Stable Isotope-Labeled Lipidomics to Unravel the Heterogeneous Development Lipotoxicity

 , ,
, , 

Abstract
:1. Introduction
2. Results
2.1. The Effect of Saturation in Free Fatty Acids
2.2. Differential Lipidomics Profiling between Palmitic Acid- and Palmitoleic Acid-Treated HepG2 Cells
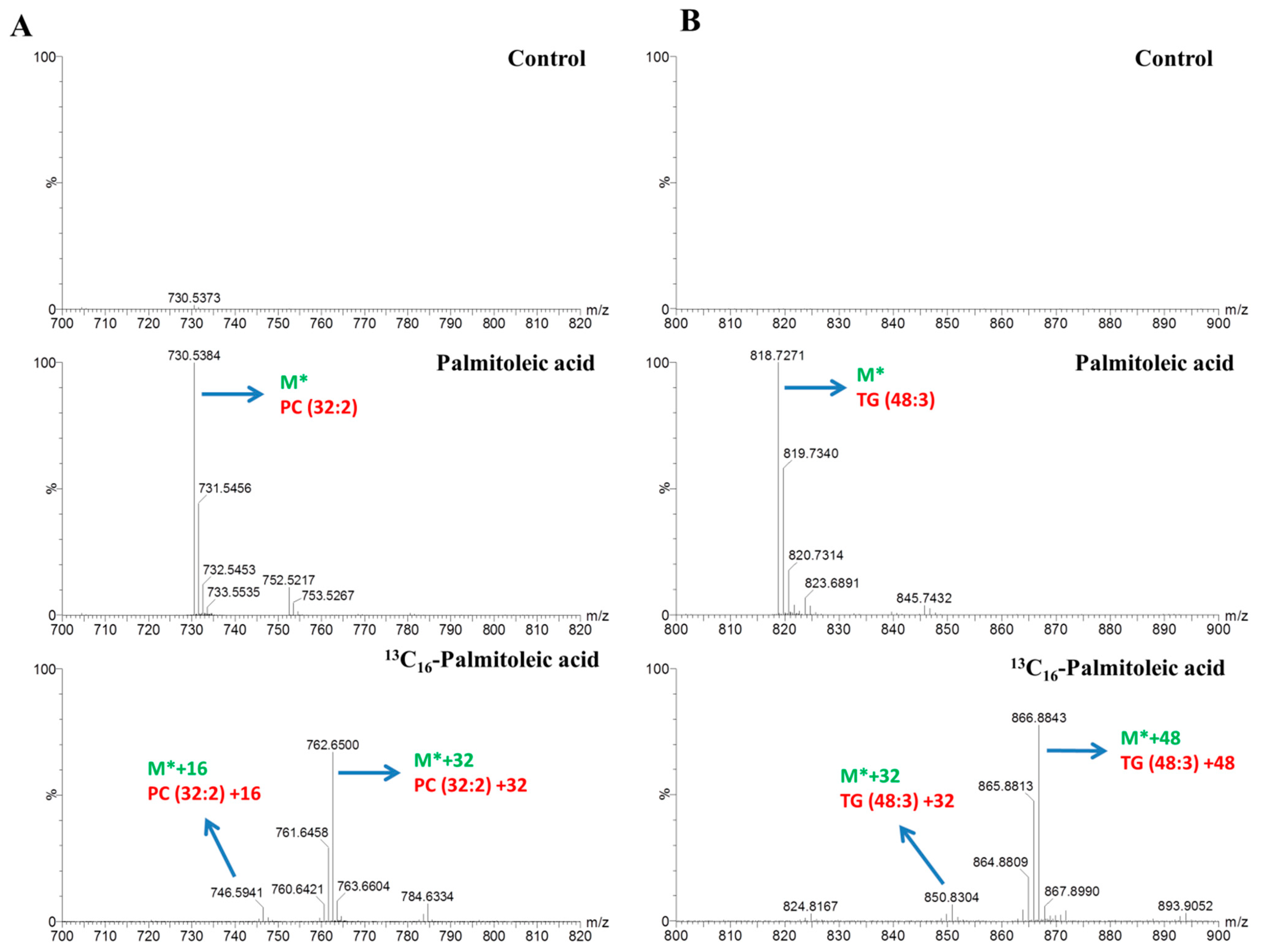
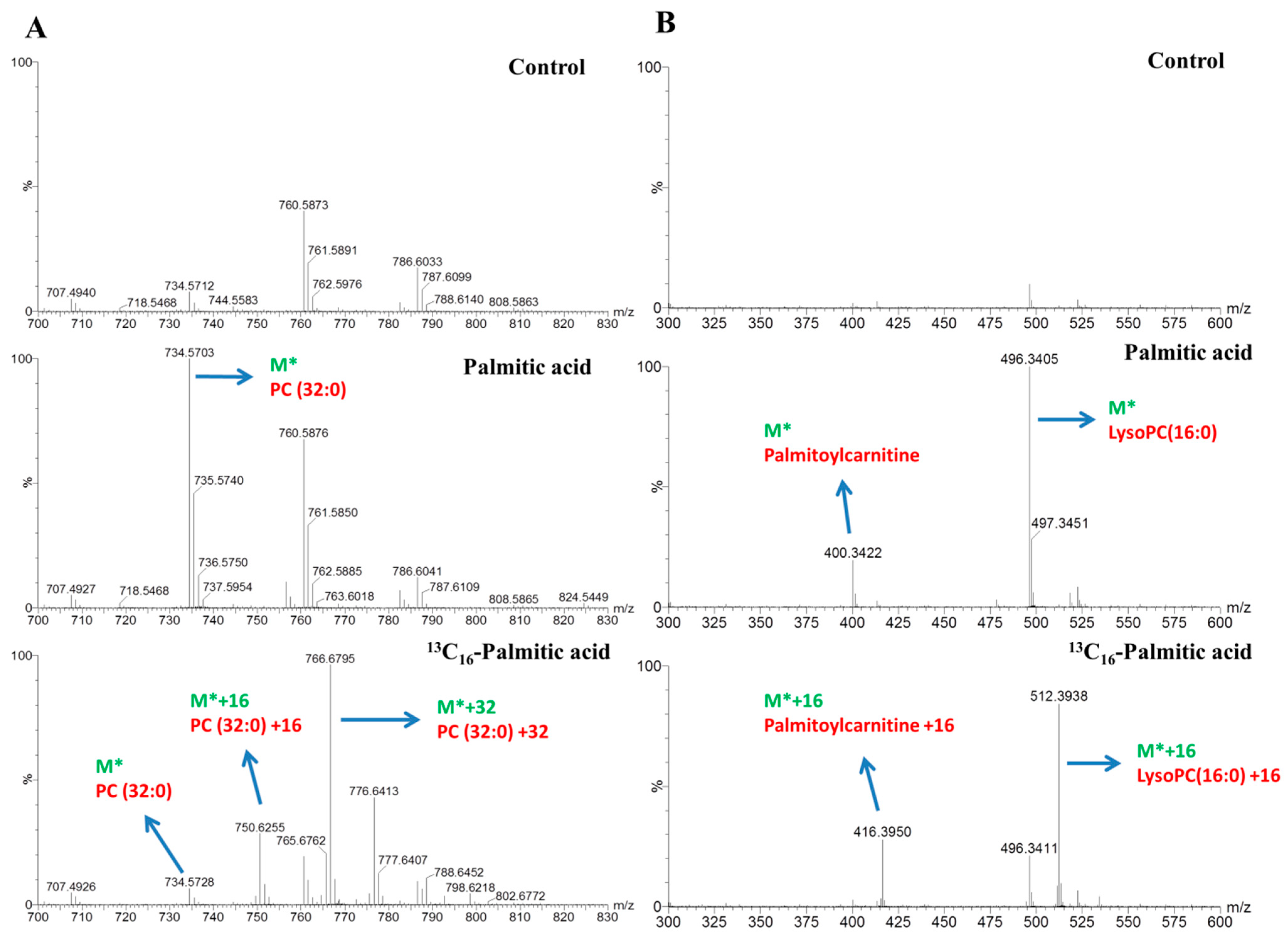
2.3. Lipids Dynamic Changes in 13C16-Palmitic Acid- and 13C16-Palmitoleic Acid-Treated HepG2 Cells
2.4. Mapping of Lipid Metabolism in Palmitic Acid- and Palmitoleic Acid-Treated HepG2 Cells
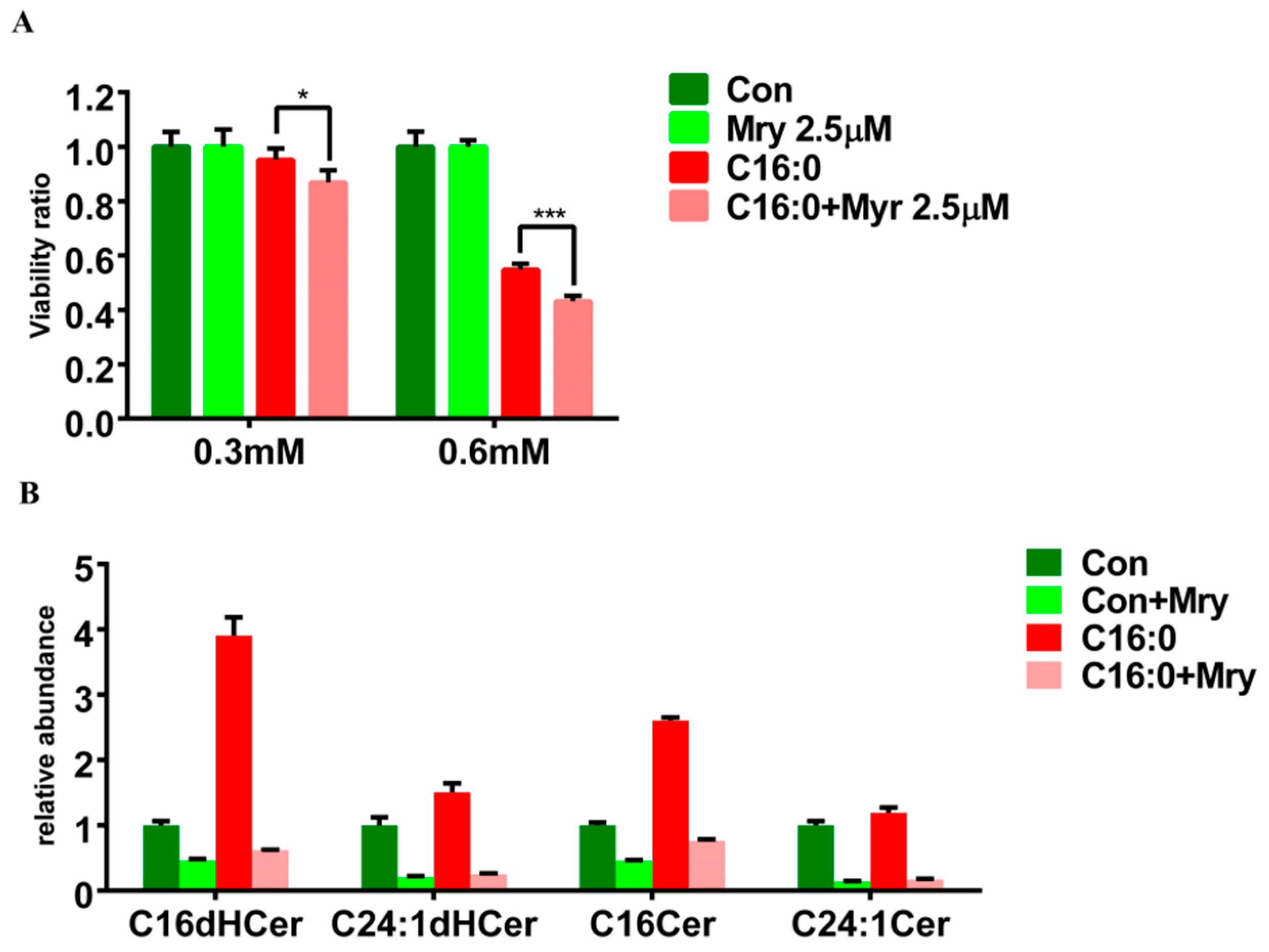
2.5. Aggravating Cell Death through the Inhibition of De Novo Sphingolipid Synthesis
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Free Fatty Acid Treatment
4.3. Cell Viability Assay
4.4. BODIPY (493/503) Staining
4.5. Relative mRNA expression
4.6. Western Blot Analysis
4.7. Sample Preparation for Lipidomics
4.8. Profiling and Identification of Lipid Species Using Liquid Chromatography System Coupled with Time-Of-Flight Mass Spectrometry
4.9. Relative Quantification of Targeted Ceramide Species Using Liquid Chromatography System Coupled with Electrospray Ionization Tandem Mass Spectrometry
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 2008, 9, 162–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.W.; Arendt, B.M.; Hillyer, L.M.; Fung, S.K.; McGilvray, I.; Guindi, M.; Allard, J.P. Plasma phospholipids and fatty acid composition differ between liver biopsy-proven nonalcoholic fatty liver disease and healthy subjects. Nutr. Diabetes 2016, 6, e220. [Google Scholar] [CrossRef] [PubMed]
- Lamaziere, A.; Wolf, C.; Quinn, P.J. Perturbations of lipid metabolism indexed by lipidomic biomarkers. Metabolites 2012, 2, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lechon, M.J.; Donato, M.T.; Martinez-Romero, A.; Jimenez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Gorden, D.L.; Myers, D.S.; Ivanova, P.T.; Fahy, E.; Maurya, M.R.; Gupta, S.; Min, J.; Spann, N.J.; McDonald, J.G.; Kelly, S.L.; et al. Biomarkers of NAFLD progression: A lipidomics approach to an epidemic. J. Lipid Res. 2015, 56, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef] [PubMed]
- Tolman, K.G.; Dalpiaz, A.S. Treatment of non-alcoholic fatty liver disease. Ther. Clin. Risk Manag. 2007, 3, 1153–1163. [Google Scholar] [PubMed]
- German, J.B.; Gillies, L.A.; Smilowitz, J.T.; Zivkovic, A.M.; Watkins, S.M. Lipidomics and lipid profiling in metabolomics. Curr. Opin. Lipidol. 2007, 18, 66–71. [Google Scholar] [PubMed]
- Zhao, Y.Y.; Cheng, X.L.; Lin, R.C. Lipidomics applications for discovering biomarkers of diseases in clinical chemistry. Int. Rev. Cell Mol. Biol. 2014, 313, 1–26. [Google Scholar] [PubMed]
- Li, J.; Hoene, M.; Zhao, X.; Chen, S.; Wei, H.; Haring, H.U.; Lin, X.; Zeng, Z.; Weigert, C.; Lehmann, R.; et al. Stable isotope-assisted lipidomics combined with nontargeted isotopomer filtering, a tool to unravel the complex dynamics of lipid metabolism. Anal. Chem. 2013, 85, 4651–4657. [Google Scholar] [CrossRef] [PubMed]
- Blachnio-Zabielska, A.U.; Persson, X.M.; Koutsari, C.; Zabielski, P.; Jensen, M.D. A liquid chromatography/tandem mass spectrometry method for measuring the in vivo incorporation of plasma free fatty acids into intramyocellular ceramides in humans. Rapid Commun. Mass Spectrom. 2012, 26, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Perez, J.M.; Roddy, T.P.; Shah, V.; McLaren, D.G.; Wang, S.P.; Jensen, K.; Vreeken, R.J.; Hankemeier, T.; Johns, D.G.; Previs, S.F.; et al. Identifying static and kinetic lipid phenotypes by high resolution UPLC-MS: Unraveling diet-induced changes in lipid homeostasis by coupling metabolomics and fluxomics. J. Proteom. Res. 2011, 10, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Zhang, B.; Tang, Y.J. Application of stable isotope-assisted metabolomics for cell metabolism studies. Metabolites 2014, 4, 142–165. [Google Scholar] [CrossRef] [PubMed]
- Kluepfel, D.; Bagli, J.; Baker, H.; Charest, M.P.; Kudelski, A. Myriocin, a new antifungal antibiotic from Myriococcum albomyces. J. Antibiot. 1972, 25, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Kozutsumi, Y.; Nakamura, S.; Fujita, T.; Kawasaki, T. Serine Palmitoyltransferase Is the Primary Target of a Sphingosine-Like Immunosuppressant, Isp-1/Myriocin. Biochem. Biophys. Res. Commun. 1995, 211, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.R.; Liu, J.; Plumeri, D.; Cao, Y.B.; He, T.; Lin, L.; Li, Y.; Jiang, Y.Y.; Li, J.; Shang, J. Lipotoxicity in HepG2 cells triggered by free fatty acids. Am. J. Transl. Res. 2011, 3, 284–291. [Google Scholar] [PubMed]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab. 2012, 23, 365–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Tang, W.J.; Yin, J.J.; Zhou, B.J. Signal transductions and nonalcoholic fatty liver: A mini-review. Int. J. Clin. Exp. Med. 2014, 7, 1624–1631. [Google Scholar] [PubMed]
- De Larichaudy, J.; Zufferli, A.; Serra, F.; Isidori, A.M.; Naro, F.; Dessalle, K.; Desgeorges, M.; Piraud, M.; Cheillan, D.; Vidal, H.; et al. TNF-alpha- and tumor-induced skeletal muscle atrophy involves sphingolipid metabolism. Skelet. Muscle 2012, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braunersreuther, V.; Viviani, G.L.; Mach, F.; Montecucco, F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, M.H.; Baranova, A.; Collantes, R.; Ranard, B.; Stepanova, M.; Bennett, C.; Fang, Y.; Elariny, H.; Goodman, Z.; Chandhoke, V.; et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 27, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Cheng, M.L.; Hua, Y.S.; Wu, Y.H.; Lin, H.R.; Liu, H.Y.; Ho, H.Y.; Chiu, D.T. Glucose 6-phosphate dehydrogenase knockdown enhances IL-8 expression in HepG2 cells via oxidative stress and NF-kappaB signaling pathway. J. Inflamm. 2015, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, R.; Hildebrand, D.F. Biosynthesis and metabolic engineering of palmitoleate production, an important contributor to human health and sustainable industry. Prog. Lipid Res. 2012, 51, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, A.M.; Roizen, M.F.; Martinez, L. Purified palmitoleic acid for the reduction of high-sensitivity C-reactive protein and serum lipids: A double-blinded, randomized, placebo controlled study. J. Clin. Lipidol. 2014, 8, 612–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Law, B.A.; Liao, X.; Moore, K.S.; Southard, A.; Roddy, P.; Ji, R.; Szulc, Z.; Bielawska, A.; Schulze, P.C.; Cowart, L.A. Lipotoxic very-long-chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J. 2018, 32, 1403–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win, S.; Than, T.A.; Le, B.H.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Ruiz, I.; Solis-Munoz, P.; Fernandez-Moreira, D.; Munoz-Yague, T.; Solis-Herruzo, J.A. In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in nonalcoholic steatohepatitis. Dis. Models Mech. 2015, 8, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Sampey, B.P.; Freemerman, A.J.; Zhang, J.; Kuan, P.F.; Galanko, J.A.; O’Connell, T.M.; Ilkayeva, O.R.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; et al. Metabolomic profiling reveals mitochondrial-derived lipid biomarkers that drive obesity-associated inflammation. PLoS ONE 2012, 7, e38812. [Google Scholar] [CrossRef] [PubMed]
- McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines-old actors auditioning for new roles in metabolic physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Al-Bakheit, A.; Traka, M.; Saha, S.; Mithen, R.; Melchini, A. Accumulation of Palmitoylcarnitine and Its Effect on Pro-Inflammatory Pathways and Calcium Influx in Prostate Cancer. Prostate 2016, 76, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.Y.; Rabkin, S.W. Palmitate-induced cardiac apoptosis is mediated through CPT-1 but not influenced by glucose and insulin. Am. J. Physiol. Heart C 2002, 282, H717–H725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Y.M.; Yanase, T.; Nishi, Y.; Tanaka, A.; Saito, M.; Jin, C.H.; Mukasa, C.; Okabe, T.; Nomura, M.; Goto, K.; et al. Saturated FFAs, palmitic acid and stearic acid, induce apoptosis in human granulosa cells. Endocrinology 2001, 142, 3590–3597. [Google Scholar] [CrossRef] [PubMed]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide Is Metabolized to Acylceramide and Stored in Lipid Droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, C.L.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V., Jr. Thematic review series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Man, W.C.; Miyazaki, M.; Chu, K.; Ntambi, J. Colocalization of SCD1 and DGAT2: Implying preference for endogenous monounsaturated fatty acids in triglyceride synthesis. J. Lipid Res. 2006, 47, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Cardaci, S.; Jerby, L.; MacKenzie, E.D.; Sciacovelli, M.; Johnson, T.I.; Gaude, E.; King, A.; Leach, J.D.; Edrada-Ebel, R.; et al. Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat. Commun. 2015, 6, 6001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spangenburg, E.E.; Pratt, S.J.; Wohlers, L.M.; Lovering, R.M. Use of BODIPY (493/503) to visualize intramuscular lipid droplets in skeletal muscle. J. Biomed. Biotechnol. 2011, 2011, 598358. [Google Scholar] [CrossRef] [PubMed]
- Loudet, A.; Burgess, K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Boil. Chem. 1957, 226, 497–509. [Google Scholar]
- Abbott, S.K.; Jenner, A.M.; Mitchell, T.W.; Brown, S.H.; Halliday, G.M.; Garner, B. An improved high-throughput lipid extraction method for the analysis of human brain lipids. Lipids 2013, 48, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. Increasing lipidomic coverage by selecting optimal mobile-phase modifiers in LC-MS of blood plasma. Metabolomics 2016, 12, 34. [Google Scholar] [CrossRef]
- Kasumov, T.; Huang, H.; Chung, Y.M.; Zhang, R.; McCullough, A.J.; Kirwan, J.P. Quantification of ceramide species in biological samples by liquid chromatography electrospray ionization tandem mass spectrometry. Anal. Biochem. 2010, 401, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| POS | 4 h | 8 h | 16 h | |
|---|---|---|---|---|
| % | % | % | ||
| Palmitoylcarnitine | M* | - | - | - |
| M* + 16 | 99.5 | 99.3 | 99.5 | |
| LysoPC (16:0) | M* | - | - | - |
| M* + 16 | 61.1 | 78.4 | 84.0 | |
| PC (32:0) | M* | - | - | - |
| M* + 16 | 24.1 | 23.1 | 23.5 | |
| M* + 32 | 69.8 | 74.2 | 74.3 | |
| Cer (d18:0/16:0) | M* | - | - | - |
| M* + 16 | - | 2.5 | 9.1 | |
| M* + 32 | - | 97.1 | 90.7 | |
| Cer (d18:1/16:0) | M* | - | - | - |
| M* + 16 | 98.0 | 74.8 | 53.2 | |
| M* + 32 | - | 24.4 | 44.3 | |
| SM (d18:1/16:0) | M* | - | - | - |
| M* + 32 | 0.2 | 2.7 | 5.5 | |
| DG (32:0) | M* | - | - | - |
| M* + 16 | 8.1 | 11.6 | 12.7 | |
| M* + 32 | 91.8 | 88.4 | 87.3 | |
| TG (52:2) | M* | - | - | - |
| M* + 16 | 3.1 | 8.6 | 15.3 | |
| NEG | % | % | % | |
| LysoPA (16:0)# | M* | - | - | - |
| M* + 16 | 92.1 | 92.1 | 92.4 | |
| LysoPE (16:0) | M* | - | - | - |
| M* + 16 | 61.3 | 81.7 | 84.8 | |
| PI (32:0) | M* | - | - | - |
| M* + 32 | 88.3 | 99.5 | 99.4 | |
| PE (32:0) | M* | - | - | - |
| M* + 32 | 98.7 | 99.7 | 99.7 | |
| POS | 4 h | 8 h | 16 h | |
|---|---|---|---|---|
| % | % | % | ||
| PC (32:2) | M* | - | - | - |
| M* + 16 | 6.7 | 5.8 | 4.9 | |
| M* + 32 | 93.1 | 94.2 | 95.1 | |
| DG (32:2) | M* | - | - | - |
| M* + 16 | - | - | - | |
| M* + 32 | - | 99 | 99.6 | |
| TG (48:3) | M* | - | - | - |
| M* + 32 | 3.8 | 4.3 | 6.4 | |
| M* + 48 | 96.2 | 95.7 | 93.6 | |
| PI (34:1) | M* | - | - | - |
| M* + 16 | - | 95.9 | 99.3 | |
| PE (34:2) | M* | - | - | - |
| M* + 16 | 58.6 | 75.8 | 63 | |
| M* + 32 | 7.2 | 20 | 35.3 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, L.-M.; Tang, H.-Y.; Lynn, K.-S.; Huang, C.-Y.; Ho, H.-Y.; Cheng, M.-L. Stable Isotope-Labeled Lipidomics to Unravel the Heterogeneous Development Lipotoxicity. Molecules 2018, 23, 2862. https://doi.org/10.3390/molecules23112862
Shih L-M, Tang H-Y, Lynn K-S, Huang C-Y, Ho H-Y, Cheng M-L. Stable Isotope-Labeled Lipidomics to Unravel the Heterogeneous Development Lipotoxicity. Molecules. 2018; 23(11):2862. https://doi.org/10.3390/molecules23112862
Chicago/Turabian StyleShih, Lu-Min, Hsiang-Yu Tang, Ke-Shiuan Lynn, Cheng-Yu Huang, Hung-Yao Ho, and Mei-Ling Cheng. 2018. "Stable Isotope-Labeled Lipidomics to Unravel the Heterogeneous Development Lipotoxicity" Molecules 23, no. 11: 2862. https://doi.org/10.3390/molecules23112862
APA StyleShih, L.-M., Tang, H.-Y., Lynn, K.-S., Huang, C.-Y., Ho, H.-Y., & Cheng, M.-L. (2018). Stable Isotope-Labeled Lipidomics to Unravel the Heterogeneous Development Lipotoxicity. Molecules, 23(11), 2862. https://doi.org/10.3390/molecules23112862