Ligand-Based Pharmacophore Modeling Using Novel 3D Pharmacophore Signatures



Abstract
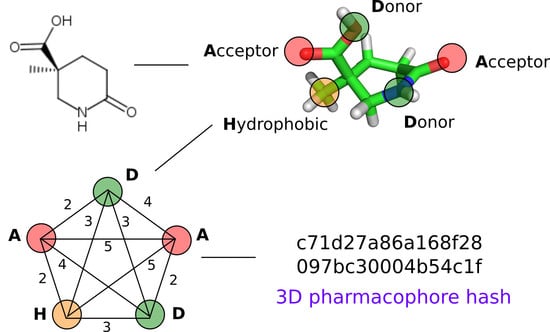
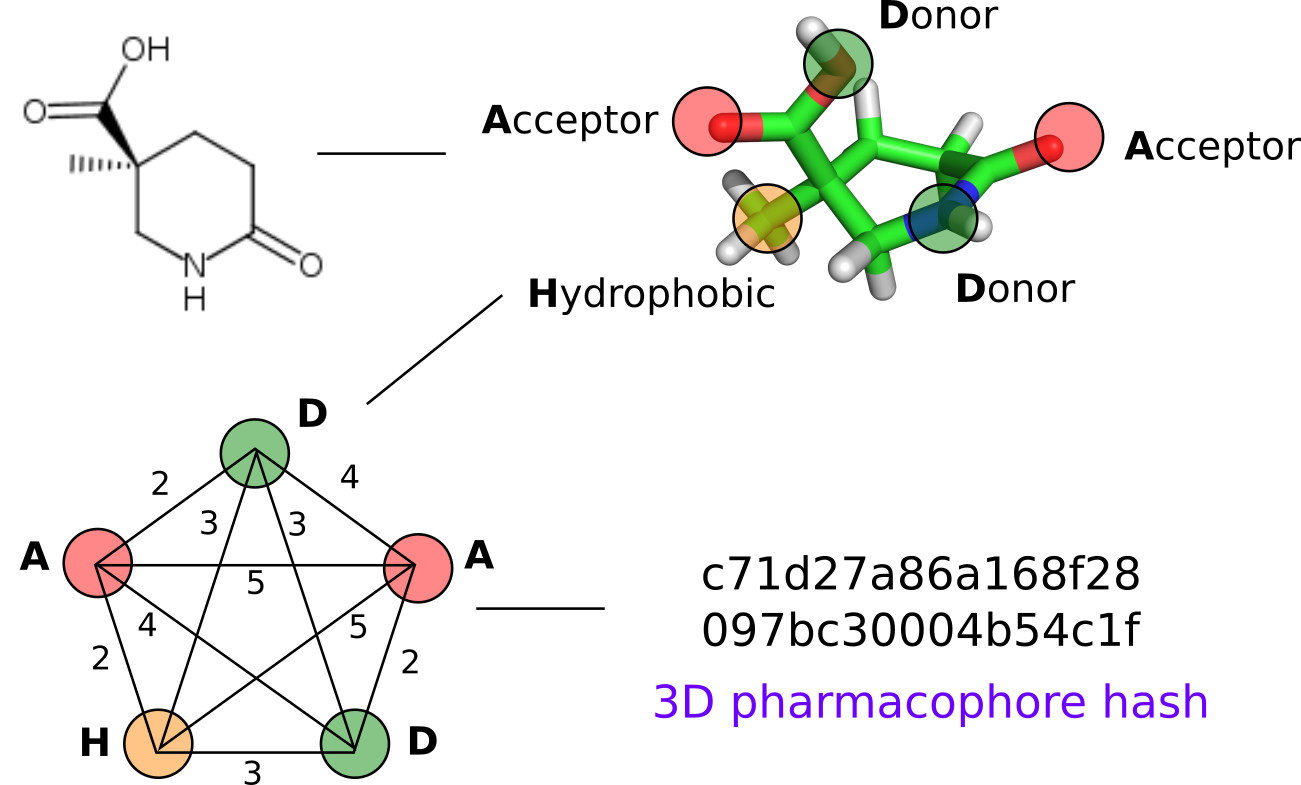
1. Introduction
2. Materials and Methods
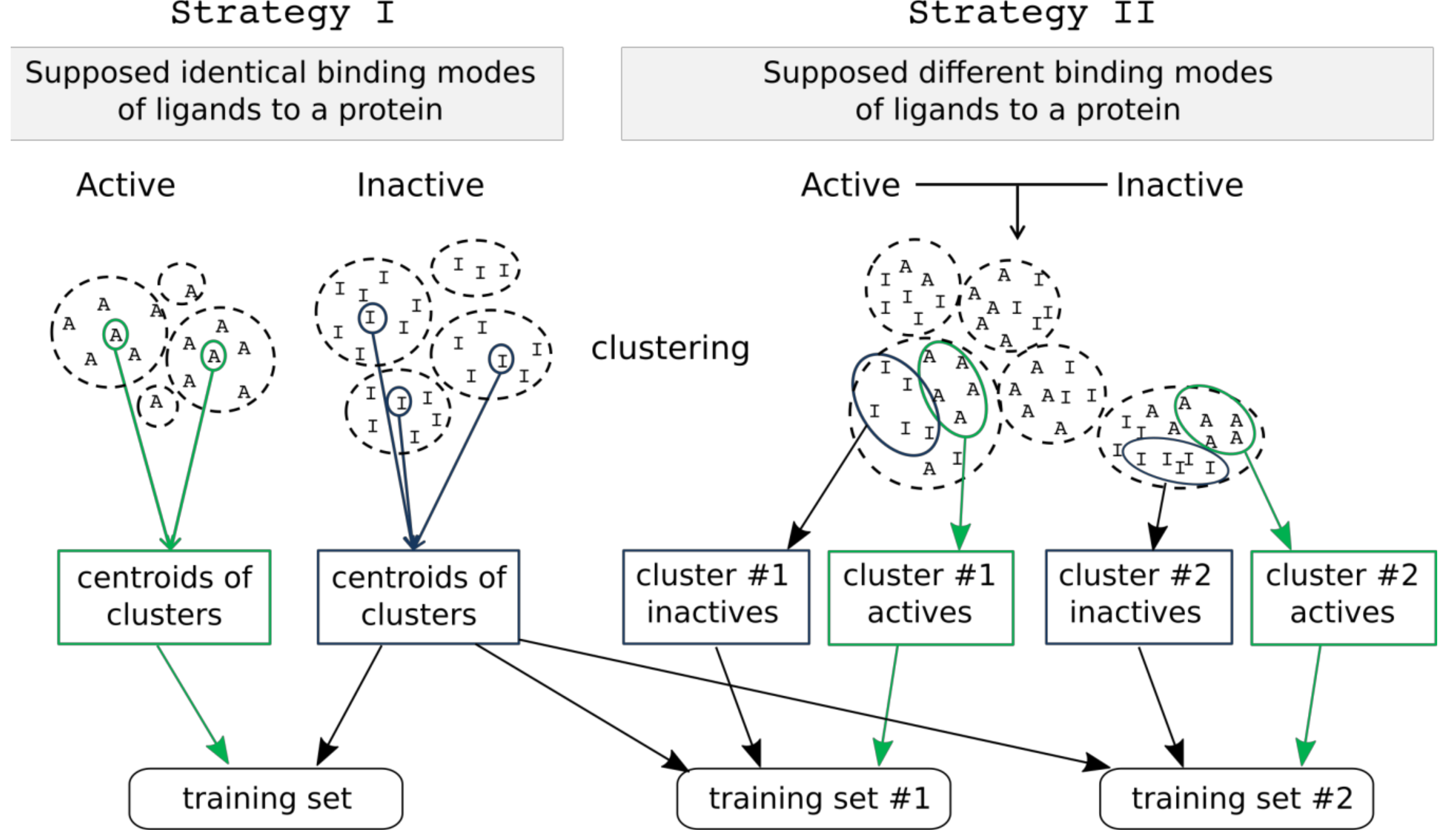
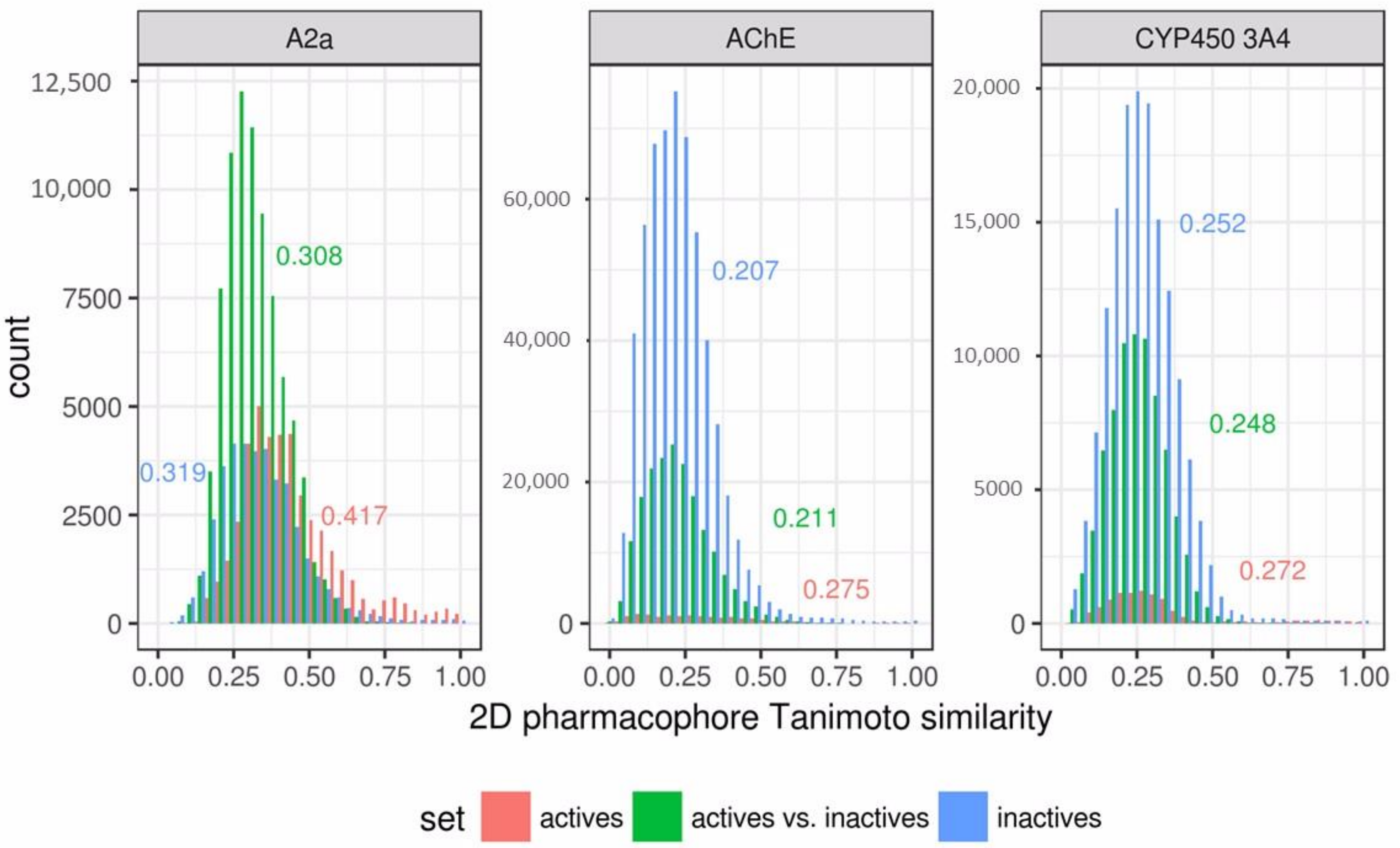
2.1. Data Sets
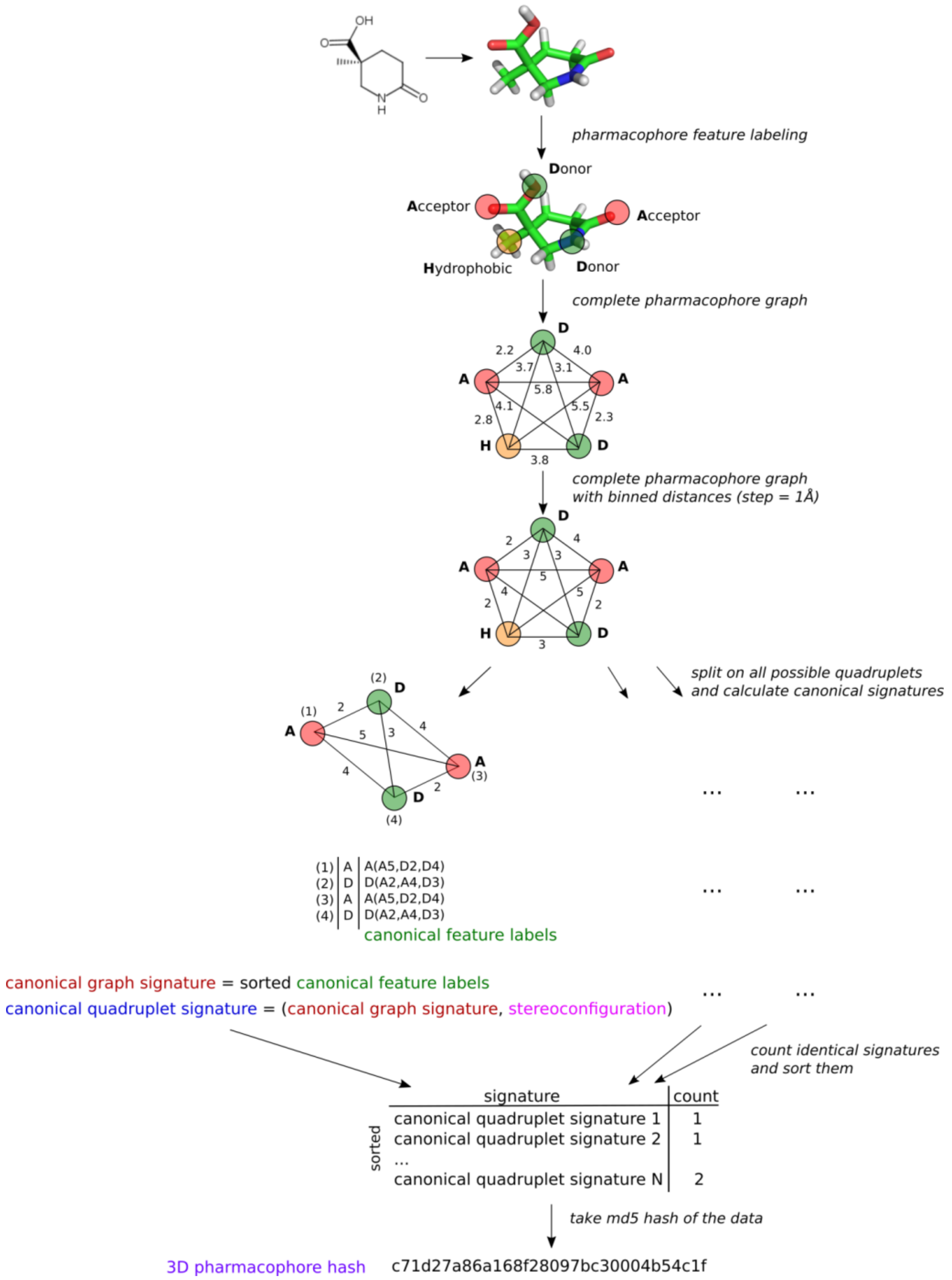
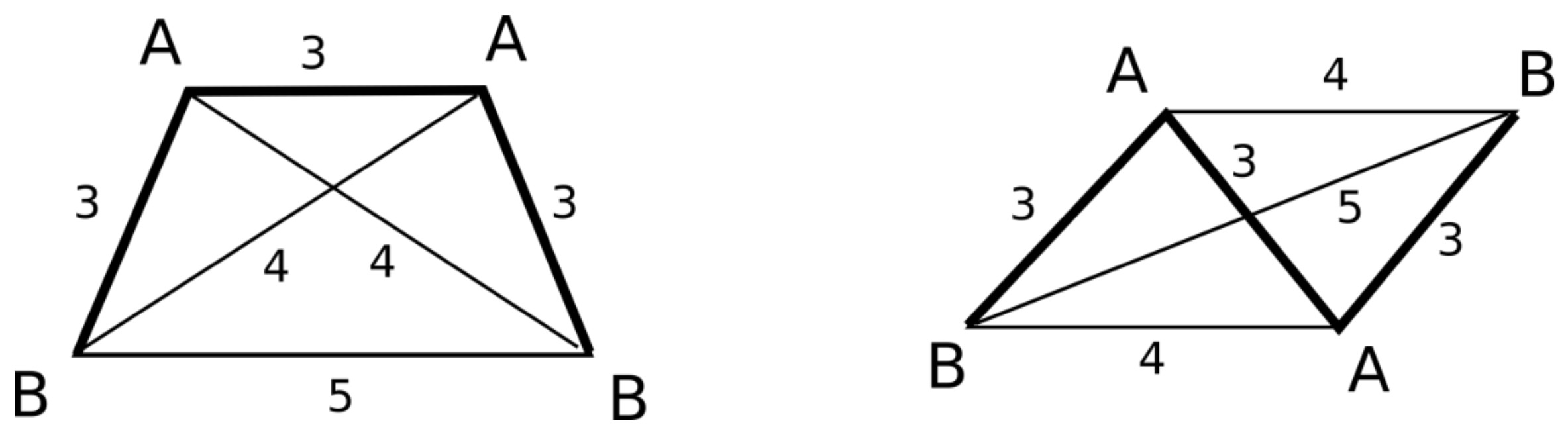
2.2. 3D Pharmacophore Signature Representation
- a)
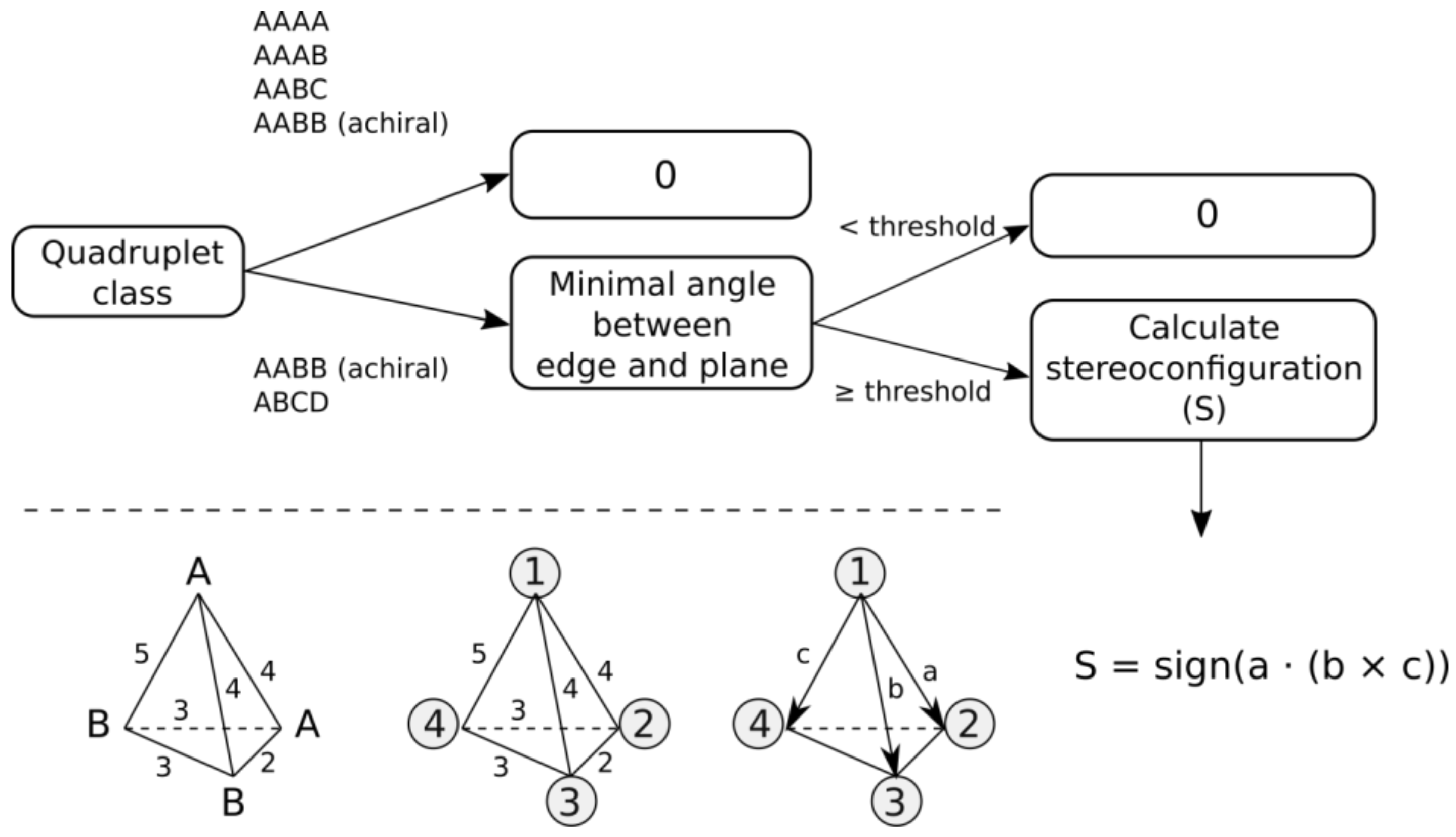
- AAAA system, where all features have identical canonical identifiers. This means that four features have identical labels and pairwise binned distances (features create a regular tetrahedron). A quadruplet belonging to this system is achiral.
- b)
- AAAB system, where three features have identical canonical identifiers (A) and one feature has a different one (B). This system corresponds to the trigonal pyramid and is achiral.
- c)
- AABC system, where two features have identical canonical identifiers (A) and two features have different ones (B and C). This system is achiral, because there is a plane of symmetry going through the center of AA distance and B and C features.
- d)
- AABB system, where pairs of features have identical canonical identifiers (A and B). This system can be chiral or achiral depending on distances between pairs of vertices. The achiral one would have a plane of symmetry, whereas the chiral one represents the case of axial chirality.
- e)
- ABCD system, where all features have distinct canonical identifiers. This system is chiral.
2.3. 3D Ligand-Based Pharmacophore Modeling
2.3.1. Training and Test Set Formation
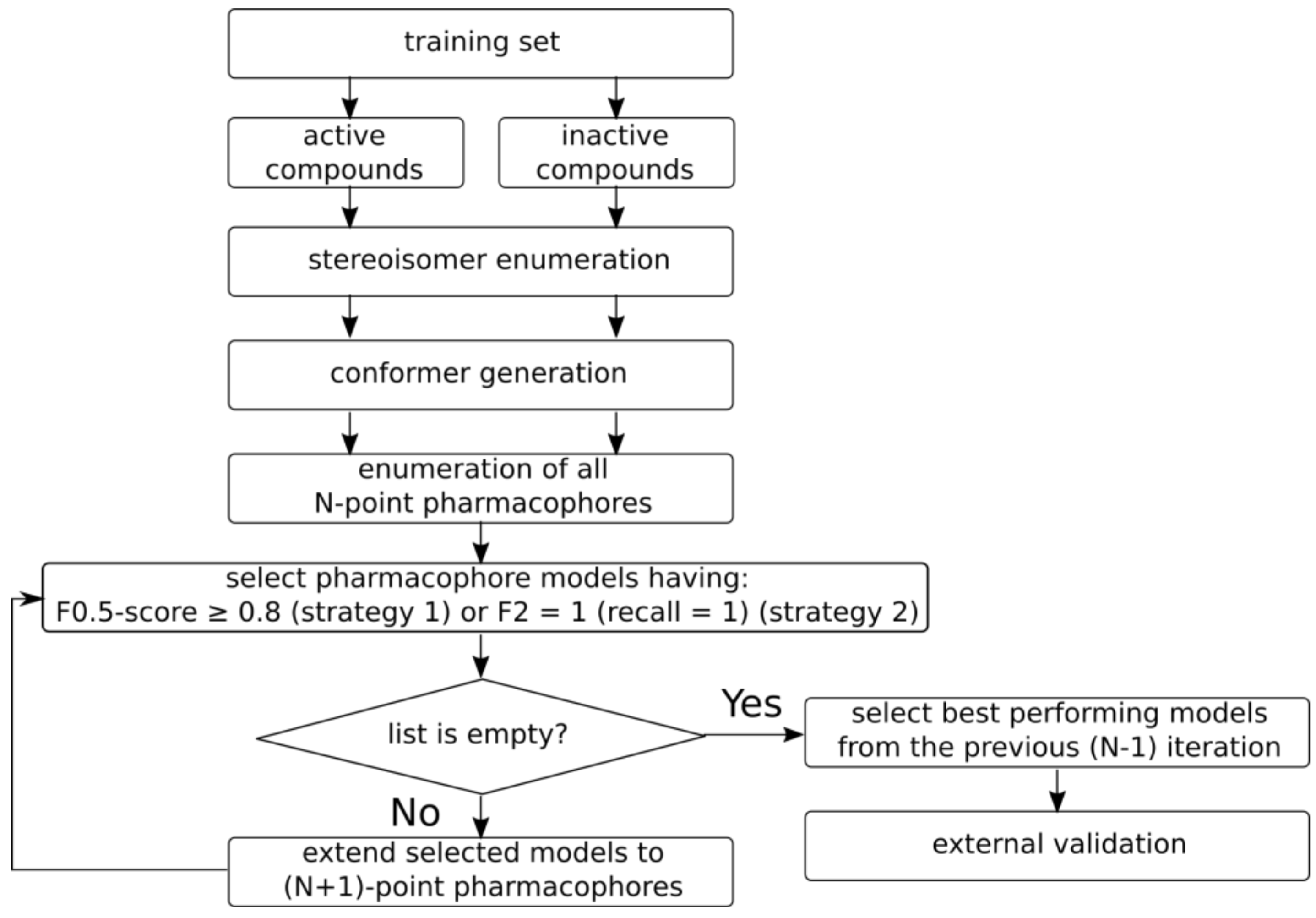
2.3.2. Model Development and Selection
2.3.3. Database Screening Using Pharmacophore Models
2.3.4. Model Quality Assessment
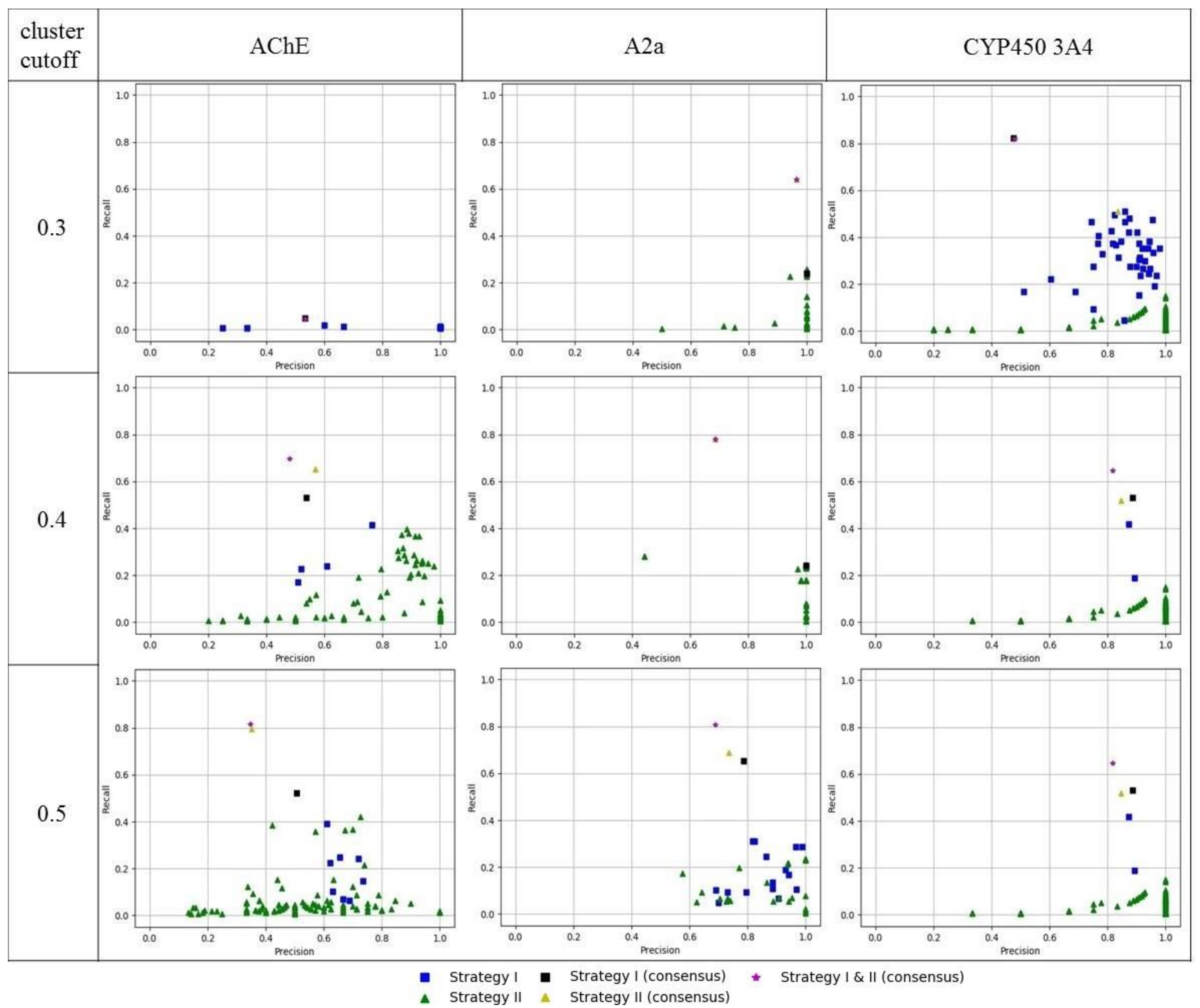
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schuster, D.; Nashev, L.G.; Kirchmair, J.; Laggner, C.; Wolber, G.; Langer, T.; Odermatt, A. Discovery of Nonsteroidal 17β-Hydroxysteroid Dehydrogenase 1 Inhibitors by Pharmacophore-Based Screening of Virtual Compound Libraries. J. Med. Chem. 2008, 51, 4188–4199. [Google Scholar] [CrossRef] [PubMed]
- Hinsberger, S.; Hüsecken, K.; Groh, M.; Negri, M.; Haupenthal, J.; Hartmann, R.W. Discovery of Novel Bacterial RNA Polymerase Inhibitors: Pharmacophore-Based Virtual Screening and Hit Optimization. J. Med. Chem. 2013, 56, 8332–8338. [Google Scholar] [CrossRef] [PubMed]
- Krautscheid, Y.; Senning, C.J.Å.; Sartori, S.B.; Singewald, N.; Schuster, D.; Stuppner, H. Pharmacophore Modeling, Virtual Screening, and in Vitro Testing Reveal Haloperidol, Eprazinone, and Fenbutrazate as Neurokinin Receptors Ligands. J. Chem. INF 2014, 54, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Polishchuk, P.G.; Samoylenko, G.V.; Khristova, T.M.; Krysko, O.L.; Kabanova, T.A.; Kabanov, V.M.; Kornylov, A.Y.; Klimchuk, O.; Langer, T.; Andronati, S.A.; et al. Design, Virtual Screening, and Synthesis of Antagonists of αIIbβ3 as Antiplatelet Agents. J. Med. Chem. 2015, 58, 7681–7694. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.; Schuster, D. Methods for generating and applying pharmacophore models as virtual screening filters and for bioactivity profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Jones, G. GAPE: An Improved Genetic Algorithm for Pharmacophore Elucidation. J. Chem. INF 2010, 50, 2001–2018. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Monecke, P.; Hessler, G.; Stützle, T.; Exner, T.E. pharmACOphore: Multiple Flexible Ligand Alignment Based on Ant Colony Optimization. J. Chem. INF 2010, 50, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Patel, Y.; Gillet, V.J.; Bravi, G.; Leach, A.R. A comparison of the pharmacophore identification programs: Catalyst, DISCO and GASP. J Comput. Aid. Mol. Des. 2002, 16, 653–681. [Google Scholar] [CrossRef]
- Martin, Y.C.; Bures, M.G.; Danaher, E.A.; DeLazzer, J.; Lico, I.; Pavlik, P.A. A fast new approach to pharmacophore mapping and its application to dopaminergic and benzodiazepine agonists. J Comput. Aid. Mol. Des. 1993, 7, 83–102. [Google Scholar] [CrossRef]
- Wolber, G.; Dornhofer, A.A.; Langer, T. Efficient overlay of small organic molecules using 3D pharmacophores. J. Comput. Aid. Mol. Des. 2006, 20, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Richmond, N.J.; Abrams, C.A.; Wolohan, P.R.N.; Abrahamian, E.; Willett, P.; Clark, R.D. GALAHAD: 1. Pharmacophore identification by hypermolecular alignment of ligands in 3D. J Comput. Aid. Mol. Des. 2006, 20, 567–587. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [PubMed]
- Schreyer, A.M.; Blundell, T. USRCAT: Real-time ultrafast shape recognition with pharmacophoric constraints. J. Cheminformatics 2012, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Koes, D.R.; Camacho, C.J. Pharmer: Efficient and Exact Pharmacophore Search. J. Chem. INF 2011, 51, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Documentation 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Butina, D. Unsupervised Data Base Clustering Based on Daylight’s Fingerprint and Tanimoto Similarity: A Fast and Automated Way To Cluster Small and Large Data Sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
Sample Availability: No samples of the compounds are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | Number of Actives | Number of Inactives | Total Number of Compounds |
|---|---|---|---|
| AChE | 176 (pIC50 ≥ 8) | 1070 (pIC50 ≤ 6) | 1246 |
| CYP450 3A4 | 138 (pIC50 ≥ 7) | 548 (pIC50 ≤ 5) | 686 |
| A2a | 293 (pKi/pKd/pIC50 ≥ 7) | 279 (pKi/pKd/pIC50 ≤ 5) | 574 |
| Data Set/Cluster Cutoff | Total Number of Clusters | Number of Active/Inactive Compounds in the Training Set (Strategy I) | Number of Training Sets (Strategy II) |
|---|---|---|---|
| AChE / 0.3 | 393 | 12/60 | 9 |
| AChE / 0.4 | 280 | 12/62 | 11 |
| AChE / 0.5 | 197 | 7/52 | 7 |
| A2a / 0.3 | 139 | 13/13 | 12 |
| A2a / 0.4 | 95 | 11/12 | 10 |
| A2a / 0.5 | 59 | 6/14 | 5 |
| CYP 3A4 / 0.3 | 293 | 8/23 | 7 |
| CYP 3A4 / 0.4 | 233 | 7/27 | 7 |
| CYP 3A4 / 0.5 | 154 | 8/27 | 7 |
| AChE | A2a | CYP450 3A4 | |
|---|---|---|---|
| strategy I | 5 | 6–8 | 6 |
| strategy II | 5–9 | 5–10 | 7–9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutlushina, A.; Khakimova, A.; Madzhidov, T.; Polishchuk, P. Ligand-Based Pharmacophore Modeling Using Novel 3D Pharmacophore Signatures. Molecules 2018, 23, 3094. https://doi.org/10.3390/molecules23123094
Kutlushina A, Khakimova A, Madzhidov T, Polishchuk P. Ligand-Based Pharmacophore Modeling Using Novel 3D Pharmacophore Signatures. Molecules. 2018; 23(12):3094. https://doi.org/10.3390/molecules23123094
Chicago/Turabian StyleKutlushina, Alina, Aigul Khakimova, Timur Madzhidov, and Pavel Polishchuk. 2018. "Ligand-Based Pharmacophore Modeling Using Novel 3D Pharmacophore Signatures" Molecules 23, no. 12: 3094. https://doi.org/10.3390/molecules23123094
APA StyleKutlushina, A., Khakimova, A., Madzhidov, T., & Polishchuk, P. (2018). Ligand-Based Pharmacophore Modeling Using Novel 3D Pharmacophore Signatures. Molecules, 23(12), 3094. https://doi.org/10.3390/molecules23123094