Identification and Characterization of NTB451 as a Potential Inhibitor of Necroptosis



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. NTB451 Inhibits TNF-Induced Necroptosis
2.2. NTB451 Prevents TNF-α-Mediated Necroptosome Formation, Resulting in Inhibition of MLKL Phosphorylation and Oligomerization
2.3. NTB451 Inhibits the Necroptosis by Targeting RIPK1
2.4. Molecular Docking and Molecular Dynamics (MD) Simulations Demonstrate NTB451 Interacts with RIPK1
2.5. NTB451 Suppresses Toll-Like Receptor 3 (TLR3)- and TLR4-Mediated Necroptotic Cell Death
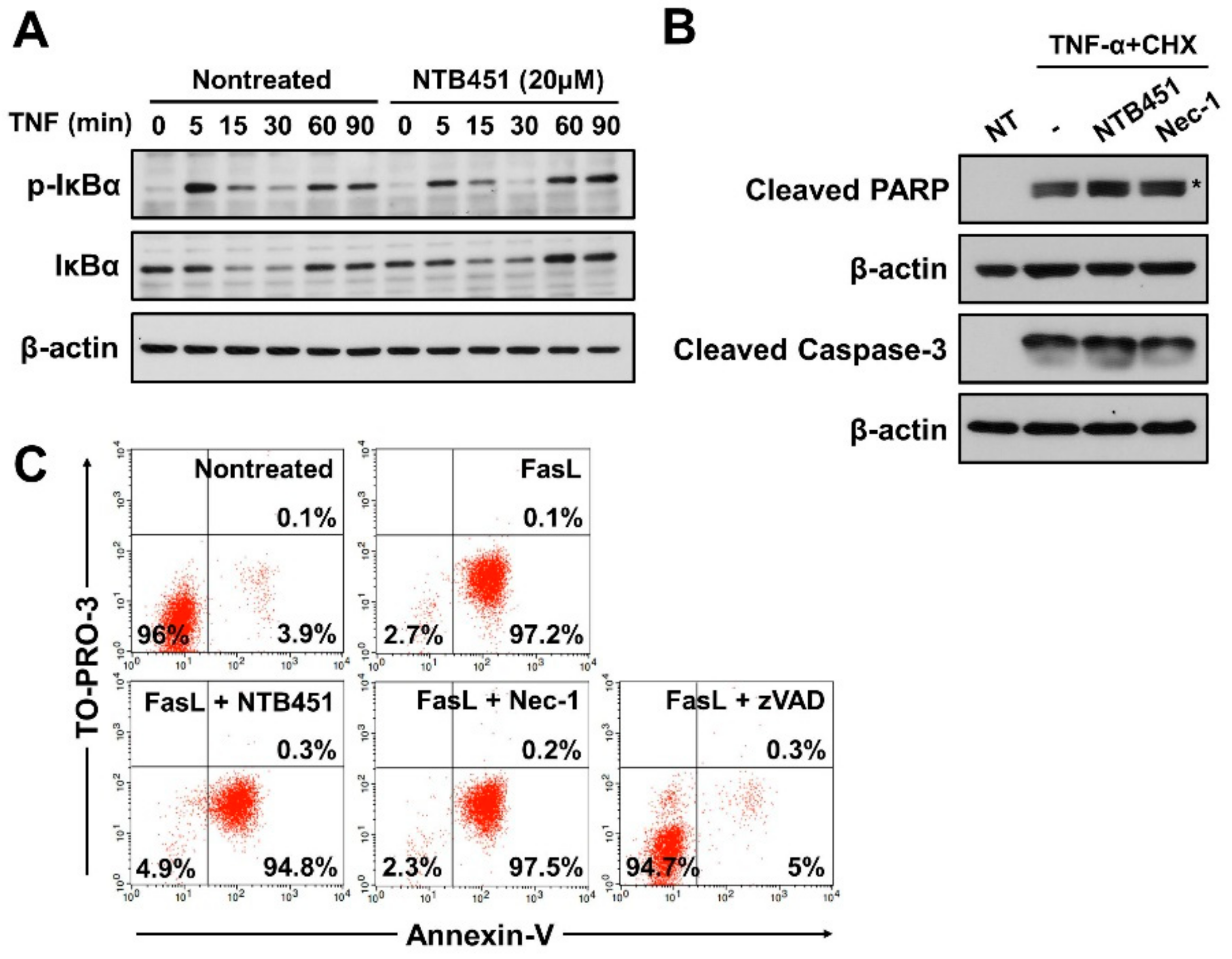
2.6. NTB451 Does not Affect TNF-Induced Apoptosis or Nuclear Factor-κB (NF-κB) Signaling
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Antibodies
4.4. Screening of the Chemical Library
4.5. Lactate Dehydrogenase (LDH) Assay and Half-Maximal Inhibitory Concentration (IC50) Determination
4.6. Immunoblotting
4.7. Co-Immunoprecipitation Assay
4.8. Triton X-100 Fractionation
4.9. siRNA-Mediated Gene Knockdown
4.10. DARTS Assay
4.11. Molecular Docking Simulations
4.12. Molecular Dynamics (MD) Simulations
4.13. FACS Analysis
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kopalli, S.R.; Kang, T.B.; Koppula, S. Necroptosis Inhibitors as Therapeutic Targets in Inflammation Mediated Disorders—A Review of the Current Literature and Patents. Expert Opin. Ther. Pat. 2016, 26, 1239–1256. [Google Scholar] [CrossRef] [PubMed]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in Development, Inflammation and Disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-Like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Jouan-Lanhouet, S.; Riquet, F.; Duprez, L.; Vanden Berghe, T.; Takahashi, N.; Vandenabeele, P. Necroptosis, In Vivo Detection in Experimental Disease Models. Semin. Cell Dev. Biol. 2014, 35, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 Signaling: A Beautiful Pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Vanlangenakker, N.; Bertrand, M.J.; Bogaert, P.; Vandenabeele, P.; Vanden Berghe, T. TNF-Induced Necroptosis in L929 Cells Is Tightly Regulated by Multiple TNFR1 Complex I and II Members. Cell. Death Dis. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular Mechanisms of Necroptosis: An Ordered Cellular Explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Christofferson, D.E.; Yuan, J. Necroptosis as an Alternative Form of Programmed Cell Death. Curr. Opin. Cell Biol. 2010, 22, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a Molecular Signaling Network that Regulates a Cellular Necrotic Cell Death Pathway. Cell 2008, 135, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 Necrosome Forms a Functional Amyloid Signaling Complex Required for Programmed Necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, D.A.; Weinlich, R.; Brown, S.; Guy, C.; Fitzgerald, P.; Dillon, C.P.; Oberst, A.; Quarato, G.; Low, J.; Cripps, J.G.; et al. Characterization of RIPK3-Mediated Phosphorylation of the Activation Loop of MLKL during Necroptosis. Cell Death Differ. 2016, 23, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma Membrane Translocation of Trimerized MLKL Protein Is Required for TNF-Induced Necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.M.; Dobson, R.C.; Webb, A.I.; et al. Activation of the Pseudokinase MLKL Unleashes the Four-Helix Bundle Domain to Induce Membrane Localization and Necroptotic Cell Death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; De Zen, F.; Weinberg, J.; Kunzendorf, U.; Krautwald, S. Programmed Necrosis in Acute Kidney Injury. Nephrol. Dial. Transplant. 2012, 27, 3412–3419. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical Inhibitor of Nonapoptotic Cell Death with Therapeutic Potential for Ischemic Brain Injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting Macrophage Necroptosis for Therapeutic and Diagnostic Interventions in Atherosclerosis. Sci. Adv. 2016, 2, e1600224. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, Y.; Bai, G.; Li, H. Necrostatin-1 Ameliorates Symptoms in R6/2 Transgenic Mouse Model of Huntington’s Disease. Cell. Death Dis. 2011, 2, e115. [Google Scholar] [CrossRef] [PubMed]
- Gunther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, M.J.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 Regulates TNF-Alpha-Induced Epithelial Necroptosis and Terminal Ileitis. Nature 2011, 477, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Vucur, M.; Reisinger, F.; Cardenas, D.V.; Roderburg, C.; Koppe, C.; Kreggenwinkel, K.; Schneider, A.T.; Bartneck, M.; Neumann, U.P.; et al. A Positive Feedback Loop between RIP3 and JNK Controls Non-Alcoholic Steatohepatitis. EMBO Mol. Med. 2014, 6, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; Le Moigne-Muller, G.; Van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL Induces Necroptosis Involving RIPK1/RIPK3-Dependent PARP-1 Activation. Cell Death Differ. 2012, 19, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 Kinase as a Specific Cellular Target of Necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Brasen, J.H.; Himmerkus, N.; Liu, S.; Huber, T.B.; Kunzendorf, U.; Krautwald, S. Rip1 (Receptor-Interacting Protein Kinase 1) Mediates Necroptosis and Contributes to Renal Ischemia/Reperfusion Injury. Kidney Int. 2012, 81, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Zhu, H.; Song, Z.; Gong, Y.; Wang, F.; Wang, W.; Zheng, Z.; Yu, Z.; Gu, Q.; Xu, X.; et al. Necrostatin-1 Protects Photoreceptors from Cell Death and Improves Functional Outcome After Experimental Retinal Detachment. Am. J. Pathol. 2012, 181, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.S.; Yi, T.L.; Zhang, S.; Xu, Z.W.; Yu, Z.Q.; Sun, H.T.; Yang, C.; Tu, Y.; Cheng, S.X. Hypoxia-Inducible Factor-1 Alpha Is Involved in RIP-Induced Necroptosis Caused by In Vitro and In Vivo Ischemic Brain Injury. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, J.X.; Feng, J.M.; Wang, Y.; Li, X.H.; Chen, X.X.; Su, Y.; Shen, Y.Y.; Chen, Y.; Xiong, B.; Yang, C.H.; et al. The B-Raf(V600E) Inhibitor Dabrafenib Selectively Inhibits RIP3 and Alleviates Acetaminophen-Induced Liver Injury. Cell. Death Dis. 2014, 5, e1278. [Google Scholar] [CrossRef] [PubMed]
- Fauster, A.; Rebsamen, M.; Huber, K.V.; Bigenzahn, J.W.; Stukalov, A.; Lardeau, C.H.; Scorzoni, S.; Bruckner, M.; Gridling, M.; Parapatics, K.; et al. A Cellular Screen Identifies Ponatinib and Pazopanib as Inhibitors of Necroptosis. Cell. Death Dis. 2015, 6, e1767. [Google Scholar] [CrossRef] [PubMed]
- Lomenick, B.; Hao, R.; Jonai, N.; Chin, R.M.; Aghajan, M.; Warburton, S.; Wang, J.; Wu, R.P.; Gomez, F.; Loo, J.A.; et al. Target Identification using Drug Affinity Responsive Target Stability (DARTS). Proc. Natl. Acad. Sci. USA 2009, 106, 21984–21989. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-Like Receptors Activate Programmed Necrosis in Macrophages through a Receptor-Interacting Kinase-3-Mediated Pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-Like Receptor 3-Mediated Necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Verma, I.M.; Stevenson, J.K.; Schwarz, E.M.; Van Antwerp, D.; Miyamoto, S. Rel/NF-Kappa B/I Kappa B Family: Intimate Tales of Association and Dissociation. Genes Dev. 1995, 9, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, F.; Schenk, B.; Martens, S.; Vandenabeele, P.; Fulda, S. Sorafenib Inhibits Therapeutic Induction of Necroptosis in Acute Leukemia Cells. Oncotarget 2017, 8, 68208–68220. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Hassannia, B.; Vandenabeele, P. An Outline of Necrosome Triggers. Cell. Mol. Life Sci. 2016, 73, 2137–2152. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Saitoh, M.; Katoh, T.; Seki, T.; Bigi, S.V.; Shimizu, Y.; Ishii, T.; Okai, T.; Kuno, M.; Hattori, H. Discovery of 7-Oxo-2, 4, 5, 7-Tetrahydro-6 H-Pyrazolo [3–C] Pyridine Derivatives as Potent, Orally Available, and Brain-Penetrating Receptor Interacting Protein 1 (RIP1) Kinase Inhibitors: Analysis of Structure-Kinetic Relationships. J. Med. Chem. 2018, 61, 2384–2409. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.B.; Oh, G.S.; Scandella, E.; Bolinger, B.; Ludewig, B.; Kovalenko, A.; Wallach, D. Mutation of a Self-Processing Site in Caspase-8 Compromises its Apoptotic but not Its Nonapoptotic Functions in Bacterial Artificial Chromosome-Transgenic Mice. J. Immunol. 2008, 181, 2522–2532. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.E.; Schuchmann, M.; Luria, V.; Chiannilkulchai, N.; Beckmann, J.S.; Mett, I.L.; Rebrikov, D.; Brodianski, V.M.; Kemper, O.C.; Kollet, O.; et al. Targeted Disruption of the Mouse Caspase 8 Gene Ablates Cell Death Induction by the TNF Receptors, Fas/Apo1, and DR3 and Is Lethal Prenatally. Immunity 1998, 9, 267–276. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harbor Protoc. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pai, M.Y.; Lomenick, B.; Hwang, H.; Schiestl, R.; McBride, W.; Loo, J.A.; Huang, J. Drug Affinity Responsive Target Stability (DARTS) for Small-Molecule Target Identification. Methods Mol. Biol. 2015, 1263, 287–298. [Google Scholar] [PubMed]
- Wu, G.; Robertson, D.H.; Brooks III, C.L.; Vieth, M. Detailed Analysis of Grid-Based Molecular Docking: A Case Study of CDOCKER—A CHARMm-Based MD Docking Algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2017. [Google Scholar]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2015, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
Sample Availability: NTB451, (4-({[5-(4-aminophenyl)-4-ethyl-4H-1,2,4-triazol-3-yl]sulfanyl}methyl)-N-(1,3-thiazol-2-yl)benzamide) is commercially available. |






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
In, E.-J.; Lee, Y.; Koppula, S.; Kim, T.-Y.; Han, J.-H.; Lee, K.-H.; Kang, T.-B. Identification and Characterization of NTB451 as a Potential Inhibitor of Necroptosis. Molecules 2018, 23, 2884. https://doi.org/10.3390/molecules23112884
In E-J, Lee Y, Koppula S, Kim T-Y, Han J-H, Lee K-H, Kang T-B. Identification and Characterization of NTB451 as a Potential Inhibitor of Necroptosis. Molecules. 2018; 23(11):2884. https://doi.org/10.3390/molecules23112884
Chicago/Turabian StyleIn, Eun-Jung, Yuno Lee, Sushruta Koppula, Tae-Yeon Kim, Jun-Hyuk Han, Kwang-Ho Lee, and Tae-Bong Kang. 2018. "Identification and Characterization of NTB451 as a Potential Inhibitor of Necroptosis" Molecules 23, no. 11: 2884. https://doi.org/10.3390/molecules23112884
APA StyleIn, E.-J., Lee, Y., Koppula, S., Kim, T.-Y., Han, J.-H., Lee, K.-H., & Kang, T.-B. (2018). Identification and Characterization of NTB451 as a Potential Inhibitor of Necroptosis. Molecules, 23(11), 2884. https://doi.org/10.3390/molecules23112884