1. Introduction
The emergence of antibiotic-resistant bacterial strains calls for new anti-bacterial strategies. One of them consists of inhibiting virulent factors in particular lectins involved in bacterial adhesion and/or biofilm formation using monovalent glycomimetics [
1] or multivalent glycoclusters. Lectins generally display multiple carbohydrate recognition domains (CRD) and high avidity ligands can be obtained through multivalent contact thanks to the glycocluster effect [
2,
3,
4,
5]. The extent of the glycoside effect depends on both the structure of the CRDs and the topology of the glycocluster.
Pseudomonas aeruginosa (PA) is a Gram-negative opportunistic bacterium involved in severe infections of the respiratory and/or urinary tracts, skin and eyes [
6]. It is one of the most prevalent nosocomial bacterial pathogens [
7,
8] and it is the major cause of cystic fibrosis patient mortality. The threat of PA infection stems from its ability to develop antibiotic resistance and to protect itself by forming a biofilm leading to chronic inflammation and eventually to death, despite aggressive antibiotic therapy. One strategy consists of targeting PA virulence factors in order to reduce its aggressiveness and/or to increase the efficacy of the host immune system for pathogen clearance. Virulence factors are molecules (secreted or surface-bound) that trigger bacterial adherence to tissue, biofilm formation or mitigate host defenses [
9].
The soluble homotetrameric fucophilic lectin LecB of PA is classified as a virulent factor and is involved in biofilm formation, host/bacteria and bacteria/bacteria interaction, cytotoxicity and inhibition of ciliated removal [
10]. It consists of four subunits of 11.7 kDa (114 amino-acids). Two calcium atoms are structurally present in the binding sites. These two calcium atoms are coordinated by the fucose upon binding, and they are responsible for the unusual sub-micromolar dissociation constant observed for a monomeric ligand [
11]. Indeed, dissociation constants (
Kd) for FucOMe are 0.43 µM for LecB from PAO1 [
12,
13], 2.2 µM for LecB from PA7 [
14], and 0.2 µM for LecB from PA14 [
12]. Furthermore,
N-fucosyl amide derivatives showed
Kd values between 1.2 to 2.1 µM [
15]. To a lesser extent, it also displays binding to
l-galactose,
d-arabinose and
d-mannose [
11].
d-galactose is not recognized. Cinnamide and sulfonamide derivatives of mannose were found to be inhibitors of LecB with a
Kd value of 18.5 and 3.3 µM respectively [
16]. More complex glycans are also bound, provided that terminal fucoses or mannoses are available. Binding to Lewis
a (Le
a) [
17], sialyl-Lewis
a [
18], antigen H [
19], 3-fucosyllactose [
17], LNFP-II, LNnFP-V, Lewis
x (Le
x) [
20], sialyl-Lewis
x [
18] and mannan [
21] have been reported. However, slight differences between different isolates were observed. Lewis
a had dissociation constants of 70, 210 and 2003 nM for LecB issued from PA14 [
12], PAO1 [
20] and PA7 [
14], respectively. Sialylation of Lewis
a seems to only slightly affect the binding to PAO1-LecB and does not affect at all the binding of PA7-LecB [
14].
Aiming at inhibiting LecB, synthetic efforts have been devoted to the synthesis of Lewis
a mimics and multivalent ligands. Indeed, it was demonstrated that the dimer αFuc-1-4GlcNAc, a truncated version of Lewis
a, also displayed a high affinity for LecB (290–310 nM) [
22]. Therefore, heterocycle α-L-fucoside ligands mimicking the dimer were synthesized and screened toward LecB. An isoxazole derivative was found to be as potent as Lewis
a [
22].
Alternatively, multivalent ligands can take advantage of the so-called glycocluster effect [
2] to reach high affinities. Various scaffolds have been studied, such as oligoethylene [
23], penta-erythritylphosphodiester [
24,
25], cyclopeptide [
26], carbohydrate-centered ligands [
25,
27,
28], peptide [
29,
30], calix [
4] arene [
31], fullerene [
32], Pillar [
5] arene [
33] and photo-switchable Janus dendrimer. [
34]. To date, the highest affinity reached was less than 30 nM using an hexadecavalent fucose cluster [
29]. To the best of our knowledge, these multivalent molecules only contain fucose. However, the synthesis of multivalent oligosaccharide ligands taking advantage of both multivalency and the high affinity of LecB for several oligosaccharides has only been described by Marotte et al. They reported the synthesis of divalent and trivalent clusters of Fuc-1-4GlcNAc and reached dissociation constants in the 100 nM range [
23]. A 2:1 LecB/ligand stoichiometry was measured by ITC suggesting that a divalent interaction occurred.
Herein, with the aim to find oligosaccharide of high affinity to LecB and then to synthesize oligosaccharide clusters of higher affinity to LecB that could compete with the natural endogenous ligands of the lectin, we developed a double screening strategy. First, 156 natural oligosaccharides exhibiting different carbohydrate compositions and degree of polymerization were qualitatively classified thanks to a competitive assay on microarray. Then, the best epitopes were introduced on different scaffolds leading to oligoglycoclusters exhibiting different topologies and valences (1, 3 or 4). To perform their screening, the oligoglycoclusters were conjugated to a DNA sequence allowing their immobilization by DNA directed immobilization (DDI) on a DNA array. The resulting glycocluster array was used to assess the affinity of the oligoglycoclusters to LecB by a direct readout of the fluorescence signal in comparison to the corresponding monovalent ligand.
2. Results and Discussion
We have previously reported a microarray-based IC
50 assay to screen the binding of immobilized fucosylated clusters to LecB using fucose as a competitor [
27]. A similar assay can be used to probe the binding of the 156 sugar candidates (see
supporting information S1). To this end, we selected a Cy3-oligonucleotide-fucosylated cluster exhibiting eight fucoses
F1 (
Figure 1). The IC
50 value of
F1 with respect to fucose, used as inhibitor, is 16 µM [
27]. This IC
50 value corresponds to the concentration of fucose required to reduce 50% of LecB interaction with the glycocluster.
F1 was immobilized on a DNA chip, thanks to its DNA sequence. The distance between two
F1 on the surface was estimated to be nearly 50 nm [
35]. Therefore, the lectin LecB can only bind to one
F1 glycocluster at the time. The binding of immobilized
F1 to LecB was competed with each of the 156 glycans at four different concentrations (0.1, 1.0, 5.0 and 15 µM). For each glycan and each concentration, the alexa 647-LecB fluorescent signal was recorded and averaged over 16 spots, leading to four-point competition histograms (
Figure 2 and
Figure S2). 100% signal (see
Figure 2) corresponded to the fluorescent signal measured with a reference assay under which no competition could occur. Glycans able to compete with
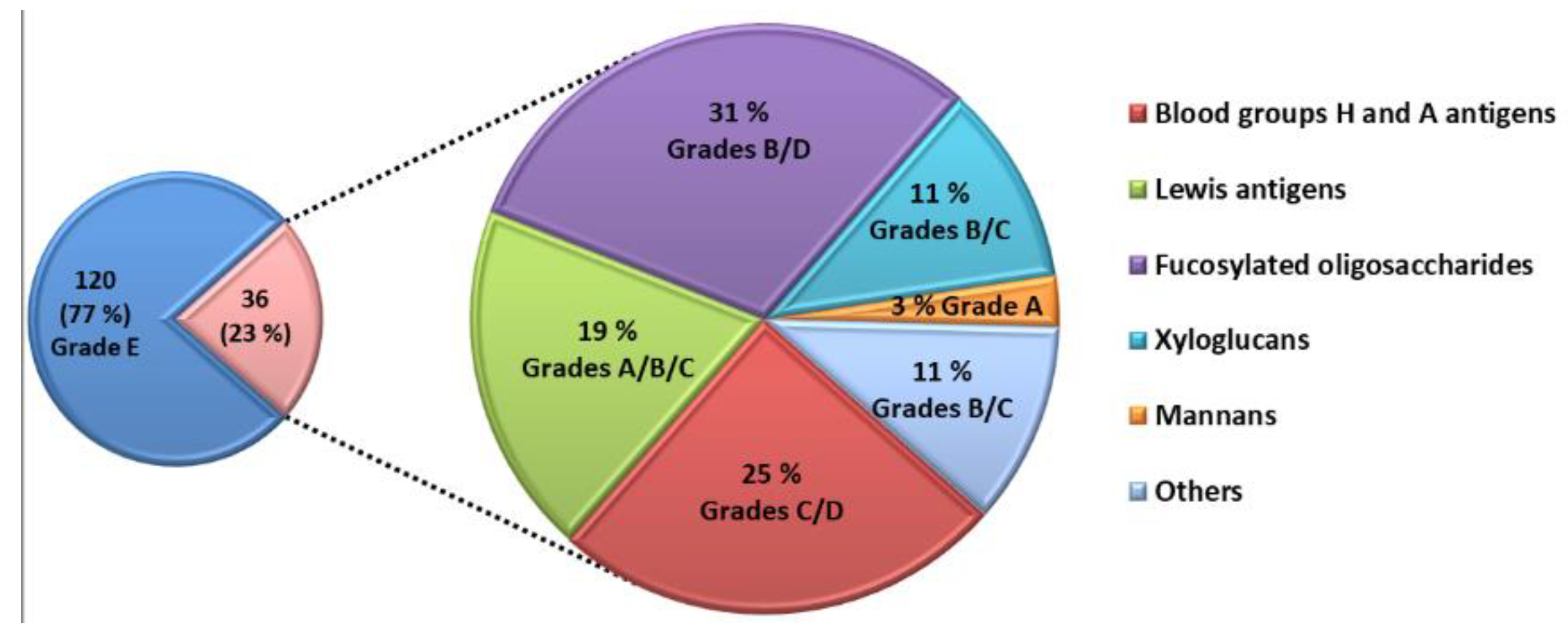
F1 to bind LecB lead to a decrease of the fluorescent signal. Therefore, for a given glycan concentration, strong binding glycans lead to stronger decrease of the fluorescent signal so to lower percentages. Accordingly, tested glycans were classified qualitatively into 5 groups (A, B, C, D and E) depending to their ability to reduce the measured fluorescent signal at the four different concentrations (
Figure 2 and
Table 1). Grade A are stronger competitor than grade B which are stronger than grade C, and so on. A Grade A was given for oligosaccharides that were able to reduce 80 to 40% of the fluorescent signal at a concentration of 0.1 µM and to reduce it by more than 90% at concentrations of 1, 5 and 15 µM. Grade B indicated a decrease of fluorescence by 70% to 90% at 1 µM and more than 90% at 5 and 15 µM. Grade C corresponded to a decrease of 50 to 70% for 1 µM, around 80% for 5 µM and more than 90% at 15 µM. D indicated a decrease of less than 50% for 1 µM and 80% at 5 and 15 µM. When no inhibition was observed for the four concentrations tested, the grade was E.
Such a screening has two main advantages: The oligosaccharides do not need to be derivatized for their immobilization and their surface densities do not have to be taken into consideration, as they remain in solution. Indeed, as underlined by several authors [
36,
37], the binding of lectins with immobilized carbohydrates depends on their intrinsic affinity, but also on their surface density. Indeed, high surface densities can result in multiple contacts and, similar to a cluster effect, can increase the measured avidity (surface cluster effect).
In our case, the surface density of
F1 cannot result in a surface cluster effect [
35] and its surface density is similar for each assay (535 nm fluorescent issued from surface-bound
F1 deviated by less than 15%). Furthermore, the competing oligosaccharides are in solution. Therefore, the classified binding is solely related to the intrinsic binding of the oligosaccharides with LecB.
The competitive assay was able to identify 36 natural glycans interacting with LecB (
Figure 3). The majority of these glycans belong to the families of A and H blood group antigens, Lewis antigens and fucosylated glycans. They were produced by bacterial fermentation. Five glycans interacting with LecB are extracted from biomass such as xyloglucans and mannans.
Table S2 (Supporting Information) gives the structure and the grade obtained for each glycan interacting with LecB. Several parameters seem to drive this interaction. All glycans, except the mannan (Id. 151), contain at least one terminal fucose residue at their non-reducing end or on their side chain, which allows the interaction with LecB. The interaction of the mannan polysaccharide with LecB is mediated by
d-mannose on its side chain as suggested by Zinger-Yosovich [
21]. Indeed, LecB is able to interact with both monosaccharide,
l-fucose and
d-mannose. Despite the fact that the interaction of LecB with the
d-mannose was described to be 25 times weaker than the one with the
l-fucose, the mannan obtained grade A [
17]. Furthermore, oligosaccharides containing one fucose on their side chain with an α1-2 glycosidic bond obtained the grade of D or C, the ones with an α1-3glycosidic bond, the grade of C or B, and finally, those with an α1-4 glycosidic bond, the grade of A. Therefore, LecB seems to preferentially bind Fucα1-4 > Fucα1-3 > Fucα1-2 oligosaccharides. Then, the presence of a Fucα1-2 close to the non-reducing end of the oligosaccharide seems to disturb the interaction between Fucα1-3/4 and LecB. For example, Galβ1-3(Fucα1-4)GlcNAcβ1-3Gal (Grade A, Id.30) interacted more strongly with LecB than Fucα1-2Galβ1-3(Fucα1-4)GlcNAcβ1-3Gal (Grade C, Id. 32), despite the fact that two fucose motifs are present in the latter structure. Finally, as described in the literature, LecB preferentially interacts with Lewis
a antigens (Grade A, Id. 30 and 31) and 3-Fucosyllactose (3FL) oligosaccharides (Grade B, Id. 33 to 37) [
18,
38]. The co-cristal LecB/Lewis
a (Galβ1-3(Fucα1-4)GlcNAc) showed that all monosaccharides of Lewis
a interact with the surface of LecB. The authors have shown that, in addition to a network of hydrogen bonds between fucose and LecB, the GlcNAc residue makes an hydrogen bond between its
O-6 atom and the Asp96 of LecB, and that both GlcNAc and Gal residues establish additional interactions with LecB through the bridging of water molecules [
20]. In comparison to Lewis
a (Grade A, Id. 30), Lewis
x antigen displays a weaker binding for LecB (Grade C, Id. 26). Perret et al. have also shown by molecular simulation that in the case of Lewis
x antigen,
N-acetyl moiety of the glucose was at the same position as the
O-6 atom of GlcNAc of Lewis
a antigen, which seems to hinder the additional hydrogen bond with Asp96 of LecB [
20]. The 3FL oligosaccharide (Id. 33) differs from Lewis
x antigen by the absence of the
N-acetyl modification on the glucose, and a stronger affinity for LecB is observed. It suggests that steric hindrance is lower for 3FL oligosaccharide than for Lewis
x antigen.
Five oligosaccharides with various grades were selected for the synthesis of oligoglycoclusters and to address the effect of clustering on their binding with LecB (
Table 1). For each family, the oligosaccharide with a lower polymerization degree was chosen.
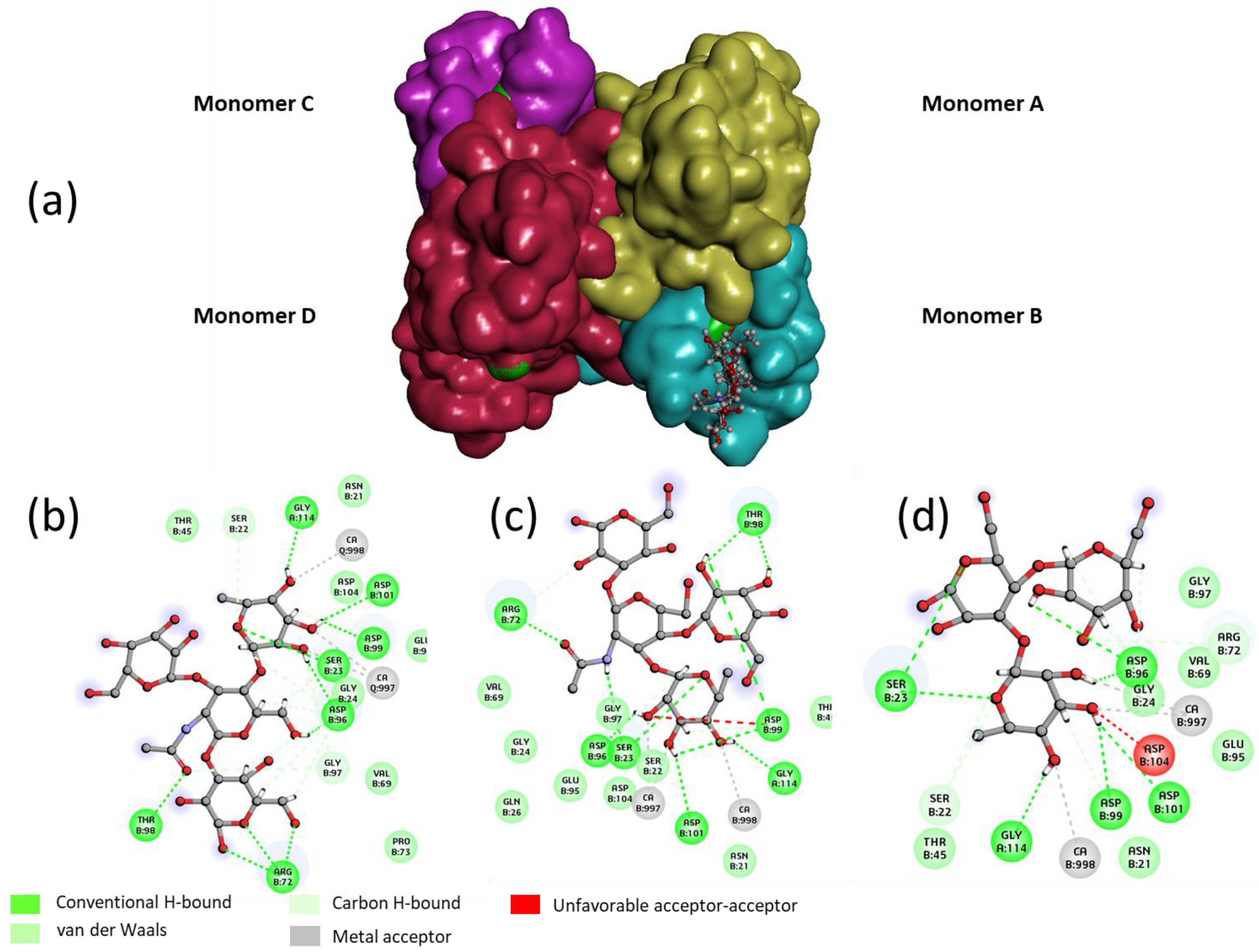
In order to rationalize the binding of Lewis
a tetrasaccharide, Lewis
x tetrasaccharide and 3-Fucosyl lactose (as examples for each grade), a docking study was performed with the tetrameric LecB (
Figure 4). An empirical calculation of the potential energy of interaction (ΔE) between LecB and Lewis
a (Id. 30), Lewis
x (id 26) and 3-Fucosyllactose (Id. 33) [
25] gave a value of −98, −81 and −67 kcal/mol respectively. Similarly to the screening on microarray and to the published results of Perret [
20], Lewis
a was found as the most potent ligand for LecB.
For the three oligosaccharides, the coordination involves the OH at position 2, 3 and 4 of the fucose as hydrogen bond (hb) donors and the oxygen of the ring as an hb acceptor (see
Table 2). The O
1, O
ring and O
6 of the galactose residue of the non-reducing are involved in the binding as hb acceptors. In the case of the Lewis
x and 3FL, only the OH at the 2 position seems to be involved in an interaction with the lectin, suggesting that the stabilization of the complex between the Lewis
a/LecB versus Lewis
x/LecB or 3FL/LecB is mainly due to the three contributions of the non-reducing galactose of Lewis
a (
Table 2 and
Figure 4).
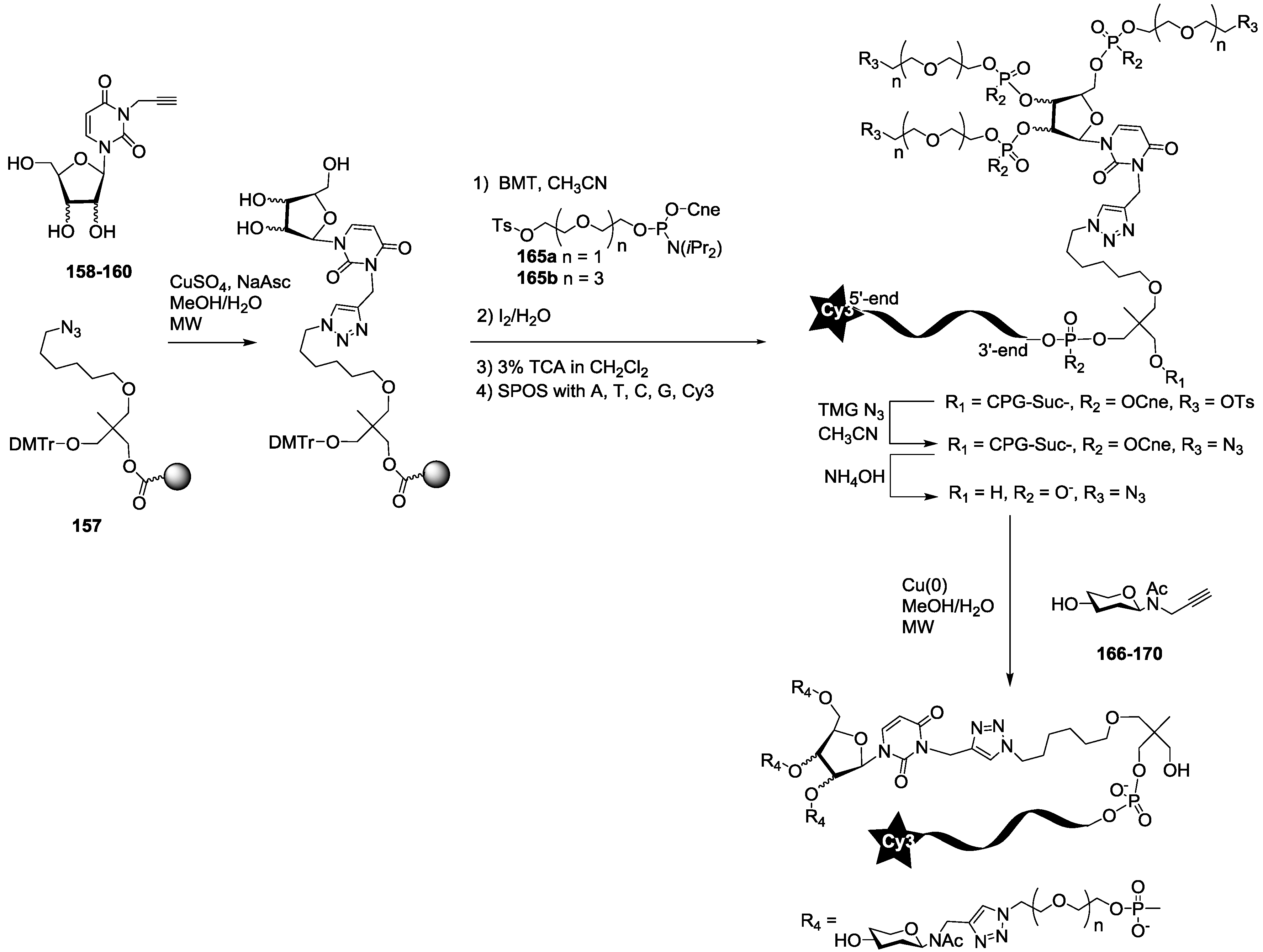
For the synthesis of oligoglycoclusters, six different centers were used as scaffolds: Three pentoses (arabinose, xylose and ribose) and three hexoses (glucose, galactose and mannose). These different cores allowed the effect of valence (trivalent for Ara, Xylo and Ribo vs. tetravalent for Glc, Gal and Man) and topology to be explored. Furthermore, two linkers, corresponding to diethyleneglycol and tetraethyleneglycol, were used to study the impact of the length between the core and the oligosaccharides on the affinity to LecB. The oligosaccharide cluster-oligonucleotide conjugates were synthesized by a combination of phosphoramidite chemistry and Cu(I) catalyzed azide alkyne cycloaddition (CuAAC) [
39]. The oligoglycoclusters were tagged with a Cy3-DNA sequence and immobilize on a DNA chip by duplex formation with their immobilized complementary sequence. Their affinity for LecB was then evaluated.
For this purpose, we first prepared the different building blocks corresponding to an azide solid support
157 [
40], trihydroxylated alkyne scaffolds prepared from arabino-, ribo- and xylo- furanosyl uracils (
158–
160) [
28], tetrahydroxylated scaffolds corresponding to propargyl-galactoside, glucoside and mannoside (
161–
163) [
27] (
Figure 5), two different linkers corresponding to tosyl-di- or tetra-ethyleneglycol phosphoramidites (
165a–
b) (
Scheme 1, see
supporting information S3 for NMR characterization) and the five
N-acetyl-propargyl oligosaccharides
166–
170 selected from the first screening (
Scheme 2, see
supporting information S4 for NMR and MALDI-TOF characterizations). Both tosyl-linkers (
165a–
b) were obtained by phosphitylation of di- and tetra-ethyleneglycols (
164a–
b). The five selected oligosaccharides were converted as alkyne derivatives by means of a treatment with an excess of propargylamine in methanol to afford the resulting glycosylamines. Then the nitrogen was
N-acetylated using acetic anhydride to avoid any further hydrolysis [
41].
The synthesis of the polyazide scaffold oligonucleotide conjugates was performed on a solid support allowing their efficient, automated and rapid synthesis at about 200 to 300 nmol scale. Finally, the conjugation with the five oligosaccharides
166–
170 was performed in solution allowing a divergent strategy, since only 20 nmol of scaffold were used for each. Thus, the synthesis required six steps (
Scheme 3 and
Scheme 4). Firstly, polyhydroxylated scaffolds (
2–
7) were immobilized on the azide solid support
157 by CuAAC. Secondly, each free hydroxyl was phosphorylated with tosyl-di- or tetra-ethylene glycol phosphoramidites (
165a–
b). Thirdly, the DNA sequence was elongated and labelled with Cy3 fluorescent dye. Fourthly, tosyl groups were substituted by azide using tetramethylguanidine azide. Finally, an ammonia treatment afforded the Cy3-oligonucleotides bearing a tri- or tetra-azidolinker scaffold. After HPLC purification on a C
18 reverse-phase column, each construction was aliquoted to 20 nmol. The final CuAAC was carried out on a 20 nmol scale with the different
N-acetyl-propargyl oligosaccharides
166–
170 affording the expected oligosaccharide cluster-Cy3-oligonucleotide conjugates
G1–
G12.
Each of them was analyzed by HPLC and characterized by MALDI-TOF mass spectrometry (See
Supporting Information S5). As a result, 12 oligoglycocluster-Cy3-oligonucleotides exhibiting three or four oligoglycosides with di- or tetra-ethyleneglycol arms were obtained for Lewis
a tetrasaccharide, Lewis
b tetrasaccharide, Lewis
x tetrasaccharide and 3FL, while for sialyl-Lewis
x pentasaccharide only two oligoglycocluster-oligonucleotides built from ribose and mannose core with tetra-ethyleneglycol arms were synthesized.
In order to evaluate the effect of multivalence of the oligosaccharide clusters, we also conjugated each oligosaccharide
166–
170 only once on an azide Cy3-oligonucleotide (
Scheme 5). The Cy3 oligonucleotide was synthesized from the azide solid support
157 by standard phosphoramidite chemistry, and the resulting monoazide oligonucleotide was conjugated in solution as described above with the different
N-acetyl-propargyl oligosaccharides
166–
170 affording the mono-oligoglyco-oligonucleotides (M-series).
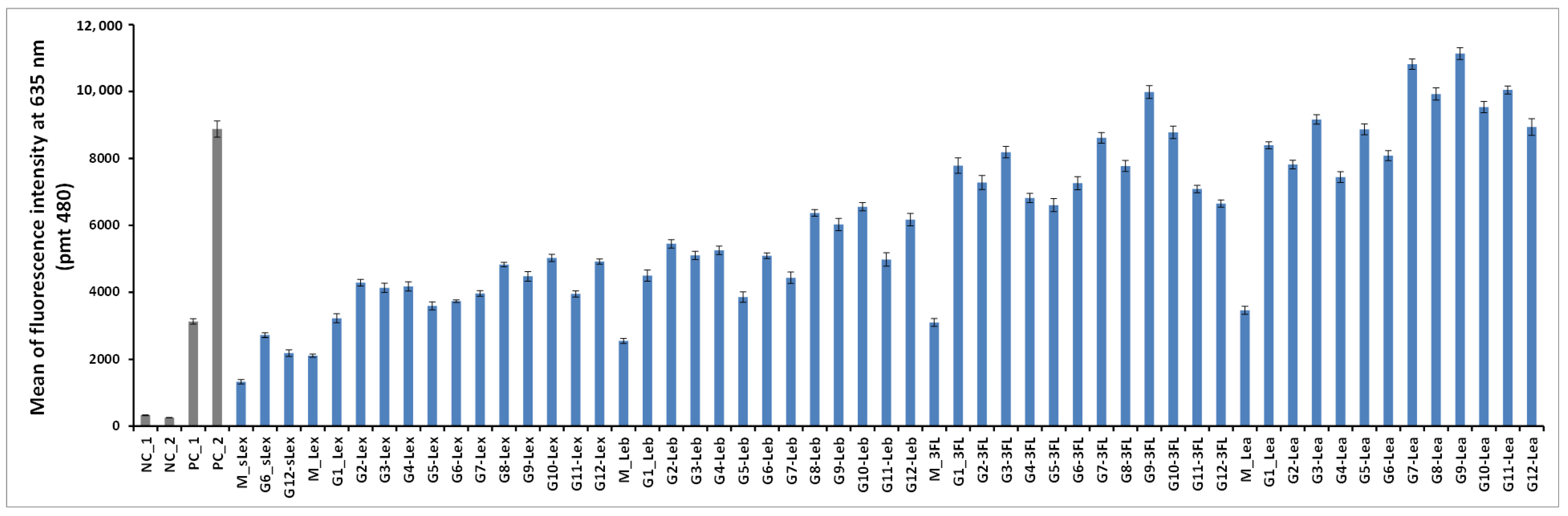
The 50 oligoglycoclusters were tested for their binding toward LecB (
Figure 6) displaying Lewis
a tetrasaccharide (G1-Le
a to G12-Le
a), 3-Fucosyllactose (G1-3FL to G12-3FL), Lewis
x tetrasaccharide (G1-Le
x to G12-Le
x), Lewis
b tetrasaccharide (G1-Le
b to G12-Le
b) and sialyl-Lewis
x pentasaccharide (G6-sLe
x and G12-sLe
x), as well as the five monovalent oligosaccharides (M-Le
a, M-3FL, M-Le
x, M-Le
b and M-sLe
x). For a given carbohydrate, oligoglycoclusters varied by their linkers (EG
2 or EG
4), their valences (3 or 4) and their topologies.
For the latest parameter, the topology of oligoglycoclusters was varied using different carbohydrate-centered scaffolds (i.e. pentofuranose: Arabinose, xylose, ribose; hexapyranose: Glucose, galactose, and mannose). Two negative controls, NC_1 and NC_2, corresponded respectively to a single oligonucleotide immobilized through an amide bond and a
d-galactocluster immobilized by DDI. Both negative controls exhibited a very low alexa 647 fluorescent signal (below 350 a.u.), illustrating the very low non-specific adsorption of LecB on the surface and its specificity towards fucose moieties. Two positive controls, PC_1 and PC_2, corresponding to a monofucoside (PC_1) and a mannose-centered tetravalent fucoside (PC_2) were used. (
Figure 7). Oligoglycocluster immobilization deviated by less than 18%.
According to the Alexa 647 fluorescence signal obtained with monovalent constructions (M-oligosaccharide series), the affinity of LecB increases as follows Le
x < Le
x < Le
b < 3FL < Le
a. The same trend was observed with the oligoglycoclusters (
Figure 6). This ranking corresponds to the one observed with the free oligosaccharides, confirming the relevance of the first screening by competition (
Table S2, in Supporting Information).
In all cases, clustering led to an increase of binding (M < G1–12). On average, tetravalent clusters (
G7 to
G12) gave a slightly higher Alexa 647 fluorescent signal than trivalent clusters (
G1 to
G6). Furthermore, for each oligosaccharide, the best tetravalent cluster was higher than the best trivalent cluster. However, for each oligosaccharide, the best trivalent cluster was better than some tetravalent clusters. This result shows that the topology seems to be more important than the valence on the binding to LecB, as previously observed with fucoclusters [
25].
Concerning the influence of the length of the linker, our study showed that a shorter linker (EG2) improves the binding of LecB for oligoglycoclusters bearing Lea tetrasaccharide and 3FL (G1, G3, G5, G7, G9, G11), while the opposite was observed for oligoglycoclusters bearing Lex and Leb tetrasaccharides, respectively. In the case of pentose core (G1 to G6), the binding to LecB is slightly influenced by the type of the furanoside core.
The data shows that the combination of xylose with an EG2 linker (G3) core is preferred, while with the EG4 linker, the arabinose core leads to a higher binding-(G2). For hexose core (G7 to G12), whatever the linker, the galactoside core (G9–G10) gave the highest fluorescent signals, while the mannoside core (G11–G12) leads to mainly the lowest ones. Strong differences of fluorescent signal were observed between the hexose cores for 3FL and Lea tetrasaccharide.
Previous results on the binding of hexose-centered glycoclusters made with four L-fucose showed that a mannose core was preferred in comparison with the galactose and glucose cores [
27]. Therefore, the interaction of a lectin with a set of oligoglycoclusters convolutes the core and the carbohydrate residue, leading to an avidity profile that can be viewed as a fingerprint of the lectin.
The three best oligosaccharide clusters targeting LecB were found for G9-Lea, G7-Lea and G9-3FL, all built with an EG2 linker and galactoside core for G9 and glucoside core for G7.
In order to ensure the effect of the carbohydrate epitope and the core, the
Kd values of the
G1,
G3,
G7 and
G9 series were measured for the two best oligosaccharides, such as Le
a and 3FL. As expected, all the oligoglycoclusters exhibited a high affinity for LecB. The differences between the
Kd values of each glycocluster were not significant, around 50 ± 7 nM for Le
a-clusters and around 60 ± 11 nM for 3FL-clusters. This data shows a cluster effect, whatever the topology of the oligoglycoclusters. So it was not possible to draw further quantitative conclusions according to the shape of the glycoclusters. However, these glycoclusters exhibit a better affinity for LecB than the previously synthesized mannose-centered fucocluster PC_2 (
Kd 153 nM) [
25].
Finally, aiming at getting insight on the possible multiple contact between the lectin LecB and the following glycoclusters
G7-3FL,
G7-Lea,
G9-3FL and
G9-Lea, have been investigated using docking techniques and molecular simulations (namely Monte Carlo computations), as previously described [
25].
An empirical potential energy of interaction ΔE was calculated for the six different possible combinations for the simultaneous binding of the Lea or 3FL epitopes displayed at the tip of two branches attached at the pyranoside core. The mean ΔE (kcal/mol) value among the six is given for each glycocluster and ranked as follow: G9-Lea: −262.9, G7-Lea: −253.7, G9-3FL: −224.6 and G7-3FL: −205.6.
For the "best" glycocluster
G9-Lea, a careful inspection of atomic interactions with LecB was performed and is shown in
Figure 8. The simultaneous contact of both binding sites of monomer A and D seems to be energetically possible. However, it seems that the two loops, V69-P73 and D96-D99, generate geometrical hindrance and these loops have to be avoided by the two interacting branches. According to our results, longer linkers, such as EG4 (
G10-Lewisa), did not improve the binding (see
Figure 6).
3. Materials and Methods
3.1. Synthesis of β-NAc-Propargyl-Oligoglycoside from Free Oligosaccharides
Starting oligosaccharides were obtained by biofermentation or extracted from biomass (Elicityl catalog:
www.elicityl-oligotech.com). Other reagents (propargylamine, Ac
2O, solvents) were purchased from Thermo Fisher Scientific (Illkirch, France).
The starting free oligosaccharides were converted into oligoglycosylamines by condensation with propargylamine; and in a second step, made non-hydrolysable by selective
N-acylation, as previously described [
41]. Briefly, each starting oligosaccharide (1 eq) was suspended in a pure propargylamine (9 eq.)/methanol mixture. The mixture was stirred at 43 °C for 16 h, to obtain a transient oligoglycosylamine. After completion of the reaction, monitored by thin-layer chromatography, the excess of propargylamine was removed by co-evaporation with MeOH/toluene (1:1,
v/v). The residual solid was then suspended in a solution of MeOH and Ac
2O (2:1,
v/v, 1mL) that corresponds to a large excess of Ac
2O (10 to 30 eq.). The mixture was stirred at room temperature until total consumption of oligoglycosylamine (checked by TLC) (around 2 h). Next, the acetylated oligoglycosylamine was recovered by co-evaporation with toluene/MeOH (1:1,
v/v), solubilized in water, and the solution was concentrated and freeze-dried. To avoid any undesired
O-acetylation, the product was treated by the following additional step. It was dissolved in anhydrous MeOH and MeONa was added to obtain a final MeONa concentration of 0.03 M. The mixture was left for 2 h at room temperature. Water was added, and the solution was brought to pH 7 with aqueous HCl. Finally, the mixture was concentrated. Finally, the final β-
NAc-propargyl-oligoglycoside with a purity of 95% was obtained by C
18 reverse-phase chromatography. The structure was validated by
1H-NMR (Avance III 400 MHz, Bruker, Billerica, MA, USA) and MALDI-TOF (Autoflex speed, Bruker, Billerica, MA, USA) analysis.
3.2. Cy3-Oligonucleotide Oligoglycoclusters Synthesis
Azide solid support
157 [
39,
40],
N3-propargyl pentofuranosyl uracils
158–160 [28] and propargyl-hexopyranoside
161–
163 [
27] were previously synthesized. Tosylated diethyleneglycol
164a [43] (3.0 g, 11.54 mmol) was co-evaporated three times with dry acetonitrile and then dissolved in dry CH
2Cl
2 (20 mL). A few molecular beads of molecular sieve (3Å) were added and diisopropylethylamine (DIEA) (3 mL, 17.3 mmol) under argon. After 15 min, 2-cyanoethyl-
N,N-diisopropylchlorophosphoramidite (3 g, 12.7 mmol) was added and the solution was stirred at room temperature for 3 h. Then water was added (1 mL) and after 10 min diluted with 200 mL of CH
2Cl
2 and washed with a saturated solution of NaHCO
3 (2 × 200 mL), dried with Na
2SO
4 and concentrated. The crude product was eluted from a column of silica gel (110 g) with cyclohexane/EtOAc (9:1 to 7:3, containing 4% of Et
3N) to afford tosylated diethyleneglycol phosphoramidite
165a, 4.30 g, 81%.
1H- NMR (CDCl
3, 300 MHz): δ 1.17 (t,
J = 6.8 Hz, 12H, 4× CH
3); 2.45 (s, 3H, CH
3); 2.64 (t,
J = 6.0 Hz, 2H, CH
2CN); 3.56 (m, 2H, 2× CHMe
2); 3.59 (m, 2H, POCH
2); 3.89 (m, 6H, OCH
2 ); 4.15 (t,
J = 4.8 Hz, 2H, SO
3CH
2); 7.35 (d, 2H,
J = 8.1 Hz, Ar); 7.79 (d, 2H,
J = 8.1 Hz, Ar).
13C-NMR (CDCl
3, 75 MHz): δ.15.08 (
CH
2CN), 15.17 (CH
3Ar), 19.35, 19.40, 19.44 (4×
CH
3CH), 37.80 and 37.93 (
CHMe
2), 53.17 and 53.41 (PO
CH
2CH
2CN), 57.22 and 57.45 (C5, PO
CH
2CH
2), 63.42 (C7), 64.08 (C8), 66.08 and 66.18 (C6, POCH
2CH
2), 112.6 (CN), 122.7 and 124.6 (ArH), 127.7 (Ar-S), 139.6 (
Ar-C).
31P-NMR (CDCl
3, 121 MHz): δ 148.65. HRMS (ESI/Q-TOF): Calcd. for C
20H
34N
2O
6PS 461.1875 [M + H]
+; found 461.1877.
The same protocol was applied to tosylated tetraethyleneglycol
164b [44] (3.3 g, 9.48 mmol) affording 4.04 g (78%%) of phosphoramidite derivative
165b.
1H-NMR (CDCl
3, 300 MHz): δ 1.16 (dd,
J = 4.1 and 6.6 Hz, 12H, 4× CH
3); 2.43 (s, 3H, CH
3); 2.63 (t, 2H,
J = 6.4 Hz, CH
2CN); 3.52 (m, 2H, 2× CHMe
2); 3.72 (m, 2H, POCH
2); 3.88 (m, 14H, OCH
2 ); 4.14 (t,
J = 4.8 Hz, 2H, SO
3CH
2); 7.32 (d, 2H,
J = 8.1 Hz, Ar); 7.77 (d, 2H,
J = 8.1 Hz, Ar).
13C-NMR (CDCl
3, 75 MHz): δ.22.26 and 22.34 (
CH
2CN), 23.6 (CH3Ar), 26.50, 26.57, 26.60, 26.67 (4×
CH
3CH), 44.95 and 45.12 (
CHMe
2), 60.37 and 60.61 (PO
CH
2CH
2CN), 64.46 and 64.69 (C5, PO
CH
2CH
2), 70.65, 71.25, 72.50, 72.55, 72.64, 72.72 (C7-C12 OCH
2), 73.15 and 73.24, (C6, POCH
2CH
2), 119.8 (CN), 130.0 and 131.8 (ArH), 135.0 (Ar-S), 146.8 (
Ar-C).
31P-NMR (CDCl
3, 121 MHz): δ 148.59. HRMS (ESI/Q-TOF): Calcd. for C
24H
42N
2O
8PS 549.2400 [M + H]
+; found 549.2393.
3.3. 3’-Oligoglycoside-5’-Cy3-Oligonucleotide Synthesis
The oligonucleotide synthesis on solid support was performed on a 381A or 394 DNA synthesizer from Applied Biosystems (Foster City, CA, USA). Reactions under microwave activation were performed on an Monowave 300 system (Anton Paar, Graz, Austria). Solutions of Cap A, Cap B and iodide were purchased from Link Technologies as well as the commercial solid supports. Cap A: Acetic anhydride/pyridine/THF, 1:1:8, v/v/v; Cap B: 10% N-methylimidazole in THF; Oxidizer solution: 0.1 M iodide in THF/pyridine/H2O, 78:20:2, v/v/v. Detritylation solution: 3% trichloroacetic acid (TCA) in CH2Cl2 and dry CH3CN for DNA synthesis were purchased from Biosolve (Dieuze, France). Cy3-amidite was purchased from GE Healthcare. All oligoglycooligonucleotides were purified and analyzed by C18 reversed-phase HPLC (Macherey-Nagel, Nucleodur 100-3 C18 ec, 4.6 × 75 mm, 3 µM or Nucleodur 100-7 C18 ec, 8 × 125 mm, 7 µm) on an Ultimate 3000 system (Dionex, Sunnyvale, CA, USA) with a Reodyne (Rohnert Park, CA, USA) injector and a UV DAD 3000 detector. Oligonucleotides were dosed by UV-Vis spectrophotometry at 550 nm on a 300 Bio UV-Vis spectrophotometer (Varian Cary, Victoria, Australia).
3.4. General Procedure for Immobilization of Polyol Scaffolds (158–163) on an Azide Solid Support (157) by Cu(I)-Catalyzed Alkyne Azide 1,3-Dipolar Cycloaddition
An aqueous solution of propargyl derivatives 158–163 (10 µmol in 100 μL H2O), freshly prepared degassed aqueous solutions of CuSO4 (40 mM, 8 µmol, 50 μL) and sodium ascorbate (100 mM, 40 µmol, 100 μL), degassed water (400 μL) and MeOH (350 μL) were added to 5 µmol of azide solid support 157. The resulting mixture in a sealed tube G10 was heated to 60 °C for 60 min, stirring at 400 r/min, using a microwave synthesizer. The temperature was monitored with an internal infrared probe. The solution was removed, and CPG beads were washed with H2O (3 × 2 mL), MeOH (3 × 2 mL) and CH2Cl2 (3 × 2 mL), and dried affording the solid-supported polyol scaffolds.
3.5. Introduction of Linkers 165a–b, Oligonucleotide Synthesis and Cy3 Labeling
Introduction of tosyl linkers: Solid-supported polyol scaffolds (1 µmol) were treated, on a DNA synthesizer, with phosphoramidites 165a or 165b using coupling, oxidation and capping steps. For the coupling step, benzylmercaptotetrazole (BMT) was used as an activator (0.3 M in anhydrous CH3CN) and phosphoramidites 165a or 165b (0.15 M in anhydrous CH3CN) were introduced four times with a 180 s coupling time. Oxidation was performed with oxidizer solution for 15 s. Capping was applied using CapA + CapB for 120 s.
Then, the elongation of DNA sequences was carried out using a standard synthesis cycle at the 1 µmol scale by phosphoramidite chemistry. For the coupling step, BMT and commercially available nucleoside phosphoramidites (0.1 M in anhydrous CH3CN) were introduced with a 20 s coupling time and Cy3 amidite (0.06 M in anhydrous CH3CN) with a 180 s coupling time. The capping step was performed with CapA + CapB for 15 s. Each oxidation was performed with oxidizer solution for 15 s. Detritylation was performed with 3% TCA in CH2Cl2 for 65 s.
3.6. General Procedure for Azidation
The solid-supported oligonucleotides bearing the tri—or tetra-tosyl scaffold (1 µmol) were treated with a solution of tetramethylguanidinuim azide (TMG-N3) (0.4 M in CH3CN, 1 mL) for 2 h at 65 °C. The beads were washed with acetonitrile (5 × 2 mL) and dried by flushing with argon.
3.7. General Procedure for Deprotection
The CPG beads bearing modified oligonucleotides with Cy3 and azide linkers were transferred to a 4 mL screw-top vial and treated with 2 mL of concentrated aqueous ammonia overnight at rt. The supernatants were withdrawn and evaporated, affording crude oligonucleotide mono-, tri- or tetra-azide conjugates.
3.8. Purification
Each construction was purified on a C18 reverse-phase column. The pure oligonucleotide azide derivatives were aliquoted to 20 nmol for subsequent conjugation with oligosaccharides 166–170.
3.9. General Procedure for CuAAC in Solution
Method A: Mono-conjugation. In a G4 microwave tube from Anton-Paar, Oligonucleotide-hexylazide (20 nmol in 20 µL H2O) oligosaccharide 166–170 (40 nmol in 40 µL H2O), ~0.1 mg of copper nanopowder were added and the mixture was sonicated for 2 s and then stirred at 65 °C with microwave assistance for 90 min.
Method B: Tri-conjugation. In a microwave tube G4 from Anton-Paar, Oligonucleotide-pentafuranosyl triazide (20 nmol in 20 µL H2O) oligosaccharide 166–169 (100 nmol in 100 µL H2O), ~0.1 mg of copper nanopowder were added and the mixture was sonicated for 2 s and then stirred at 65 °C under microwave assistance for 90 min.
Method C: Tri-conjugation. In a G4 microwave tube from Anton-Paar, Oligonucleotide-pentafuranosyl triazide (20 nmol in 20 µL H2O), oligosaccharide 166–169 (100 nmol in 50 µL H2O), THPTA (60 nmol, 3 µL) and ~0.1 mg of copper nanopowder were added and the mixture was sonicated for 2 s and then stirred at 65 °C under microwave assistance for 60 min.
Method D: Tetra-conjugation. In a G4 microwave tube from Anton-Paar, Oligonucleotide-hexapyranosyl tetraazide (20 nmol in 20 µL H2O), oligosaccharide 166–169 (140 nmol in 140 µL H2O) and ~0.1 mg of copper nanopowder were added and the mixture was sonicated for 2 s and then stirred at 65 °C under microwave assistance for 90 min.
Method E: Tri-conjugation. In a G4 microwave tube from Anton-Paar, Oligonucleotide- hexapyranosyl tetraazide (20 nmol in 20 µL H2O), oligosaccharide 166–169 (100 nmol in 50 µL H2O), THPTA (60 nmol, 3 µL) and ~0.1 mg of copper nanopowder were added and the mixture was sonicated for 2 s and then stirred at 65 °C under microwave assistance for 60 min.
Method F: sLex Tri-conjugation. In a G4 microwave tube from Anton-Paar, Oligonucleotide-pentafuranosyl triazide (20 nmol in 20 µL H2O), oligosaccharide 170 (100 nmol in 100 µL H2O), 0.4 M TEAAc buffer pH 7.5 (20 µL) and ~0.1 mg of copper nanopowder were added and the mixture was sonicated for 2 s and then stirred at 65 °C under microwave assistance for 120 min.
Method G: sLex Tetra-conjugation. In a G4 microwave tube from Anton-Paar, Oligonucleotide-hexapyranosyl tetraazide (20 nmol in 20 µL H2O), oligosaccharide 170 (140 nmol in 140 µL H2O), 0.4 M TEAAc buffer pH 7.5 (20 µL) and ~0.1 mg of copper nanopowder were added and the mixture was sonicated for 2 s and then stirred at 65 °C under microwave assistance for 120 min.
3.10. General Work-up and Purification
The crude solutions were centrifuged and the supernatant was directly purified by C18 reverse-phase HPLC. The products were co-evaporated with water, and then lyophilized.
MALDI-TOF mass spectra were recorded on a Voyager DE mass spectrometer (Applied Biosystems, Foster City, CA, USA) equipped with a nitrogen laser. MALDI conditions were: accelerating voltage 24,000 V; guide wire 0.05% of the accelerating voltage; grid voltage 94% of the accelerating voltage; delay extraction time 500 ns. 1 µL of sample was mixed with 5 µL of a saturated solution of HPA in acetonitrile/water (1:1, v/v) containing 10% of ammonium citrate and a few beads of DOWEX 50W-X8 ammonium sulfonic acid resin were added. Then, 1 µL of the mixture was placed on a plate and dried at room temperature and pressure.
3.11. General Procedure for Microarray Production
All chemicals were provided by Sigma-Aldrich (Saint Quentin Fallavier, France). When they are purchased from other providers, their origin is then specified.
3.12. Microstructured Glass Slide Fabrication
Microarrays were made from Nexterion glass D slides (Schott). They were microstructured with 40 square wells (3 mm width, 102 ± 1 µm depth, with 4.5 mm spacings between each well). The slide microstructuring was performed using xurography and oxide-buffered etch (BOE): (Ammonium fluoride/hydrofluoric acid mixture)/(37% hydrochloric acid)/(Ultra-Pure water, 18.2 MΩ); 1:2:2 (v/v/v).
The slides were washed with a piranha mixture (30% H2O2/ 97% H2SO4; 1:2 (v/v)) for 30 min in an ultrasound bath and rinsed with water (3 × 5 min). A structured vinyl adhesive mask (40 squares: side 3 mm, spacing 4.5 mm) was stuck on each slide and slides were incubated in the BOE mixture for 2 h at room temperature under agitation. Then, the slides were rinsed with water (3 × 5 min) and the masks were removed from the slides.
3.13. Microarray Silanization
The microstructured slides were functionalized by silanization using a gas phase protocol previously described [
45]. Briefly, the microstructured slides were cleaned with a piranha solution (30 min), rinsed and heated at 150 °C for 2 h under vacuum (10
−1 mbar). After introduction of the
tert-butyl-11-(dimethylamino)silylundecanoate (TDSUM), it was vaporized under reduced pressure and allowed to react for 12 h at pressure ranging between 4 × 10
−1 and 10
−1 mbar and the temperature was set at 145 °C. The slides were washed with tetrahydrofuran (THF) and then water (10 min, under ultrasound). The
tert butyl ester functionalized slides were then converted into the corresponding acid by incubation for 7 h in glacial formic acid at room temperature. After washing with dichloromethane and water (10 min, under ultrasound), acid functions were activated for amine coupling using
N-hydroxysuccinimide (NHS, 0.1 M) and
N,N’-diisopropylcarbodiimide (0.1 M) in dry THF overnight at room temperature. The slides were finally rinsed with THF and dichloromethane (10 min, under ultrasound).
3.14. Amino-Modified Oligonucleotides Immobilization
Two amino-modified oligonucleotides (sequence c1 and c2) (Eurogentec, Seraing, Belgium) were used. Each oligonucleotide was prepared at 25 µM in phosphate-buffered saline (PBS) 10× (pH 8.5) and spotted using the sciFLEXARRAYER s3 piezo-electric system (Scienion, Berlin, Germany). Thirty two spots (16 spots per amino-modified oligonucleotide) of 0.3 nL were spotted at the bottom of each well of the microarray. The substitution reaction between the amino-modified oligonucleotides and NHS-activated microarray surface was performed for 3 h under water-saturated atmosphere and overnight in normal atmosphere to allow for the gentle drying of the spots. To remove the excess of oligonucleotides, the slides were washed for 30 min at 70 °C in sodium dodecyl sulfate (SDS) 0.1% and rinsed with water.
To avoid nonspecific adsorption, the slides were incubated for 2 h with bovine serum albumin (BSA) at 4% in PBS 1× (pH 7.4). Then, the slides were washed in 0.05% PBS 1×-Tween20 (3 × 3 min), in PBS 1× (3 × 3 min) and rinsed with water. BSA blocking was repeated following the same procedure, after the oligoglycocluster immobilization.
3.15. Oligoglycocluster Immobilization
Cy3-oligonucleotide oligoglycoclusters
F1 [
46] (
Figure 1) exhibiting a complementary sequence of sequence c1 and c2 respectively were prepared at 1 µM in PBS 1× (pH 7.4). In each microwell, 1.5 µL of the oligoglycocluster mixture was incubated for DNA-directed immobilization (DDI) for 3 h at 37 °C in a water-saturated atmosphere. The slides were washed with saline sodium citrate (SSC) 2× SDS 0.1% at 51 °C for 1 min, SSC 2× for 5 min and rinsed with water. Hence, in each microwell, both oligoglycoclusters were immobilized with 16 repetition spots.
3.16. General Procedure for the Screening of a Library of Glycans
A library of 156 sugars extracted from biomass or produced by bacterial fermentation (see
Supporting Information) issued from the OligoTech
® (Elicityl) was screened in a competitive assay. The binding of the 156 carbohydrates was screened toward the lectin LecB (PAO1, Elicityl). Lectin was labeled as previously described [
47] using the Alexa Fluor
® 647 Microscale Protein Labelling Kit (Invitrogen).
On each slide, nine oligosaccharides and α-l-fucose (as a control) were tested. The inhibitory effect of each oligosaccharide was determined at four concentrations 0.1, 1.0, 5.0 and 15 µM and at 0.5, 2.5, 5.0 and 7.5 mg/L for polymers: Aliginate, galacturonan, galactomannan, glucomannan, mannan, ulvan, xylan and the glucan. To this end, the labeled LecB lectin (0.12 µM final concentration), BSA (2%, final concentration), CaCl2 (7.5 µM, final concentration) in PBS 1× (pH 7.4) were mixed with the solutions of oligosaccharides (from the OligoTech®, Crolles, France). For each concentration, 1.5 µL of the mixture was poured into a microwell, where F1 was previously immobilized. The slides were incubated for 3 h at 37 °C in a water-saturated atmosphere. In each microwell, a competition between the two types of glycoclusters (F1) and oligosaccharide occurred to bind LecB. The microarrays were washed with 0.02% PBS-Tween20 for 5 min at room temperature. The fluorescence signal emitted by the complex glycocluster/Alexa 647-lectin was detected and an average of the mean fluorescence signal of 16 spots per glycocluster was calculated. The percentage of inhibition was calculated as follows: The alexa-647 fluorescent signal for a given sugar was normalized by the Alexa 647 fluorescent signal measured at 0.1 µM of fucose. Under these conditions, no inhibition of the interaction between LecB and the two fucosylated clusters by fucose is observed (IC50 10 and 20 µM for fucose).
3.17. General Procedure for the Screening of Oligoglycoclusters
3.17.1. Probing Oligoglycocluster-LecB Interaction on a Microarray
The interaction of LecB with the oligoglycoclusters was measured with Alexa 647 –labeled LecB. For the screening of the library of oligonucleotide oligoglycoclusters bearing several oligosaccharides (sLex pentasaccharide, Lex tetrasaccharide, Leb tetrasaccharide, 3FL and Lea tetrasaccharide), two oligoglycoclusters were immobilized per well by DDI with 32 repetition spots. In order to assess the relative surface densities of the oligoglycoclusters, the slides were scanned at 532 nm (excitation of Cy3) using the Axon microarray scanner, GenePix 4100A software package. The fluorescence signal of each oligoglycocluster was determined as the average of the mean fluorescence signal of 32 spots. The fluorescent signal of the oligoglycoclusters deviated by less than 15%.
1.5 µL of Alexa 647-LecB (0.12 µM, 7.5 μM CaCl2, 2% BSA in PBS 1×) were added in each well. Incubation was performed at 37 °C for 3 h in a water-saturated atmosphere. The slides were washed with 0.02% PBS-Tween20 for 5 min and rinsed with water. The slides were scanned at 635 nm (excitation of alexa 647) using the Axon microarray scanner, GenePix 4100A software package (Molecular device, San Jose, CA, USA). The Alexa 647-lectin fluorescence signal for each oligoglycocluster was determined as the average of the mean fluorescence signal of 32 spots. Error bars were calculated from these 32 spots.
3.17.2. Dissociation Constant Determination on a Microarray
A similar protocol was used for dissociation constant determination [
47,
48]. In this case, the lectin LecB concentration was varied from 1 nM to 2400 nM. The Alexa 647 fluorescent signal was recorded, and from the resulting isotherm, the
Kd values were determined using the equation:
where FI represents the fluorescent signal recorded at 635 nm for the given concentration of LecB ([LecB]) and FI
max is the maximum fluorescent signal obtained at 635 nm.
Kd value determination on a microarray deviated by less than 10% [
35].
3.18. In-Silico Molecular Docking
The lectin structure file was retrieved from the RCSB Protein Data Bank website (PDB code 1UZV). Docking experiments were performed with the GOLD software (The Cambridge Crystallographic Data Centre, Cambridge, UK). The six heavy atoms of the ligand fucose ring found in the structure file were used as a scaffold in the active site. The rest of the ligand was considered as flexible during the docking process. Side chains of the following residues in the vicinity of the active site of the same monomer, S22, S23, T45, E95, D96, T98, D99, N100, D101 and D104 were also defined as flexible during the docking procedure. For each ligand, 10 poses that are energetically reasonable were kept while searching for the correct binding mode of the ligand. The decision to keep a trial pose is based on a computed ligand-receptor interaction energy (score) of that pose. The ChemPLP fitness scoring function is used to rank the poses. The ChemPLP scoring function is the default in Gold version 5.2 used here. Additionally, an empirical potential energy of interaction ΔE for the ranked complexes is evaluated using the simple expression (2):
For that purpose, the Spectroscopic Empirical Potential Energy function SPASIBA and the corresponding parameters are used [
49,
50]. Molecular graphics and analysis were performed using the Discovery Studio Visualizer 4.0 software (Accelrys, San Diego, CA, USA).

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}