Construction of a Quantitative Acetylomic Tissue Atlas in Rice (Oryza sativa L.)



 and
and 
Abstract
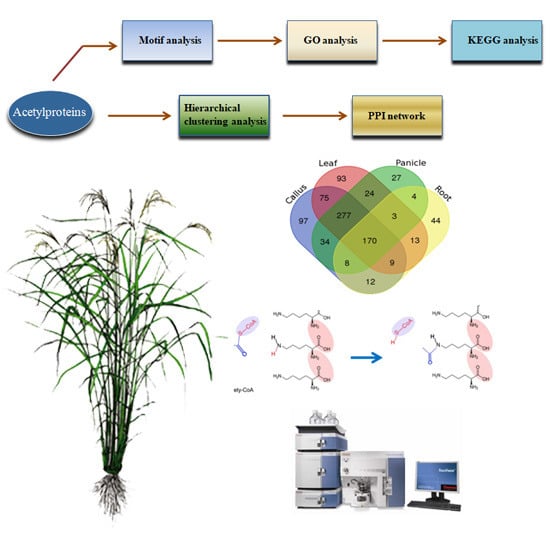
1. Introduction
2. Results
2.1. Profiling the Acetylsites and Acetylproteins on Various Rice Tissues
2.2. Motif Analysis of the Acetylsite Flanking Sequences
2.3. Functional Features of Rice Acetylproteins
2.4. Differentially Acetylated (DA) Peptides among Rice Tissues
2.5. Protein-Protein Interaction (PPI) Analysis of Acetylproteins
3. Discussion
3.1. PKA Profiling in Rice
3.2. Acetylproteins in Rice Callus
3.3. PKA on Chloroplast Development and Photosynthesis Proteins in Leaf
3.4. PKA May Regulate Flowering and Pollen Fertility in Rice Panicle
3.5. Acetylation on Root Proteins
4. Materials and Methods
4.1. Collection and Preparation of Plant Materials
4.2. Protein Extraction
4.3. Western Blotting
4.4. Protein Digestion and Acetylpeptide Enrichment
4.5. LC–MS/MS Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rao, R.S.; Thelen, J.J.; Miernyk, J.A. Is Lys-Nvarepsilon-acetylation the next big thing in post-translational modifications? Trends Plant Sci. 2014, 19, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Mirsky, A.E. Acetylation and Methylation of Histones and their Possible Role in the Regulation of RNA Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Ramirez-Torres, A.; Encarnacion-Guevara, S. Lysine acetylation and cancer: A proteomics perspective. J. Proteom. 2017, 150, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Batta, K.; Das, C.; Gadad, S.; Shandilya, J.; Kundu, T.K. Reversible acetylation of non histone proteins: Role in cellular function and disease. Sub-Cell. Biochem. 2007, 41, 193–212. [Google Scholar]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Song, L.; Liang, W.; Mu, P.; Wang, S.; Lin, Q. Comprehensive profiling of lysine acetylproteome analysis reveals diverse functions of lysine acetylation in common wheat. Sci. Rep. 2016, 6, 21069. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Peng, X.; Cheng, Z.; Liu, W.; Wang, G.L. A comprehensive catalog of the lysine-acetylation targets in rice (Oryza sativa) based on proteomic analyses. J. Proteom. 2016, 138, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [PubMed]
- Lee, J.; Manning, A.J.; Wolfgeher, D.; Jelenska, J.; Cavanaugh, K.A.; Xu, H.; Fernandez, S.M.; Michelmore, R.W.; Kron, S.J.; Greenberg, J.T. Acetylation of an NB-LRR Plant Immune-Effector Complex Suppresses Immunity. Cell Rep. 2015, 13, 1670–1682. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L. Substrate and Functional Diversity of Lysine Acetylation Revealed by a Proteomics Survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, J.; Lin, S.; Deng, W.; Zhang, Y.; Xue, Y. PLMD: An updated data resource of protein lysine modifications. J. Genet. Genom. 2017, 44, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Finkemeier, I.; Laxa, M.; Miguet, L.; Howden, A.J.; Sweetlove, L.J. Proteins of diverse function and subcellular location are lysine acetylated in Arabidopsis. Plant Physiol. 2011, 155, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Konig, A.C.; Hartl, M.; Boersema, P.J.; Mann, M.; Finkemeier, I. The mitochondrial lysine acetylome of Arabidopsis. Mitochondrion 2014, 19, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Melo-Braga, M.N.; Verano-Braga, T.; Leon, I.R.; Antonacci, D.; Nogueira, F.C.; Thelen, J.J.; Larsen, M.R.; Palmisano, G. Modulation of protein phosphorylation, N-glycosylation and Lys-acetylation in grape (Vitis vinifera) mesocarp and exocarp owing to Lobesia botrana infection. Mol. Cell. Proteom. 2012, 11, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Smith-Hammond, C.L.; Hoyos, E.; Miernyk, J.A. The pea seedling mitochondrial Nepsilon-lysine acetylome. Mitochondrion 2014, 19, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Smith-Hammond, C.L.; Swatek, K.N.; Johnston, M.L.; Thelen, J.J.; Miernyk, J.A. Initial description of the developing soybean seed protein Lys-N(epsilon)-acetylome. J. Proteom. 2014, 96, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Chen, W.; Zhao, Y.; Ruan, S.; Zhang, H.; Yan, C.; Jin, L.; Cao, L.; Zhu, J.; Ma, H.; et al. Global analysis of lysine acetylation in strawberry leaves. Front. Plant Sci. 2015, 6, 739. [Google Scholar] [CrossRef] [PubMed]
- Salvato, F.; Havelund, J.F.; Chen, M.; Rao, R.S.; Rogowska-Wrzesinska, A.; Jensen, O.N.; Gang, D.R.; Thelen, J.; Moller, I.M. The potato tuber mitochondrial proteome. Plant Physiol. 2014, 164, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Jing, D.; Kong, L.; Zhang, J.; Yang, F.; Zhang, H.; Wang, J.; Zhang, S. Global Lysine Acetylome Analysis of Desiccated Somatic Embryos of Picea asperata. Front. Plant Sci. 2016, 7, 1927. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Deng, X.; Wang, J.; Zhu, G.; Cao, H.; Yuan, L.; Yan, Y. First Comprehensive Proteome Analyses of Lysine Acetylation and Succinylation in Seedling Leaves of Brachypodium distachyon L. Sci. Rep. 2016, 6, 31576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, D.; Chang, Y.; You, C.; Li, X.; Dai, X.; Weng, Q.; Zhang, J.; Chen, G.; Li, X.; et al. Non-random distribution of T-DNA insertions at various levels of the genome hierarchy as revealed by analyzing 13,804 T-DNA flanking sequences from an enhancer-trap mutant library. Plant J. 2007, 49, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Huber, S.C. Lysine Acetylation Is a Widespread Protein Modification for Diverse Proteins in Arabidopsis. Plant Physiol. 2011, 155, 1769–1778. [Google Scholar] [CrossRef] [PubMed]
- Nallamilli, B.R.; Edelmann, M.J.; Zhong, X.; Tan, F.; Mujahid, H.; Zhang, J.; Nanduri, B.; Peng, Z. Global analysis of lysine acetylation suggests the involvement of protein acetylation in diverse biological processes in rice (Oryza sativa). PLoS ONE 2014, 9, e89283. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hou, Y.; Qiu, J.; Li, Z.; Zhao, J.; Tong, X.; Zhang, J. A Quantitative Acetylomic Analysis of Early Seed Development in Rice (Oryza sativa L.). Int. J. Mol. Sci. 2017, 18, 249–265. [Google Scholar]
- He, D.; Wang, Q.; Ming, L.; Damaris, R.N.; Yi, X.; Cheng, Z.; Yang, P. Global Proteome Analyses of Lysine Acetylation and Succinylation Reveal the Widespread Involvement of both Modification in Metabolism in the Embryo of Germinating Rice Seed. J. Proteome Res. 2016, 15, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ye, J.; Ma, H.; Lu, P. Proteomic analysis of lysine acetylation provides strong evidence for acetylated proteins involved in plant meiosis and tapetum function. Plant J. 2018, 93, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Lv, Y.; Mujahid, H.; Edelmann, M.J.; Zhao, H.; Peng, X.; Peng, Z. Proteome-wide lysine acetylation identification in developing rice (Oryza sativa) seeds and protein co-modification by acetylation, succinylation, ubiquitination, and phosphorylation. Biochim. Biophys. Acta 2017, 1866, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Liu, S.; Chen, C.; Zhu, J.; Yang, X.; Yong, Z.; Rui, G.; Liu, X.; Gong, Z. Global Proteome Analysis Links Lysine Acetylation to Diverse Functions in Oryza sativa. Proteomics 2017, 18. [Google Scholar] [CrossRef]
- Zhou, H.; Finkemeier, I.; Guan, W.; Tossounian, M.A.; Wei, B.; Young, D.; Huang, J.; Messens, J.; Yang, X.; Zhu, J. Oxidative stress-triggered interactions between the succinyl- and acetyl-proteomes of rice leaves. Plant Cell Environ. 2018, 41, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; De la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.F.; Schwartz, D. Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinform. 2011, 15–24. [Google Scholar] [CrossRef]
- Qiu, J.; Hou, Y.; Tong, X.; Wang, Y.; Lin, H.; Liu, Q.; Zhang, W.; Li, Z.; Nallamilli, B.R.; Zhang, J. Quantitative phosphoproteomic analysis of early seed development in rice (Oryza sativa L.). Plant Mol. Biol. 2016, 90, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Walley, J.W.; Shen, Z.; Mcreynolds, M.R.; Schmelz, E.A.; Briggs, S.P. Fungal-induced protein hyperacetylation in maize identified by acetylome profiling. Proc. Natl. Acad. Sci. USA 2018, 115, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M.; Füßl, M.; Boersema, P.J.; Jost, J.O.; Kramer, K.; Bakirbas, A.; Sindlinger, J.; Plöchinger, M.; Leister, D.; Uhrig, G. Lysine acetylome profiling uncovers novel histone deacetylase substrate proteins in Arabidopsis. Mol. Syst. Biol. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Liew, C.C.; Yip, C.C. Acetylation of reticulocyte ribosomal proteins at time of protein biosynthesis. Proc. Natl. Acad. Sci. USA 1974, 71, 2988–2991. [Google Scholar] [CrossRef] [PubMed]
- Kamita, M.; Kimura, Y.; Ino, Y.; Kamp, R.M.; Polevoda, B.; Sherman, F.; Hirano, H. N(α)-Acetylation of yeast ribosomal proteins and its effect on protein synthesis. J. Proteom. 2011, 74, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Tozawa, Y.; Hasegawa, H.; Terakawa, T.; Wakasa, K. Characterization of rice anthranilate synthase alpha-subunit genes OASA1 and OASA2. Tryptophan accumulation in transgenic rice expressing a feedback-insensitive mutant of OASA1. Plant Physiol. 2001, 126, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.R.; Khanday, I.; Majhi, B.B.; Veluthambi, K.; Vijayraghavan, U. Auxin-Responsive OsMGH3, a Common Downstream Target of OsMADS1 and OsMADS6, Controls Rice Floret Fertility. Plant Cell Physiol. 2011, 52, 2123–2135. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Y.; Liu, L.; Wang, C.; Gan, T.; Zhang, Z.; Wang, Y.; Wang, D.; Niu, M.; Long, W. Isolation and characterization of a spotted leaf 32 mutant with early leaf senescence and enhanced defense response in rice. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, Y.; Rahman, M.L.; Cho, S.H.; Kim, Y.S.; Koh, H.J.; Yoo, S.C.; Paek, N.C. The rice faded green leaf locus encodes protochlorophyllide oxidoreductaseB and is essential for chlorophyll synthesis under high light conditions. Plant J. 2013, 74, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Hu, S.; Wu, L.; Ge, C.; Cui, Y.; Chen, P.; Wang, X.; Xu, J.; Ren, D.; Dong, G. White stripe leaf 12 (WSL12), encoding a nucleoside diphosphate kinase 2 (OsNDPK2), regulates chloroplast development and abiotic stress response in rice (Oryza sativa L.). Mol. Breed. 2016, 36. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Karki, S.; Coe, R.A.; Bagha, S.; Khoshravesh, R.; Balahadia, C.P.; Ver, S.J.; Tapia, R.; Israel, W.K.; Montecillo, F. Targeted Knockdown of GDCH in Rice Leads to a Photorespiratory Deficient Phenotype Useful as a Building Block for C4 Rice. Plant Cell Physiol. 2016, 57. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Hu, H.; Ren, H.; Kong, Y.; Lin, H.; Guo, J.; Wang, L.; He, Y.; Ding, X.; Grabsztunowicz, M. LIGHT-INDUCED RICE1 Regulates Light-Dependent Attachment of LEAF-TYPE FERREDOXIN-NADP + OXIDOREDUCTASE to the Thylakoid Membrane in Rice and Arabidopsis. Plant Cell 2016, 28, 712–728. [Google Scholar] [CrossRef] [PubMed]
- Magorzata, A.; Krzysztof, G.; Robert, L.; Wojciech, G.; Przemyslaw, C.M.; Sebastian, S.; Weronika, S.; Rienk, V.G.; Grzegorz, J. Excitation energy transfer and charge separation are affected in Arabidopsis thaliana mutants lacking light-harvesting chlorophyll a/b binding protein Lhcb3. J. Photochem. Photobiol. B 2015, 153, 423–428. [Google Scholar]
- Lee, S.; Kim, J.; Han, J.J.; Han, M.J.; An, G. Functional analyses of the flowering time gene OsMADS50, the putative SUPPRESSOR OF OVEREXPRESSION OF CO 1/AGAMOUS-LIKE 20 (SOC1/AGL20) ortholog in rice. Plant J. Cell Mol. Biol. 2010, 38, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Cho, L.H. OsMADS50 and OsMADS56 function antagonistically in regulating long day (LD)-dependent flowering in rice. Plant Cell Environ. 2010, 32, 1412–1427. [Google Scholar]
- Li, X.; Gao, X.; Wei, Y.; Deng, L.; Ouyang, Y.; Chen, G.; Li, X.; Zhang, Q.; Wu, C. Rice APOPTOSIS INHIBITOR5 coupled with two DEAD-box adenosine 5′-triphosphate-dependent RNA helicases regulates tapetum degeneration. Plant Cell 2011, 23, 1416–1434. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhao, H.; Ruan, W.; Deng, M.; Wang, F.; Peng, J.; Luo, J.; Chen, Z.; Yi, K. ABNORMAL INFLORESCENCE MERISTEM1 Functions in Salicylic Acid Biosynthesis to Maintain Proper Reactive Oxygen Species Levels for Root Meristem Activity in Rice. Plant Cell 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Hiei, Y.; Komari, T.; Kubo, T. Transformation of rice mediated by Agrobacterium tumefaciens. Plant Mol. Biol. 1997, 35, 205–218. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification | Intensity on Average | ||||||
|---|---|---|---|---|---|---|---|
| Protein Accession | Protein Names | Modified Sequence | Protein Annotation | Cal | Lea | Pan | Root |
| Q9XJ60 | MADS50 | _LEALETYK(ac)R_ | MADS-box transcription factor 50 | 1 | |||
| Q10N21 | APX1 | _PLVEK(ac)YAADEK_ | l-ascorbate peroxidase 1, cytosolic | 1.9 | 1.93 | 1.32 | 0.5 |
| Q8W3D9 | PORB | _ELLADLTSSDYPSK(ac)R_ | Protochlorophyllide reductase B | 1 | |||
| Q69RJ0 | Fd-GOGAT1 | _TDILK(ac)AK_ | Ferredoxin-dependent glutamate synthase | 0.38 | 1.83 | ||
| P30298 | SUS2 | _IYEK(ac)YTWK_ | Sucrose synthase 2 | 0.78 | 3.83 | 0.82 | 1.41 |
| Q0IMS5 | WSL12 | _NVVHGSDSPDNGK(ac)R_ | Nucleoside diphosphate kinase | 0.82 | 2.27 | ||
| Q2RAK2 | OsPK1 | _VFNQDLYFK(ac)R_ | Pyruvate kinase | 2.41 | 0.6 | 1.46 | |
| Q5JK84 | API5 | _DFLLK(ac)PELLR_ | DEAD-box ATP-dependent RNA helicase | 1.57 | |||
| P31924 | SUS1 | _IEEK(ac)YTWK_ | Sucrose synthase 1 | 1.48 | 0.91 | 2.1 | 1.09 |
| Q93X08 | UGP | _TNPSNPSIELGPEFK(ac)K_ | UDP-glucose pyrophosphorylase protein | 1.38 | 2.21 | 0.79 | 1.56 |
| A3C6G9 | GDCSH | _YTK(ac)HCEEEDAH_ | Glycine cleavage system H protein | 0.97 | 0.03 | ||
| Q53LQ0 | PDIL1-1 | _SPEDATNLIDDK(ac)K_ | Protein disulfide isomerase-like 1-1 | 0.55 | 2.84 | 1.09 | |
| Q0D9D0 | SBE1 | _CLIEK(ac)HEGGLEEFSK_ | Glycoside Hydrolase | 1.29 | 0.78 | 0.83 | |
| Q43009 | SUS3 | _AEK(ac)HLAGITADTPYSEFHHR_ | Sucrose synthase 3 | 2.08 | |||
| Q5Z8Y4 | USP | _THGAISEFVNPK(ac)YTDSTK_ | UDP-sugar pyrophosphorylase | 1.79 | 3.59 | 1.07 | |
| Q69V57 | OsAld-Y | _(ac)SAFVGK(ac)YADELIK_ | Fructose-bisphosphate aldolase | 2.1 | 1.32 | 0.34 | |
| Q8W1L6 | AIM1 | _YTK(ac)HCEEEDAH_ | Peroxisomal fatty acid beta-oxidation | 5.16 | 2.57 | 1.24 | |
| Q7GD79 | RAN2 | _LTYK(ac)NVPTWHR_ | GTP-binding nuclear protein Ran-2 | 0.96 | 1.8 | 1.16 | |
| Species | Tissue | Treatment | Acetylsites | Acetylproteins | Reference |
|---|---|---|---|---|---|
| Oryza sativa | Callus, root, leaf and panicle | 1536 | 890 | This study | |
| Oryza sativa | Seed | 1003 | 692 | [28] | |
| Oryza sativa | Pistil and developing seeds | 1817 | 972 | [25] | |
| Oryza sativa | Suspension cells | 60 | 44 | [24] | |
| Oryza sativa | Young seedling | 1337 | 716 | [7] | |
| Oryza sativa | Leaf | Oxidative stress | 1669 | 1024 | [30] |
| Oryza sativa | Seedling | 1353 | 866 | [29] | |
| Oryza sativa | Anther | 1354 | 676 | [27] | |
| Oryza sativa | Seed | Imbibition | 699 | 389 | [26] |
| Zea may | Seedling | Fungus | 2791 | 912 | [35] |
| Triticum aestivum | Leaf | 416 | 277 | [6] | |
| Glycine max | Seed | 400 | 245 | [17] | |
| Arabidopsis | Mitochondria of leaf | 243 | 120 | [14] | |
| Arabidopsis | Chloroplast of leaf | 91 | 74 | [13] | |
| Arabidopsis | Leaf | Deacetylase inhibitor | 2057 | 1022 | [36] |
| Arabidopsis | Root, leaf, shoot, flower and silique | 64 | 57 | [23] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Wang, Y.; Bello, B.K.; Ajadi, A.A.; Tong, X.; Chang, Y.; Zhang, J. Construction of a Quantitative Acetylomic Tissue Atlas in Rice (Oryza sativa L.). Molecules 2018, 23, 2843. https://doi.org/10.3390/molecules23112843
Li Z, Wang Y, Bello BK, Ajadi AA, Tong X, Chang Y, Zhang J. Construction of a Quantitative Acetylomic Tissue Atlas in Rice (Oryza sativa L.). Molecules. 2018; 23(11):2843. https://doi.org/10.3390/molecules23112843
Chicago/Turabian StyleLi, Zhiyong, Yifeng Wang, Babatunde Kazeem Bello, Abolore Adijat Ajadi, Xiaohong Tong, Yuxiao Chang, and Jian Zhang. 2018. "Construction of a Quantitative Acetylomic Tissue Atlas in Rice (Oryza sativa L.)" Molecules 23, no. 11: 2843. https://doi.org/10.3390/molecules23112843
APA StyleLi, Z., Wang, Y., Bello, B. K., Ajadi, A. A., Tong, X., Chang, Y., & Zhang, J. (2018). Construction of a Quantitative Acetylomic Tissue Atlas in Rice (Oryza sativa L.). Molecules, 23(11), 2843. https://doi.org/10.3390/molecules23112843