nonhuman Primates Following Intraportal Administration of Adenoviral Vectors. Mol. Ther. 2001, 3, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An Overview of Poly(Lactic-co-Glycolic) Acid (PLGA)-Based Biomaterials for Bone Tissue Engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- Sachlos, E.; Czernuszka, J.T. Making Tissue Engineering Scaffolds Work. Review: The Application of Solid Free Form Fabrication Technology to the Production of Tissue Engineering Scaffolds. Eur. Cell. Mater. 2003, 5, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.Y.; Tsai, S.P.; Wang, D.M.; Chang, Y.N.; Hsieh, H.J. Preparation of γ-PGA/chitosan composite tissue engineering matrices. Biomaterials 2005, 26, 5617–5623. [Google Scholar] [CrossRef] [PubMed]
- Khalil, I.R.; Irorere, V.U.; Radecka, I.; Burns, A.T.H.; Kowalczuk, M.; Mason, J.L.; Khechara, M.P. Poly-γ-Glutamic Acid: Biodegradable Polymer for Potential Protection of Beneficial Viruses. Materials 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Feng, L.; Wu, X.; Xia, G.; Xu, L. Microencapsulation of Recombinant Adenovirus Within Poly-DL-Lactide-Poly (Ethylene Glycol) Microspheres for Enhanced Gene Transfection Efficiency and Inhibitory Effects on Hepatocellular Carcinoma Cells in vitro. Mol. Med. Rep. 2015, 12, 2336–2342. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.; Petch, A.; Al-Rubeai, M. Encapsulation of Viral Vectors for Gene Therapy Applications. Biotechnol. Prog. 2007, 23, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Molavi, O.; Lutsiak, M.E.; Elamanchili, P.; Kwon, G.S.; Samuel, J. Poly(d,l-Lactic-Co-Glycolic Acid) Microsphere Delivery of Adenovirus For Vaccination. J. Pharm. Sci. 2007, 10, 217–230. [Google Scholar]
- Puapermpoonsiri, U.; Spencer, J.; van der Walle, C.F. A freeze-Dried Formulation of Bacteriophage Encapsulated in Biodegradable Microspheres. Eur. J. Pharm. Biopham. 2009, 72, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Ichihara, M.; Yoshioka, Y.; Ishida, T.; Nakagawa, S.; Kiwada, H. Intravenous Administration of Polyethylene Glycol-Coated (PEGylated) Proteins and PEGylated Adenovirus Elicits an Anti-PEG Immunoglobulin M Response. Biol. Pharm. Bull. 2012, 35, 1336–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Ma, Z.; Khor, E.; Lim, L.Y. Uptake of FITC-Chitosan Nanoparticles by A549 Cells. Pharm. Res. 2002, 19, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.; Yu, H. Polyethyleneimine/Poly (γ-Glutamic Acid)/Poly (Lactide-co-Glycolide) Nanoparticles for Loading and Releasing Antiretroviral Drug. Colloids Surf. B Biointerfaces 2011, 88, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Kedia, G.; Hill, D.; Hill, R.; Radecka, I. Production of Poly-γ-Glutamic Acid by Bacillus subtilis and Bacillus licheniformis with Different Growth Media. J. Nanosci. Nanotechnol. 2010, 10, 5926–5934. [Google Scholar] [CrossRef] [PubMed]
- Kocianova, S.; Vuong, C.; Yao, Y.; Voyich, J.M.; Fischer, E.R.; DeLeo, F.R.; Otto, M. Key Role of Poly-Gamma-DL-Glutamic Acid in Immune Evasion and Virulence of Staphylococcus epidermidis. J. Clin. Investig. 2005, 115, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, H.; Natsuda, O.; Hirose, J.; Hayashi, S.; Takasaki, Y. Characteristics of a Biopolymer Flocculant Produced by Bacillus sp. PY-90. J. Ferment. Bioeng. 1995, 79, 378–380. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, Y.; Liu, W.; Lu, C.; Wang, L. Chitosan Microparticles Ionically Cross-Linked with Poly(γ-Glutamic Acid) As Antimicrobial Peptides and Nitric Oxide Delivery Systems. Biochem. Eng. J. 2015, 95, 78–85. [Google Scholar] [CrossRef]
- Ge, Y.; Zhang, Y.; He, S.; Nie, F.; Teng, G.; Gu, N. Fluorescence Modified Chitosan-Coated Magnetic Nanoparticles for High-Efficient Cellular Imaging. Nanoscale Res. Lett. 2009, 4, 287–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds γ-PGA are available from the authors. |













© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalil, I.R.; Khechara, M.P.; Kurusamy, S.; Armesilla, A.L.; Gupta, A.; Mendrek, B.; Khalaf, T.; Scandola, M.; Focarete, M.L.; Kowalczuk, M.; et al. Poly-Gamma-Glutamic Acid (γ-PGA)-Based Encapsulation of Adenovirus to Evade Neutralizing Antibodies. Molecules 2018, 23, 2565. https://doi.org/10.3390/molecules23102565
Khalil IR, Khechara MP, Kurusamy S, Armesilla AL, Gupta A, Mendrek B, Khalaf T, Scandola M, Focarete ML, Kowalczuk M, et al. Poly-Gamma-Glutamic Acid (γ-PGA)-Based Encapsulation of Adenovirus to Evade Neutralizing Antibodies. Molecules. 2018; 23(10):2565. https://doi.org/10.3390/molecules23102565
Chicago/Turabian StyleKhalil, Ibrahim R., Martin P. Khechara, Sathishkumar Kurusamy, Angel L. Armesilla, Abhishek Gupta, Barbara Mendrek, Tamara Khalaf, Mariastella Scandola, Maria Letizia Focarete, Marek Kowalczuk, and et al. 2018. "Poly-Gamma-Glutamic Acid (γ-PGA)-Based Encapsulation of Adenovirus to Evade Neutralizing Antibodies" Molecules 23, no. 10: 2565. https://doi.org/10.3390/molecules23102565
APA StyleKhalil, I. R., Khechara, M. P., Kurusamy, S., Armesilla, A. L., Gupta, A., Mendrek, B., Khalaf, T., Scandola, M., Focarete, M. L., Kowalczuk, M., & Radecka, I. (2018). Poly-Gamma-Glutamic Acid (γ-PGA)-Based Encapsulation of Adenovirus to Evade Neutralizing Antibodies. Molecules, 23(10), 2565. https://doi.org/10.3390/molecules23102565