1. Introduction
Sanguisorba officinalis L.
(S. officinalis), which is a traditional Chinese medicine (TCM), and a member of Rosaceae family, has the effects of detoxification, analgesic [
1] and hemostatic [
2]. According to the Chinese pharmacopoeia, it plays a major role in the treatment of hematochezia, bleeding hemorrhoids, bloody flux, metrorrhagia and metrostaxis, bleeding wounds, burns and scalds, and swollen carbuncles [
3]. Besides, in vivo and in vitro studies have illustrated that plants from the
S. officinalis present a wide range of pharmacological properties, including hemostatic, antioxidant [
4], anti-inflammatory [
5], antiviral [
6], antibacterial [
7,
8], anti-tumor [
9], neuroprotective and hypoglycemic activities [
10]. Simultaneously, in Chinese medical practice, many drugs (e.g., tablets and powders) that contain
S. officinalis roots have been applied to treat leukopenia, hemorrhaging and burns [
11]. It has been reported that
S. officinalis has obvious anti-tumor effect, which inhibits the growth of human leukemia cell K562, hepatoma cell HepG2, gastric cancer cell BGC823, leukemia cell L1210, cervical cancer cell Hela, and lung cancer cell H460, and induces apoptosis of human liver cancer SMMC-7721 cells [
12,
13,
14,
15]. The main chemical constituents isolated from
S. officinalis include triterpenes and their glycosides [
16,
17], tannins, flavonoids [
2,
17], etc. Triterpenes are the main hemostatic components of
S. officinalis, the pharmacological studies mainly focus on antioxidant, anti-inflammatory and anti-tumor activities [
2,
18,
19] in nearly a decade.
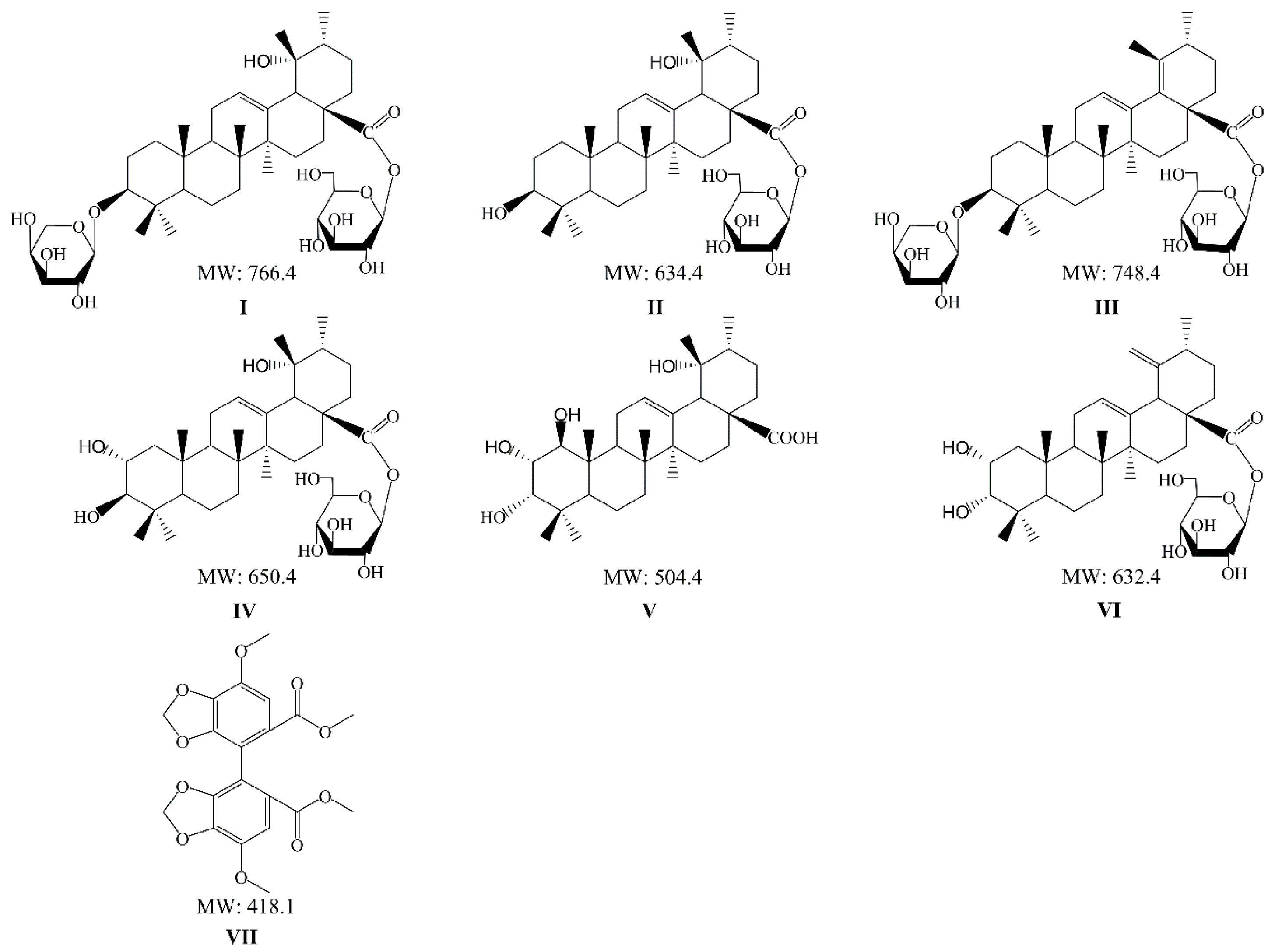
Ziyuglycoside I (ZGI) (
I), 3β,19α-dihydroxyurs-12-en-28-oic-acid 28-β-
d-glucopyranosyl ester (DGE) (
II), 3β-[(α-
l-arabinopyranosyl) oxy]-urs-12,18(19)-dien-28-oic acid β-
d-glucopyranosyl ester (AGE) (
III), rosamultin (RMU) (
IV), 1β-hydroxyeuscaphic acid (HDA) (
V) and alpinoside (APS) (
VI) (
Figure 1) are the active components of triterpenes isolated from
S. officinalis, and many studies have concentrated upon their pharmacological properties [
20]. Six triterpenes have not only common pharmacological activities, but also their own pharmacological characteristics. On the one hand, ZGI, one of the main triterpenes in
S. officinalis, was considered to play a role in eliminating free radicals and inhibiting elastase activity [
21,
22]. On the other hand, ZGI could inhibit skin wrinkles by boosting the production of collagen, not by its anti-oxidant activity [
23]. It has been reported that DGE significantly inhibited NO production [
24]. The effects of DGE on reduction of both
d-galactosamine (
d-GalN) and TNF-α-induced cytotoxicity, the viability of L929 cells and a TNF-α-sensitive cell line were examined under the presence of the constituents [
25]. Thus, DGE significantly improved cell viability. Some studies have shown that RMU possesses antioxidant and anti-apoptosis effects, which could treat H
2O
2-induced oxidative stress injury [
26]. HDA, which is one of triterpenes ingredients, could effectively attenuate the leakage of intracellular enzymes, and decrease the oxidation of proteins and the incidence of apoptosis. Thus, its remarkable hepatoprotective effect was revealed [
27].
Triterpenes have made great progress in pharmacological research, but there are few studies on pharmacokinetic aspects. Several methods have been applied to the determination of triterpenes in the past few years. For instance, Ye et al. offered an original and universally appropriate method to determine ziyuglycoside I and ziyuglycoside II in rat plasma based on LC-MS [
22]. Besides, in 2018, Li et al. developed a simple and sensitive HPLC-MS/MS method for simultaneous determination and pharmacokinetics of ziyuglycoside I and its metabolite ziyuglycoside II in rats [
28]. However, there are few reports on the simultaneous determination and pharmacokinetics of ZGI, DGE AGE, RMU, HDA and APS from
S. officinalis. Thus, the pharmacokinetics of DGE, AGE, RMU, HDA and APS are reported for the first time in this paper.
Hence, the purpose of this study was to set up a sensitive and efficient UHPLC-MS/MS method for simultaneous determination and pharmacokinetics of six analytes in rats after single oral administration of S. officinalis extract. Meanwhile, the DGE, AGE, RMU, HDA and APS are the first report in pharmacokinetic study of S. officinalis. This study could be conducive to furnish basis for clinical application of S. officinalis.
3. Discussion
Even though scholars have employed the LC-MS/MS method, the selective ion monitoring mode (SIM) is mainly used to take the place of MRM mode, there may be more interference in the SIM mode [
31]. Therefore, a sensitive and efficient UHPLC-MS/MS method was established for simultaneous determination of the six analytes in MRM mode.
It is vital for the optimization of mass spectrometry parameters to acquire steady and sensitive responses for analytes. The analysis was performed on an Agilent series 1290 UHPLC instrument (Agilent Technologies, Santa Clara, CA, USA) coupled with an Agilent Technologies 6430 mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) with an electrospray ionization (ESI) interface. The eluent was monitored using a triple quadrupole tandem mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) equipped with ESI source and operated in positive ion mode with MRM. To achieve good resolution, several different chromatographic columns including a XTerra® MS C18 (2.1 × 50 mm, 2.5 μm, Waters Technologies, Milford, MA, USA) column, an ACQUITY UPLC® HSS T3 (2.1 × 100 mm, 1.8 μm, Waters Technologies, Milford, MA, USA) column, and an Agilent Eclipse Plus C18 RRHD (2.1 × 50 mm, 1.8 μm, Agilent Technologies, Santa Clara, CA, USA) column were attempted. Finally, the Agilent Eclipse Plus C18 RRHD (2.1 × 50 mm, 1.8 μm, Agilent Technologies, Santa Clara, CA, USA) column was adopted to achieve great resolution. In summary, for the sensitive detection of six analytes, the positive mode was adopted with MRM. The fragment and collision energy were optimized to increase the sensitivity of the six analytes. The conditions of MS analysis are as follows: drying gas (N2) flow-rate, 11 L/min; drying gas temperature, 300 °C; high purity nitrogen (N2) was atomized as the nebulizing gas; and capillary voltage, 4000 V.
Chromatographic conditions were updated to meliorate peak shape, enhance signal response of six components and reduce the running time. We attempted different mobile phase systems including acetonitrile–water, and methanol–water in terms of different proportions. When methanol–water was used as mobile phase, the response of six analytes was apparently higher than that of acetonitrile–water. Various additives have a notable impact on improving the response of analytes. Different additives such as formic acid (0.1%), ammonium acetate (2 and 5 mM) and acetic acid (0.1%) were investigated. Finally, 0.1% formic acid water was selected as mobile phase to increase the peak intensity of six analytes. It is worth noticing that the high resolution of the UHPLC system increases the speed and peak capacity of the six analytes. At the same time, there was no crosstalk by adjusting all aspects. Finally, the optimized separating conditions were obtained with methanol–0.1% formic acid water as mobile phase at 30 °C and a flow rate of 0.3 mL/min. The mobile phase was as follows: solvent A was 0.1% formic acid water, and solvent B was methanol, which was delivered at a flow rate of 0.3 mL/min. The gradient elution program was as follows: 0–4.0 min, 60% to 65% B; 4.0–4.5 min, 65% to 90% B; 4.5–5.6 min, 90% B; 5.6–6.0 min, 90% to 60% B. The all run time was 6.0 min, and sample injection volume was 5 μL.
To achieve high extraction recovery and weak matrix effect, it is crucial for simultaneously and accurately analyzing the target compound to choose a reasonable sample preparation method [
32]. For sample preparation, we have tried some extraction methods such as SPE (Solide Phase Extraction), protein precipitation and LLE. We tried the protein precipitation method due to its simplicity. Nevertheless, the drawback is that the recovery of compounds is not only insufficient but also non-renewable. Moreover, SPE is time-consuming and columns are relatively expensive. Thus, LLE was selected as sample preparation method owing to its constant extraction recoveries, and negligible matrix effects. It is important for the extraction recoveries to choose a suitable organic solvent as the extract agent. Thus, several solvents have been attempted including ether, dichloromethane and ethyl acetate. Because six analytes have similar polarities, ethyl acetate is the best choice in terms of extraction efficiency and reproducibility.
It is important for the pharmacokinetic study to select reasonable internal standard. The IS should be provided with similar polarity and solubility, it should not react with analytes, and it should not interfere with compounds. In this experiment, we tested bifendate and theophylline. Eventually, bifendate was chosen as IS, which has the suitable retention time and good precision in this experiment. Meanwhile, bifendate had no interference with the analytes and may be utilized to determine the concentration of triterpenes.
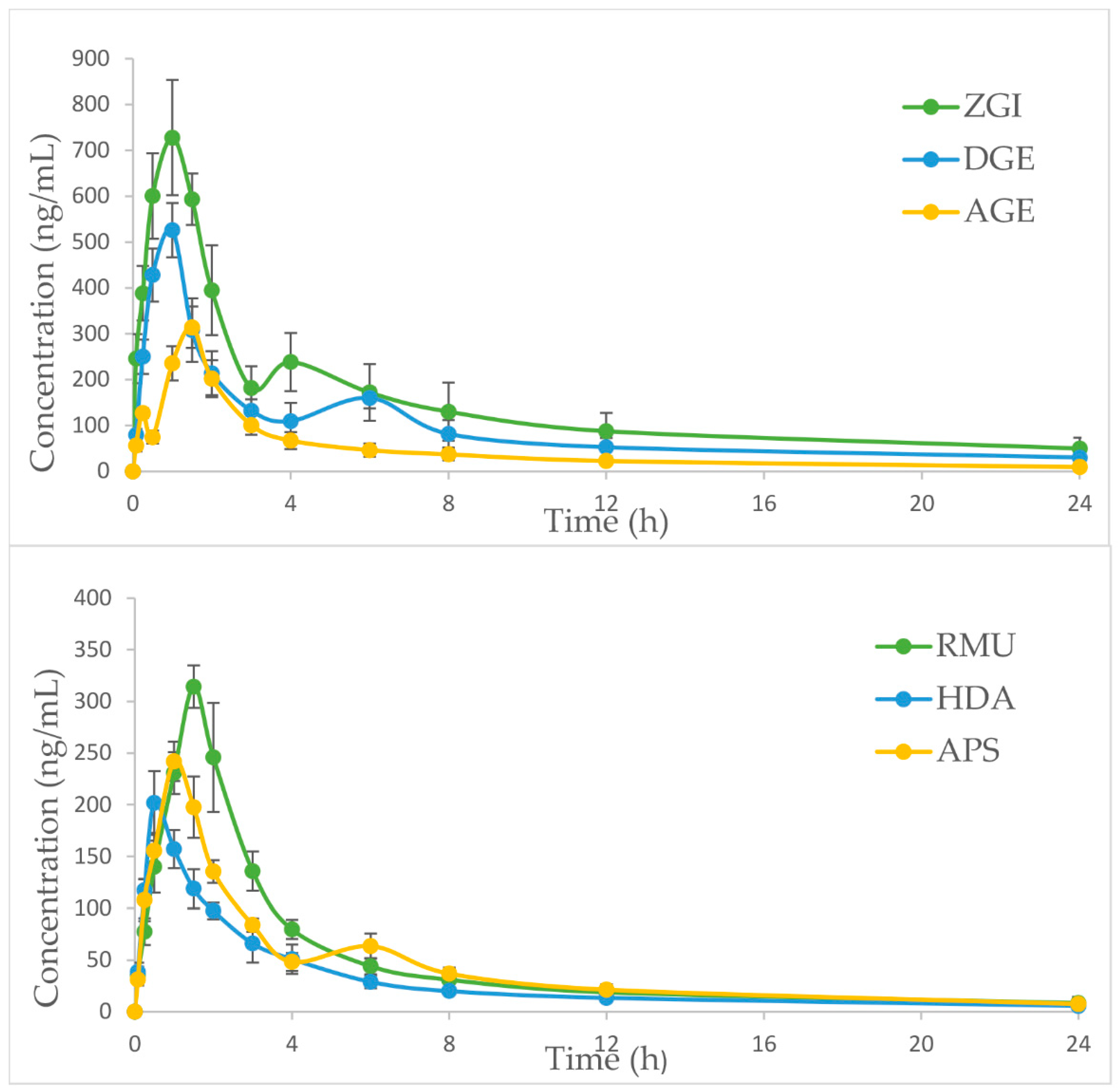
As shown in
Table 6, the pharmacokinetic process of the six analytes in
S. officinalis extract was different. The
Tmax values of ZGI, DGE, AGE, RMU, HDA and APS were 0.92 ± 0.20, 0.83 ± 0.26 and 1.33 ± 0.26, 1.58 ± 0.20, 0.58 ± 0.20 and 1.17 ± 0.26 h, respectively, after single dose administration of
S. officinalis extract, which indicated the absorbance velocity of six compounds was relatively rapid. The
Cmax values of ZGI, DGE, AGE, RMU, HDA and APS in
S. officinalis extract were 744.66 ± 85.74, 526.58 ± 64.02, 317.87 ± 47.60, 314.53 ± 22.46, 211.51 ± 11.81 and 243.21 ± 19.90 ng/mL, respectively. It may be attributed to the difference in the content of six analytes in
S. officinalis extract. Furthermore, as report goes, many herbal medicine or natural compounds separated from Chinese medicinal materials have been appraised as substrates, inhibitors and inducers of various CYP3A4, and herb–CYP interactions, the above-mentioned illustrated that it was possible to have impact on the pharmacokinetics of some compounds [
33]. Moreover, the
t1/2 of ZGI, DGE, AGE, RMU, HDA and APS were 11.63 ± 3.07, 8.99 ± 3.04, 6.86 ± 2.91, 7.32 ± 2.74, 7.35 ± 3.95 and 7.72 ± 1.22 h, respectively. It was revealed that the other five compounds were eliminated and metabolized quickly compared with ZGI. The slow elimination of ZGI may be attributed to its high content in
S. officinalis extract. Besides, according to our research results, the
t1/2 of ZGI was 11.63 h instead of 19.76 or 6.12 h, which is different from the
t1/2 of ZGI in the reported literature [
22,
28]. The difference of
t1/2 values of the ZGI may be caused by the different dosage, the way of administration and the complexity of Chinese medicine composition. Moreover, the AUC
0→t values of six compounds in
S. officinalis extract were 2879.96 ± 303.36, 2296.46 ± 416.63, 1139.15 ± 150.38, 1208.39 ± 119.71, 733.31 ± 94.08 and 1026.03 ± 73.43 ng·h/mL, respectively. The AUC
0→∞ values of six compounds were 3319.05 ± 429.07, 2661.61 ± 600.92, 1231.82 ± 192.74, 1302.86 ± 192.89, 794.41 ± 151.77 and 1112.72 ± 98.12 ng·h/mL, respectively. As shown in
Figure 4, the mean plasma concentration–time distribution curves of ZGI, DGE, AGE and APS exhibited the double-peak phenomenon during elimination phase. The first peak of ZGI, DGE and APS, which occurred at 0.5–1.5 h, the second one appeared at 4-8 h after oral administration. Compared with three compounds, the first peak of AGE appeared in 0.25 h, and the second emerged before 1.5 h. The second peak was far greater than the first peak. The double-peak phenomenon of compounds may be due to the distribution of reabsorption and entero-hepatic circulation [
34]. These results could be conducive to further explore the mechanism of triterpenes and provide effective pharmacokinetic information for
S. officinalis.
4. Materials and Methods
4.1. Chemicals and Reagents
The ZGI, DGE, AGE, RMU, HDA and APS, the purities of which were more than 98%, were refined in our laboratory (identified by NMR and MS). Bifendate (lot: 73536-69-3; purity > 98%, IS) was purchased from Chengdu Must Bio-Technology (Chengdu, Sichuan province, China). Methanol and acetonitrile (HPLC-grade) were obtained from J & K Medical (Beijing, China). Ammonium acetate was purchased from Kermel (Tianjin, China). Ultra-pure water was gained by using Milli-Q water purification system (Millipore, Molsheim, France). All other reagents including ethyl acetate, ether and dichloromethane were of analytical grade. The plasma samples were obtained from the blood of rat.
The S. officinalis was collected from the Anguo Traditional Chinese Medicine Market of Hebei and authenticated by Professor Zhenyue Wang of Heilongjiang University of Chinese Medicine in September 2016. A voucher specimen was deposited in Pharmaceutical Research Department of Harbin Medical University, China.
4.2. Preparation of S. officinalis Extract
After crushing the dried root of
S. officinalis (200 g), it was extracted by hot reflux with 2 L 70% ethanol (1:10,
w/
v) solution 2 times at 80 °C, 60 min each, and then filtrated. The combined filtrate was evaporated to steam, and the residue was dissolved in water to get a concentration equivalent to 0.05 g/mL of the
S. officinalis extract [
35]. The contents of
S. officinalis extract for
I–
VI were 50.26, 33.20, 18.01, 11.34, 22.34 and 6.91 mg/g, respectively. The results of simultaneous determination of six triterpenes from
S. officinalis extract by HPLC-ELSD are presented in the
Supplementary Materials.
4.3. Preparation of Calibration Standards and QC Samples
Standard stock solutions of I–VI were gained through dissolving each compound in methanol to yield a nominal concentration (0.24 mg/mL, 0.11 mg/mL, 0.13 mg/mL, 0.14 mg/mL, 0.13 mg/mL, and 0.13 mg/mL, respectively). Standard working solutions were prepared by appropriate dilutions of the stock solutions with methanol (6.1–2420 ng/mL for I, 4.9–1950 ng/mL for II, 1.3–533.3 ng/mL for III, 3.8–1510 ng/mL for IV, 1.5–604.0 ng/mL for V, and 5.7–2250 ng/mL for VI). The IS stock standard solution was diluted to a 1040 ng/mL working solution. Calibration standards were prepared by spiking each working stock solution at seven concentrations of: 6.1, 12.1, 24.2, 121.0, 242.0, 484.0 and 2420 ng/mL for I; 4.9, 9.8, 19.5, 97.5, 195.0, 390.0 and 1950 ng/mL for II; 1.3, 2.7, 5.3, 26.7, 53.4, 106.7 and 533.3 ng/mL for III; 3.8, 7.6, 15.1, 75.5, 151.0, 302.0 and 1510 ng/mL for IV; 1.5, 3.0, 6.0, 30.2, 60.4, 120.8 and 604.0 ng/mL for V; and 5.7, 11.3, 22.5, 112.5, 225.0, 450.0 and 2250 ng/mL for VI. Quality control (QC) samples were prepared at: 12.1, 121.0 and 1936 ng/mL for I; 9.8, 97.5 and 1560 ng/mL for II; 2.7, 26.7 and 426.67 ng/mL for III; 7.6, 75.5 and 1208 ng/mL for IV; 3.0, 30.2 and 483.2 ng/mL for V; and 11.3, 112.5, and 1800 ng/mL for VI. LLOQ of I–VI was 6.1, 4.9, 1.3, 3.8, 1.5 and 5.7 ng/mL, respectively. All solutions were immediately stored at 4 °C.
4.4. Animals Experiments
The experimental protocol was permitted by the Animal Ethics Committee of Harbin Medical University and conformed to the principles for the Care and Use of Laboratory Animals. Twelve male Sprague-Dawley rats (Weight 200 ± 20 g) were provided by the Laboratory Animal Centre of Harbin Medical University (Harbin, China). Each rat was fasted for 12 h before giving the drug and had free water supply even during the experiment. The S. officinalis extract was dissolved in water. A single dose of the S. officinalis extract (0.015 g/kg) was administrated to the rats. Blood (0.3 mL) was gained from the retinal venous plexus at 0, 0.083, 0.25, 0.5 1.0, 1.5, 2.0, 3.0, 4.0, 6.0, 8.0, 12.0 and 24.0 h after dosing. The plasma was immediately separated by centrifugation at 12000 rpm for 5 min at −4 °C.
4.5. Plasma Samples Preparation
Ten microliters of IS (1040.0 ng/mL) solution and 100 μL of methanol were added to 100 μL aliquot of plasma sample and vortexed for 30 s. The mixture was extracted with 3 mL ethyl acetate by being vortex-mixed for 1 min. The supernatant was separated and evaporated to dryness by N
2 blowing at 40 °C after centrifuging at 3800 rpm for 5 min. The residue was reconstituted with 100 μL of methanol, and then vortex-mixed for 2 min and filtered by a 0.22 μm nylon 66 organic membrane. This was followed by injection of 5 μL aliquot of the solution into the UHPLC-MS/MS system (Agilent Technologies, Santa Clara, CA, USA) [
36].
4.6. Method Validations
The selectivity, linearity, precision, accuracy, extraction recovery, matrix effect and stability were evaluated based on the FDA guidelines [
29].
4.6.1. Selectivity
The method of selectivity was used in the quantitative analysis of possible interfering substances in samples; the results show that this method was accurate and specific. All results demonstrate that no endogenous substances interfered with quantitative analysis.
4.6.2. Recovery and Matrix Effect
The extraction recovery of analytes was determined via comparing the peak areas of the six analytes from the QC samples with those obtained from blank plasma samples with the six analytes spiked into the post-extraction supernatant at three QC levels in six replicates. The matrix effect was evaluated through comparing the peak areas of analytes spiked after plasma extraction with those of standard samples. The extraction recovery and matrix effects of IS were also measured at one concentration.
4.6.3. Linearity and LLOQ
The calibration curves were constructed by plotting the peak area ratio versus the concentration of the six analytes and IS with a weighted (1/x2) least square linear regression using standard plasma samples. The lower limit of quantification (LLOQ) was defined as the lowest analytical concentration of the calibration curve with an acceptable precision (RSD) below 20% and accuracy (RE) within ±20%. The lower limit of detection (LLOD) was determined as the concentration of the analytes with a signal-to-noise ratio at 3 in the blank plasma.
4.6.4. Precision and Accuracy
The intra- and inter-day precision and accuracy were measured by testing the LLOQ sample and QC samples at three QC levels of six compounds in six replicates on three days in a row. The precision was determined and expressed as RSD and the accuracy as relative error (RE). The intra-day and inter-day precision and accuracy were within 15%, which is an acceptable requirement. The RSD of LLOQ samples should be within 20%.
4.6.5. Stability
The stability of six compounds in rat plasma including freeze and thaw stability (three freeze–thaw cycles at −20 °C), long-term stability (storage for 2 weeks at −80 °C), room temperature stability (storage for 4 h at ambient temperature), and post-preparation stability (storage for 12 h after sample preparation at 4 °C) was tested at three QC levels with five replicates at each level. All stability testing QC samples were determined by using the calibration curve of freshly prepared standard samples.
4.7. Plasma Pharmacokinetic Study
The maximum concentration (Cmax) and the time to attain it (Tmax) were observed directly from the measured data. The elimination rate constant (Ke) was calculated by linear regression of the terminal points in a semi-log plot of the plasma concentration against time. The elimination half-life (t1/2) was calculated using the formula t1/2 = 0.693/Ke. The area under plasma concentration–time curve (AUC0→t) to the last measurable plasma concentration (Ct) was estimated by using the linear trapezoidal rule. The area under the plasma concentration–time curve to time infinity (AUC0→∞) was calculated as: AUC0→∞ = AUC0→t + Ct/Ke. The pharmacokinetic parameters of six analytes were reckoned by non-compartmental analysis using DAS 2.0 (Mathematical Pharmacology Professional Committee of China, Shanghai, China).

 and
and 




{kind=link}
{kind=link}
{kind=link}
{kind=link}