Abstract
The first example of a carborane with a catecholborolyl substituent, [1-Bcat-2-Ph-closo-1,2-C2B10H10] (1), has been prepared and characterized and shown to act as the Lewis acid component of an intermolecular frustrated Lewis pair in catalyzing a Michael addition. In combination with B(C6F5)3 the C-carboranylphosphine [1-PPh2-closo-1,2-C2B10H11] (IVa) is found to be comparable with PPh2(C6F5) in its ability to catalyze hydrosilylation, whilst the more strongly basic B-carboranylphosphine [9-PPh2-closo-1,7-C2B10H11] (V) is less effective and the very weakly basic species [μ-2,2′-PPh-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (IX) is completely ineffective. Base strengths are rank-ordered via measurement of the 1J 31P-77Se coupling constants of the phosphineselenides [1-SePPh2-closo-1,2-C2B10H11] (2), [9-SePPh2-closo-1,7-C2B10H11] (3), and [SePPh2(C6F5)] (4).
1. Introduction
The recognition by Stephan and co-workers, little more than a decade ago [1], that H2 could be reversibly activated using sterically-encumbered main group Lewis acid (LA)/Lewis base (LB) pairs has given rise to the burgeoning field of frustrated Lewis pair (FLP) chemistry [2,3,4,5,6,7,8]. FLPs can co-exist on the same molecule (intramolecular FLPs) or be on different molecules (intermolecular FLPs). Since its inception, the breadth of FLP chemistry has expanded considerably, and now, as summarized in a recent review [9], impacts upon small-molecule activation, organic chemistry, radical chemistry, transition-metal chemistry, enzyme models, polymers and materials, and surface chemistry.
Carborane chemistry is a well-established and wide-ranging field with thousands of derivatives known and a huge number of diverse applications now established for carborane-containing species [10]. Thus far, however, FLP chemistry and carborane chemistry have not intersected, in spite of the fact that the carborane scaffold offers a number of unique advantages for potential FLPs including high chemical and thermal stability, the ability to act as an electron-donating or electron-accepting support dependent on the vertex substituted (with no significant difference in the steric bulk of the carborane) [11,12,13], and further tuneability of electronic (and steric) FLP properties through isomerization, cage derivatization, or substitution [10].
We now report the first examples of intermolecular carborane-supported FLP chemistry, through (i) the synthesis of a catecholborolyl (Bcat) carborane (the LA) and its catalytic activation of a Michael addition reaction in combination with PPh3, and (ii) a comparison of C- and B-carboranylphosphines (the LB) in combination with B(C6F5)3 to catalyze a hydrosilylation reaction, demonstrating the unique advantage of carborane scaffolds in tuning the strength of FLP components.
2. Results and Discussion
2.1. Synthesis and Characterization of Compound 1
Following deprotonation of [1-Ph-closo-1,2-C2B10H11] in tetrahydrofuran (THF) by nBuLi and subsequent exchange of solvent for toluene, 2-Br-1,3,2-benzodioxaborole (BcatBr) in toluene was added and the reaction mixture heated to reflux overnight. The solvent was removed and the product extracted into petroleum ether. Unreacted phenylcarborane was removed via vacuum sublimation, leaving the product [1-Bcat-2-Ph-closo-1,2-C2B10H10] (1) in a 45% isolated yield. As far as we are aware, compound 1 is the first example of a carborane with a catecholborolyl substituent. C-substituted pinacolborolyl (Bpin) carboranes are known (see, e.g., References [14,15]) (there is one recent report of a B-substituted pinacolborolyl carborane [16]) and several C-substituted diazaborolyl carboranes have been prepared, e.g., References [17,18].
Compound 1 is a moderately air-sensitive, but significantly moisture-sensitive, colorless solid, initially characterized using elemental analysis, mass spectrometry, and by 1H and 11B nuclear magnetic resonance (NMR) spectroscopies. The latter features the resonance due to the catecholborolyl B atom at δ 28.8 ppm in C6D6, easily identified by its high frequency chemical shift and lack of 1H coupling. The carboranyl boron resonances in the 11B{1H} NMR spectrum appear with relative integrals 1:1:2:2:2:2 from high frequency to low frequency suggest a molecule with time-averaged Cs molecular symmetry in solution.
Ultimately compound 1 was unambiguously characterized crystallographically. Single crystals suitable for an X-ray diffraction study were grown by cooling a solution of 1 in C6H5F and a perspective view of a single molecule is shown in Figure 1. The Cs symmetry in solution was not retained in the solid state as the Ph and Bcat units were twisted by ≈19° in a conrotatory manner with respect to the least-squares plane through atoms B100C1C2C21. The C1−C2 distance in 1, 1.6840(15) Å, sits within the range of such distances in related compounds (see, e.g., References [17,18]), whilst the C1−B100 distance, 1.5703(16) Å, was comparable to that (1.565(4) Å) in [1-Bpin-2,3,4,5,6,7,8,9,10,11-Me10-12-(4′-Br-C6H4)-closo-1-CB11]− [14] but significantly shorter than that (1.6046(19) Å) in [1-B(OMe)2-2-iPr-closo-1,2-C2B10H10] [15], the only other compounds with {BO2} fragments bonded to a carborane C atom to have been structurally characterized.
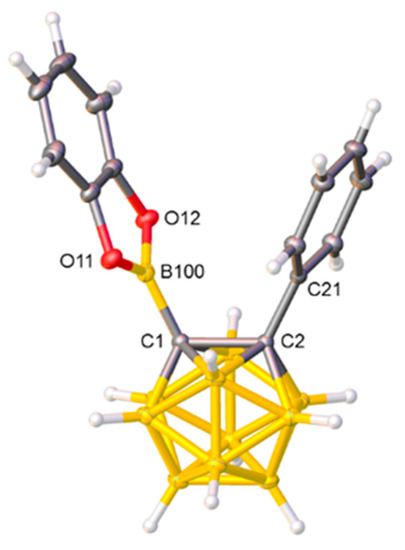
Figure 1.
Molecular structure of compound 1. Selected interatomic distances (Å): C1−C2 1.6840(15), C1−B100 1.5703(16), B100−O11 1.3744(14), B100−O12 1.3739(15), and C2−C21 1.5083(14).
2.2. Carborane-Supported Components of Intermolecular FLPs to Catalyze Michael Addition
The steric bulk and electron-withdrawing nature of the phenylcarborane fragment results in B100 being both sterically-encumbered and highly Lewis acidic, and consequently compound 1 is an ideal potential LA component of an intermolecular FLP. To investigate this, we have studied the involvement of 1 as co-catalyst in the Michael addition of dimethyl malonate (I) to 3-buten-2-one (II) to afford dimethyl 2-(3-oxobutyl) malonate (III), a classic Michael addition reaction (Scheme 1). Such reactions are known to be catalyzed by phosphines alone [19], but enhanced rates have been observed if a species with the potential to act as a Lewis acid is also present because the LA and LB form an FLP (see, e.g., References [20,21,22]).
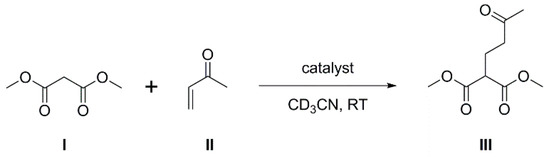
Scheme 1.
Michael addition of dimethyl malonate and 3-buten-2-one producing dimethyl 2-(3-oxobutyl) malonate.
In the presence of 10 mol% PPh3 in CD3CN at room temperature, a 1:1 mixture of I and II affords III in 43% yield after 6 h and 64% yield after 24 h (entries 1 and 2, Table 1). No reaction was observed in the absence of a catalyst nor in the presence of only the Bcat carborane 1. When, however, an intermolecular FLP of 10 mol% 1 and 10 mol% PPh3 was used as a catalyst, the yield of III was 56% after 6 h and 76% after 24 h (entries 3 and 4). This demonstrates the co-operative nature of the two components, with the enhancement in catalysis fully consistent with them acting as a frustrated Lewis pair and represents the first time that a carborane-containing species has been used as a component of an FLP. The results are at least as good as those obtained for the same reaction using either PhBpin/PPh3 as an intermolecular FLP or 1-Bpin-2-PPh2-C6H4 as an intramolecular FLP [22].

Table 1.
Results for catalysis of the Michael addition reaction in Scheme 1 by PPh3, the FLP 1/PPh3 and V. 1
An interesting alternative to using PPh3 as a catalyst for this Michael addition reaction would be to have the LB functionality on a carborane cage. However, the C-carboranylphosphines [1-PR2-closo-1,2-C2B10H11] [R = Ph (IVa), iPr (IVb), tBu (IVc) (Scheme 2)] were all inactive in catalyzing the reaction, presumably a consequence of their relatively low basicity because of the strong electron-acceptor property of the carborane when substituted at C. In contrast, it is well established that a carborane substituted at B is electron-releasing [11,12,13], and we therefore prepared the B-substituted carboranylphosphine [9-PPh2-closo-1,7-C2B10H11] (V) [13] and tested it as the single catalyst for Michael addition, finding it to be significantly more effective than PPh3 (Table 1, entries 5 and 6). Note that IVa and V are related as simple positional isomers. Replacing a Ph group in PPh3 with a C-bound carborane cage (affording IVa) reduces the basicity of the phosphine and switches off the Michael addition reaction, whilst replacing a Ph in PPh3 with a B-bound carborane cage (affording V) enhances this particular catalysis. This clearly demonstrates the potential of electronically-flexible carborane scaffolds in controlling the properties of substituents and so optimizing catalysis.
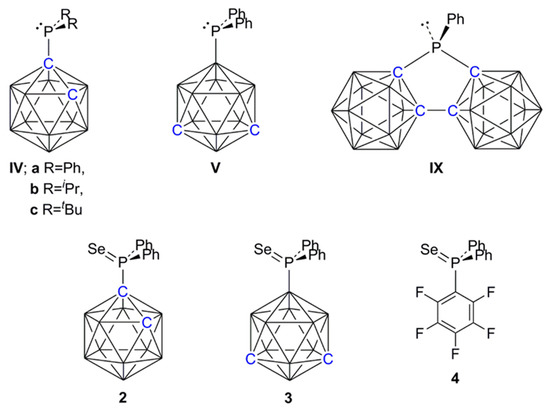
Scheme 2.
Line diagrams of key species.
2.3. Carborane-Supported Components of Intermolecular FLPs to Catalyze Hydrosilylation
Although the C-substituted carboranylphosphines IV are too weakly basic for Michael addition, they can be, as part of an FLP, highly effective in the hydrosilylation of dimethylfulvene (Scheme 3). In combination with B(C6F5)3, the weak Lewis base PPh2(C6F5) has been shown by Paradies and co-workers to catalyze this reaction effectively, whilst the strong Lewis base PtBu3 has no activity (it is assumed that this is due to the formation of the silylium salt [tBu3P-SiPh2Me][HB(C6F5)3]) [23]. This is therefore an ideal reaction in which to study the effect of controlling the basicity of the Lewis base component of the FLP and we have investigated the effectiveness of carborane supports in affording that control.
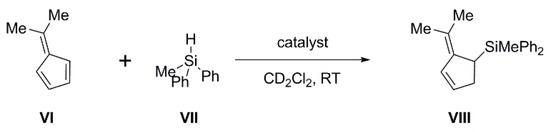
Scheme 3.
Hydrosilylation of dimethylfulvene.
In Table 2, the intermolecular FLPs formed by B(C6F5)3 with PPh2(C6F5), IVa and V are compared for their efficiency in catalyzing this reaction. The FLP B(C6F5)3/IVa was fully comparable with B(C6F5)3/PPh2(C6F5) in efficiency, both combinations producing nearly 90% product yield after only 11–12 min (entries 1 and 2), whilst the FLP from B(C6F5)3 with the more strongly basic V was inferior to both, affording the product only 80% yield after more than twice the time (entry 3). We also investigated the effect on the reaction of using the FLP formed from B(C6F5)3 and the very weakly basic bis(carboranyl)phosphine [μ-2,2′-PPh-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (IX) [24] (Scheme 2). In this case significant amounts of oligomerized product were immediately observed (entry 4), consistent with the results obtained by Paradies when no base was used [23]. These results clearly establish that this reaction was very sensitive to the strength of the Lewis base component; if the base is too strong (e.g., PtBu3) there is no catalysis [23], whilst if it is too weak (e.g., IX), the base plays no part in the chemistry and the Lewis acid catalyzes oligomerization. Between these two extremes, the base acts as an FLP with the B(C6F5)3 Lewis acid, which catalyzes hydrosilylation, with weaker bases performing somewhat better.
Table 2.
Results for catalysis of the hydrosilylation reaction in Scheme 2 using the FLPs B(C6F5)3/PPh2(C6F5), B(C6F5)3/IVa, B(C6F5)3/V, and B(C6F5)3/IX. 1
2.4. Synthesis and Characterization of Phosphineselenides 2, 3 and 4
In an attempt to understand the relative efficiencies of the phosphines PPh3, IVa, V, IX, and PPh2(C6F5) as stand-alone Lewis bases or as components of FLPs, we have attempted to rank their basicities via formation of the appropriate selenide. This is because it is well established that, in the absence of significant intra- or intermolecular H-bonding contacts, pKb of phosphines correlates almost linearly with the magnitude of the 1J 31P-77Se coupling constants of the corresponding selenide (see, e.g., References [25,26].
The selenides [1-SePPh2-closo-1,2-C2B10H11] (2, derived from IVa), [9-SePPh2-closo-1,7-C2B10H11] (3, derived from V) and [SePPh2(C6F5)] (4) were prepared in good yields using the straightforward procedure of heating to reflux the appropriate phosphine and excess Se in toluene. All three pale or colorless compounds were crystalline solids that were initially characterized using elemental analysis, mass spectrometry, and 1H and 31P{1H} NMR spectroscopies, plus 11B{1H} (for 2 and 3), 77Se (for 3), and 19F{1H} (for 4) NMR studies.
Compounds 2, 3, and 4 were also studied crystallographically, and perspective views of single molecules together with key molecular parameters are presented in Figure 2, Figure 3, and Figure 4, respectively. In 2, there appears to be a preferred orientation of the {SePPh2} fragment relative to the carborane cage with the torsion angle C2−C1−P−Se only 5.73(7)°, allowing the Se atom and the relatively protonic H bound to C2 to approach to within 2.752(17) Å, substantially less than the sum of their van der Waals radii of ≈3.10 Å [27].
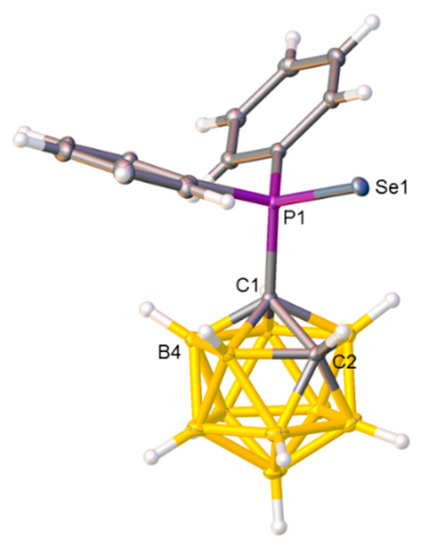
Figure 2.
Molecular structure of compound 2. Selected interatomic distances (Å): C1−C2 1.6511(14), C1−P1 1.8869(10), and P1−Se1 2.1037(3).
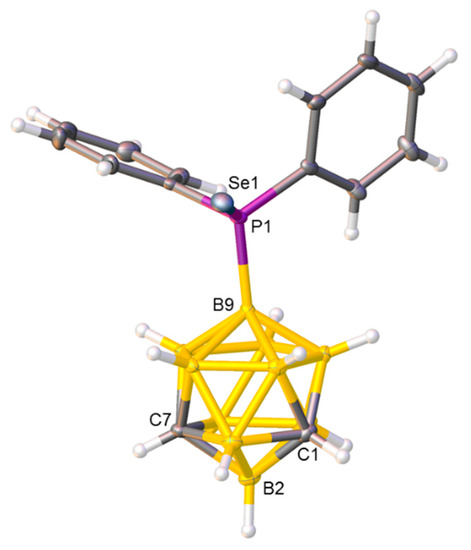
Figure 3.
Molecular structure of compound 3. Selected interatomic distances (Å): B9−P1 1.937(2) and P1−Se1 2.1196(5).
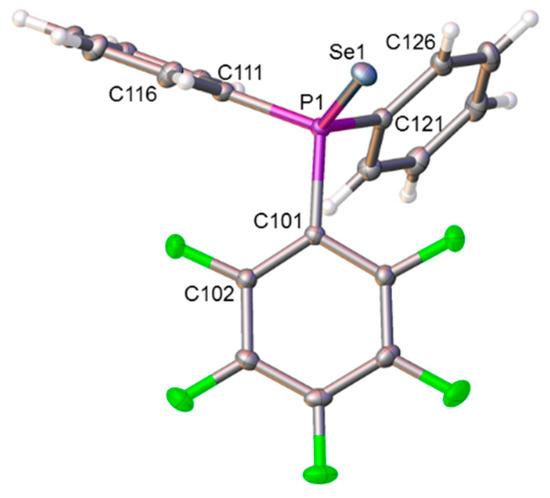
Figure 4.
Molecular structure of compound 4. Selected interatomic distances (Å): P1−Se1 2.1047(3), P1−C101 1.8346(11), P1−C111 1.8141(12), and P1−C121 1.8080(12).
In all cases, the 31P{1H} NMR shifts of the selenides 2, 3, and 4 were observed at significantly higher frequency than those of the precursor phosphine (δ 50.7 ppm vs 25.3 ppm for 2, +3.8 ppm vs −48.2 ppm for 3, +20.4 ppm vs −25.0 ppm for 4, all comparisons made in the same solvent). Most importantly, 77Se satellites reveal 1J 31P-77Se coupling constants of 799 Hz for 2, 704 Hz for 3 (confirmed using the 77Se NMR spectrum), and 774 Hz for 4. SePPh3 (1J 31P-77Se = 732 Hz) [25] and SeIX (1J 31P-77Se = 891 Hz) [24] are known species.
Based on the magnitude of these 1JPSe values, the ranking of the base strengths of the phosphines was V (most basic) > PPh3 > PPh2(C6F5) > IVa > SeIX (least basic). This is fully consistent with the observation that V acts as the best stand-alone phosphine for catalyzing the Michael addition reaction but is the worst LB component of an FLP with B(C6F5)3 in catalyzing the hydrosilylation reaction, known to be favored by less basic phosphines [23]. Comparison of the coupling constants of 2 and 3 dramatically illustrates the different basicities of carboranylphosphines substituted at C (compound IVa, weakly basic) versus those substituted at B (compound V, strongly basic). In an alternative description, starting from PPh3, notional replacement of one Ph by (C6F5) reduces the basicity (as expected), whilst notional replacement of Ph by a C-carborane cage, forming [1-PPh2-closo-1,2-C2B10H11] (IVa), reduced the basicity even further (C-substituted carborane cage is more electron-withdrawing than Ph). On the other hand, notional replacement of one Ph in PPh3 by a B-carborane cage, affording [9-PPh2-closo-1,7-C2B10H11] (V), increased the basicity (B-substituted carborane cage is less electron-withdrawing than Ph).
These results confirm that the nature of the site of substitution of a carborane significantly affects the strength of an appended Lewis base and, by extension, an appended Lewis acid. In principle, the acid and/or base strength can be further tuned by varying the size of the carborane, its structural type (closo, neutral; nido, anionic), its isomeric form, the nature of any heteroatoms present (e.g., metal fragment vertices), and the nature of additional substituents at B and/or at C. Thus, carborane scaffolds have the unique potential to offer an exceptional degree of control over the Lewis acid and/or Lewis base strength of appended groups, which is likely to be highly important in constructing useful FLP combinations for a variety of catalytic applications. Future contributions will explore further these possibilities.
3. Experimental Section
3.1. General Considerations
All experiments were performed, unless otherwise stated, under an atmosphere of dry nitrogen using standard Schlenk or glovebox techniques, with some subsequent manipulations and purifications carried out in air. Tetrahydrofuran (THF) was distilled from sodium/benzophenone, dichloromethane (DCM) from CaH2, and toluene and petroleum ether (40–60 °C, petrol) from sodium. All solvents were freeze-pump-thawed three times prior to use. Deuterated solvents were stored over 4 Å molecular sieves. [1-PPh2-closo-1,2-C2B10H11] (IVa) [28], [1-PiPr2-closo-1,2-C2B10H11] (IVb) [29], [1-PtBu2-closo-1,2-C2B10H11] (IVc) [30], [9-PPh2-closo-1,7-C2B10H11] (V) [13], [μ-2,2′-PPh-{1-(1′-1′,2′-closo-C2B10H10)-1,2-closo-C2B10H10}] (IX) [24], and [1-Ph-closo-1,2-C2B10H11] [31,32] were prepared according to the literature. All other reagents were purchased from Sigma Aldrich Ltd. (Gillingham, UK), Fluorochem Ltd. (Hadfield, UK) or Alfa Aesar (Heysham, UK) and used without further purification. NMR spectra were recorded at 298 K using a Bruker AVIII-400 spectrometer (Bruker BioSpin AG, Fallenden, Switzerland), with chemical shifts reported relative to the residual protonated solvent peaks (1H) or to external standards (11B; BF3∙OEt2). Elemental analyses were conducted using an Exeter CE-440 elemental analyser (Exeter Analytical Inc., North Chelmsford, MA, USA). Electron ionization mass spectrometry (EIMS) was carried out on a Bruker Microtof II mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) at the University of Edinburgh.
3.1.1. Synthesis and Characterization of [1-Bcat-2-Ph-closo-1,2-C2B10H10] (1)
[1-Ph-closo-1,2-C2B10H11] (300 mg, 1.36 mmol) was dried under vacuum and dissolved in anhydrous THF (20 mL). The solution was cooled to 0 °C before nBuLi (1.02 mL, 1.6M, 1.63 mmol) was added dropwise. The colorless solution turned pale pink and was stirred at 0 °C for 0.5 h before being warmed to room temperature, then heated to 65°C for 1 h. The pale-yellow solution was allowed to cool to room temperature and concentrated to dryness. Anhydrous toluene (25 mL) was added. The Schlenk tube was covered in foil and the pale-yellow solution cooled to −78 °C before the addition of a toluene solution of 2-bromo-1,3,2-benzodioxaborole (324 mg, 1.63 mmol), resulting in a purple solution and a blue precipitate. The mixture was heated to reflux overnight. The purple solution was transferred via cannula to a second Schlenk tube along with anhydrous toluene washings (2 × 20 mL) and concentrated to a purple solid. This was extracted with anhydrous petrol (2 × 50 mL) and the soluble materials evaporated to reveal a white solid. Excess phenyl carborane was removed via vacuum sublimation leaving the product [1-Bcat-2-Ph-closo-1,2-C2B10H10] (1) (205 mg, 0.61 mmol, 45%). C14H19B11O2 requires C 49.7, H 5.66; found C 49.2, H 5.66%. 1H NMR (400 MHz, C6D6): δ 7.44–7.41 (m, 2H, C6H5), 6.74–6.71 (m, 1H, C6H5), 6.67–6.61 (m, 4H), 6.50–6.53 (m, 2H). 11B{1H}[11B] NMR (128 MHz, C6D6): δ 29.0 (s, 1B, Bcat), 2.4 (s [d, JBH = 150 Hz], 1B), −2.4 (s [d, JBH = 150 Hz], 1B), −7.3 (2B), −8.3 (2B), −9.7 (2B), −10.7 (2B). EIMS: envelope centered on m/z 338.2 (M+).
3.1.2. General Synthesis and Characterization of Phosphine Selenides 2, 3, and 4
The phosphine (typically 0.25–0.5 mmol) was dissolved in toluene (typically 10–15 mL) and ≈10 equivalents of elemental selenium were added. Under N2, the reagents were heated to reflux overnight. Excess Se was filtered off and washed with DCM. The filtrate and washings were evaporated to afford essentially pure colorless or pale-colored solids.
[1-SePPh2-closo-1,2-C2B10H11] (2) Colorless, 56% yield. C14H21B10PSe requires C 41.3, H 5.20; found C 40.8, H 5.06%. 1H NMR (400 MHz, CDCl3): δ 8.28–8.22 (m, 4H, C6H5), 7.64–7.52 (m, 6H, C6H5), 4.80 (br. s, 1H, CcageH). 11B{1H} NMR (128 MHz, CDCl3): δ −0.5 (1B), −2.5 (1B), −7.2 (2B), −11.0 (2B), −12.8 (4B). 31P{1H} NMR (162.0 MHz, CDCl3): δ 50.66 (s + Se satellites, 1JPSe = 799 Hz). EIMS: envelope centered on m/z 407.1 (M+).
[9-SePPh2-closo-1,7-C2B10H11] (3) Pale yellow, 71% yield. C14H21B10PSe requires C 41.3, H 5.20; found C 42.2, H 5.35%. 1H NMR (400 MHz, CD2Cl2): δ 8.04–7.98 (m, 4H, C6H5), 7.47–7.43 (m, 6H, C6H5), 3.13 (br. s, 2H, CcageH). 11B{1H} NMR (128 MHz, C6D6): δ −3.4 (1B), −4.7 (1B), −5.9 (2B), −9.9 (1B), −11.9 (1B), −12.7 (2B), −16.3 (2B). 31P{1H} NMR (162.0 MHz, C6D6): δ 3.76 (q [1JPB = 164 Hz] + Se satellites, 1JPSe = 704 Hz). 77Se NMR (76.4 MHz, C6D6): δ −230.54 (d, 1JPSe = 704 Hz). EIMS: envelope centered on m/z 408.2 (M+).
[SePPh2(C6F5)] (4) Pale pink, 57% yield. C18H10F5PSe requires C 50.1, H 2.34; found C 50.1, H 2.34%. 1H NMR (400 MHz, CDCl3): δ 7.99–7.92 (m, 4H, C6H5), 7.58–7.47 (m, 6H, C6H5). 19F NMR (376.5 MHz, CDCl3): δ −126.9 (s, 2F, C6F5), −147.1 (s, 2F, C6F5), −158.8 (s, 1F, C6F5). 31P{1H} NMR (162.0 MHz, CDCl3): δ 20.42 (s + Se satellites, 1JPSe = 774 Hz). EIMS: envelope centred on m/z 431.9 (M+).
3.2. Crystallographic Studies
Compound 1, crystal data: C14H19B11O2, M = 338.20, monoclinic, P21/c, a = 15.0049(6), b = 6.78992(18), c = 19.0043(6) Å, β = 112.135(4)°, U = 1793.49(11) Å3, Z = 4, Dc = 1.253 Mg m−3, μ = 0.069 mm−1, F(000) = 696. 37112 data to θmax = 31.23° were collected at 120.01(10) K on a Rigaku Oxford Diffraction SuperNova diffractometer using Mo-Kα X-radiation. A total of 5441 unique reflections (Rint = 0.0424) were used to solve (using SHELXS [33]) and refine (using SHELXL [34]) the structure within the Olex2 [35] package. R1 = 0.0480, wR2 = 0.1189 for data with I ≥ 2σ(I), S (all data) = 1.073, Emax, Emin = 0.38, −0.23 eÅ−3, respectively. CCDC 1848620.
Compound 2, crystal data: C14H21B10PSe, M = 407.34, monoclinic, P21/c, a = 8.7241(4), b = 25.2149(11), c = 9.4588(4) Å, β = 111.615(2)°, U = 1934.41(15) Å3, Z = 4, Dc = 1.399 Mg m−3, μ = 2.018 mm−1, F(000) = 816. 58543 data to θmax = 34.07° were collected at 100.00(10) K on a Bruker X8 APEXII diffractometer using Mo-Kα X-radiation. A total of 7897 unique reflections (Rint = 0.0451) were used to solve and refine the structure, as for compound 1. R1 = 0.0256, wR2 = 0.0592 for data with I ≥ 2σ(I), S (all data) = 1.030, Emax, Emin = 0.48, −0.36 eÅ−3, respectively. CCDC 1848621.
Compound 3, crystal data; C14H21B10PSe, M = 407.34, orthorhombic, Pbca, a = 9.9130(5), b = 18.9783(9), c = 20.7608(10) Å, U = 3905.8(3) Å3, Z = 8, Dc = 1.385 Mg m−3, μ = 1.999 mm−1, F(000) = 1632. 51443 data to θmax = 31.49° were collected at 100.00(10) K on a Bruker X8 APEXII diffractometer using Mo-Kα X-radiation. A total of 6491 unique reflections (Rint = 0.0693) were used to solve and refine the structure, as for compound 1. R1 = 0.0389, wR2 = 0.0899 for data with I ≥ 2σ(I), S (all data) = 1.027, Emax, Emin = 0.81, −0.86 eÅ−3, respectively. CCDC 1848622.
Compound 4, crystal data; C18H10F5PSe, M = 431.19, monoclinic, P21/c, a = 16.3024(12), b = 7.2293(5), c = 14.4191(11) Å, β = 104.365(4)°, U = 1646.2(2) Å3, Z = 4, Dc = 1.740 Mg m−3, μ = 2.428 mm−1, F(000) = 848. 489984 data to θmax = 34.20° were collected at 100.00(10) K on a Bruker X8 APEXII diffractometer using Mo-Kα X-radiation. A total of 6774 unique reflections (Rint = 0.0396) were used to solve and refine the structure, as for compound 1. R1 = 0.0262, wR2 = 0.0611 for data with I ≥ 2σ(I), S (all data) = 1.056, Emax, Emin = 0.50, −0.46 eÅ−3, respectively. CCDC 1848623.
For 2 and 3, the cage C atoms were distinguished from B atoms by application of the Vertex-Centroid Distance (VCD) and Boron-Hydrogen Distance (BHD) methods [36,37,38].
4. Conclusions
The first example of a carborane with a catecholborolyl substituent, 1, was prepared and fully characterized, and was shown to enhance the catalysis of a Michael addition reaction by forming an FLP with PPh3. A variety of carboranylphosphines were tested as FLP components in combination with B(C6F5)3 as catalysts of a hydrosilylation reaction, with the strongly-basic V performing less well than the relatively weakly basic IVa, whilst the very weakly basic IX was completely ineffective. These results demonstrate that the ability to tune the Lewis acid and/or Lewis base strength of FLP components is critical in optimizing their use as catalysts and suggest that the electronic tuneability of carborane supports offers great potential in this respect.
Supplementary Materials
NMR and mass spectra of all new compounds reported. Details of the catalytic runs. Crystallographic data for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications nos. CCDC 1848620-1848623 (compounds 1–4). Copies of the data can be obtained free of charge an application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-1223-336033; email: deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk).
Author Contributions
A.B. and Z.C. synthesized and characterized the compounds and A.B. performed the catalytic studies. G.M.R. undertook the crystallographic determinations. S.M.M. and A.J.W. devised and supervised the research. A.B., S.M.M., and A.J.W. contributed to writing the paper.
Funding
This research received no external funding.
Acknowledgments
We are grateful to the EPSRC for the award of a DTP studentship supporting AB. We thank D. Ellis for the NMR spectroscopy and G.S. Nicol (University of Edinburgh) for data collection of compound 1.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Welch, G.C.; San Juan, R.R.; Masuda, J.D.; Stephan, D.W. Reversible, Metal-Free hydrogen activation. Science 2006, 314, 1124–1126. [Google Scholar] [CrossRef]
- Stephan, D.W. “Frustrated Lewis pairs”: a concept for new reactivity and catalysis. Org. Biomol. Chem. 2008, 6, 1535–1539. [Google Scholar] [CrossRef]
- Stephan, D.W. Frustrated Lewis pairs: a new strategy to small molecule activation and hydrogenation catalysis. Dalton Trans. 2009, 3129–3136. [Google Scholar] [CrossRef]
- Stephan, D.W.; Erker, G. Frustrated Lewis pairs: Metal-Free hydrogen activation and more. Angew. Chem. Int. Ed. 2010, 49, 46–76. [Google Scholar] [CrossRef]
- Stephan, D.W. “Frustrated Lewis pair” hydrogenations. Org. Biomol. Chem. 2012, 10, 5740–5746. [Google Scholar] [CrossRef]
- Stephan, D.W.; Erker, G. Frustrated Lewis pair chemistry of carbon, nitrogen and sulfur oxides. Chem. Sci. 2014, 5, 2625–2641. [Google Scholar] [CrossRef]
- Stephan, D.W. Frustrated Lewis pairs from concept to catalysis. Acc. Chem. Res. 2015, 48, 306–316. [Google Scholar] [CrossRef]
- Stephan, D.W. Frustrated Lewis pairs. J. Am. Chem. Soc. 2015, 137, 10018–10032. [Google Scholar] [CrossRef]
- Stephan, D.W. The broadening reach of frustrated Lewis pair chemistry. Science 2016, 354, 1248–1256. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Zheng, Z.; Diaz, M.; Knobler, C.B.; Hawthorne, M.F. A mercuracarborand characterized by B-Hg-B bonds: Synthesis and structure of cyclo-[(t-BuMe2Si)2C2B10H8Hg]3. J. Am. Chem. Soc. 1995, 117, 12338–12339. [Google Scholar] [CrossRef]
- Spokoyny, A.M.; Machan, C.W.; Clingerman, D.J.; Rosen, M.S.; Wiester, M.J.; Kennedy, R.D.; Stern, C.L.; Sarjeant, A.A.; Mirkin, C.A. A coordination chemistry dichotomy for icosahedral carborane-based ligands. Nat. Chem. 2011, 3, 590–596. [Google Scholar] [CrossRef]
- Spokoyny, A.M.; Lewis, C.D.; Teverovskiy, G.; Buchwald, S.L. Extremely electron-rich, boron-functionalized, icosahedral carborane-based phosphinoboranes. Organometallics 2012, 31, 8478–8481. [Google Scholar] [CrossRef]
- Janoušek, Z.; Lehmann, U.; Častulik, J.; Cisařová, I.; Michl, J. Li+-Induced σ-Bond metathesis: Aryl for methyl exchange on boron in a methylated monocarbadodecaborate anion. J. Am. Chem. Soc. 2004, 126, 4060–4061. [Google Scholar] [CrossRef]
- Svidlov, S.V.; Voloshin, Y.Z.; Yurgina, N.S.; Potapova, T.V.; Belyy, A.Y.; Ananyev, I.V.; Bubnov, Y.N. Synthesis, structure, and reactivity of C-isopropyl-ortho-carborane organoboron derivatives. Russ. Chem. Bull. Int. Ed. 2014, 63, 2343–2350. [Google Scholar] [CrossRef]
- Cheng, R.; Qiu, Z.; Xie, Z. Iridium-catalysed regioselective borylation of carboranes via direct B-H activation. Nat. Commun. 2017, 8, 14827. [Google Scholar] [CrossRef]
- Weber, L.; Kahlert, J.; Brockhinke, R.; Böhling, L.; Brockhinke, A.; Stammler, H.-G.; Neumann, B.; Harder, R.A.; Fox, M.A. Luminescence properties of C-diazaborolyl-ortho-carboranes as donor–acceptor systems. Chem. Eur. J. 2012, 18, 8347–8357. [Google Scholar] [CrossRef]
- Weber, L.; Kahlert, J.; Böhling, L.; Brockhinke, A.; Stammler, H.-G.; Neumann, B.; Harder, R.A.; Low, P.J.; Fox, M.A. Electrochemical and spectroelectrochemical studies of C-benzodiazaborolyl-ortho-carboranes. Dalton Trans. 2013, 42, 2266–2281. [Google Scholar] [CrossRef]
- Gimbert, C.; Lumbierres, M.; Marchi, C.; Moreno-Mañas, M.; Sebastián, R.M.; Vallribera, A. Michael additions catalyzed by phosphines. An overlooked synthetic method. Tetrahedron 2005, 61, 8598–8605. [Google Scholar] [CrossRef]
- Gómez-Bengoa, E.; Cuerva, J.M.; Mateo, C.; Echavarren, A.M. Michael reaction of stabilized carbon nucleophiles catalyzed by [RuH2(PPh3)4]. J. Am. Chem. Soc. 1996, 118, 8553–8565. [Google Scholar] [CrossRef]
- Saidi, M.R.; Azizi, N.; Akbari, E.; Ebrahimi, F. LiCO4/Et3N: Highly efficient and active catalyst for selective Michael addition of active methylene compounds under solvent-free condition. J. Mol. Cat. A: Chem. 2008, 292, 44–48. [Google Scholar] [CrossRef]
- Baslé, O.; Porcel, S.; Ladeira, S.; Bouhadir, G.; Bourissou, D. Phosphine-boronates: efficient bifunctional organocatalysts for Michael addition. Chem. Commun. 2012, 48, 4495–4497. [Google Scholar] [CrossRef]
- Tamke, S.; Daniliuc, C.-G.; Paradies, J. Frustrated Lewis pair catalyzed hydrosilylation and hydrosilane mediated hydrogenation of fulvenes. Org. Biomol. Chem. 2014, 12, 9139–9144. [Google Scholar] [CrossRef]
- Riley, L.E.; Krämer, T.; McMullin, C.L.; Ellis, D.; Rosair, G.M.; Sivaev, I.B.; Welch, A.J. Large, weakly basic bis(carboranyl)phosphines: an experimental and computational study. Dalton Trans. 2017, 46, 5218–5228. [Google Scholar] [CrossRef]
- Allen, D.W.; Taylor, B.F. The chemistry of heteroarylphosphorus compounds. Part 15. Phosphorus-31 nuclear magnetic resonance studies of the donor properties of heteroarylphosphines towards selenium and platinum(II). J. C. S. Dalton 1982, 51–54. [Google Scholar] [CrossRef]
- Beckmann, U.; Süslüyan, D.; Kunz, P.C. Is the 1JPSe coupling constant a reliable probe for the basicity of phosphines? A 31P NMR study. phosphorus. Sulfur Silicon Relat. Elem. 2011, 186, 2061–2070. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Kivekäs, R.; Teixidor, F.; Viñas, C.; Nuñez, R. 1-Diphenylphosphino-1,2-dicarba-closo-dodecaborane(12) at 153 K. Acta Cryst. 1995, C51, 1868–1870. [Google Scholar]
- Nuñez, R.; Viñas, C.; Teixidor, F.; Sillanpää, R.; Kivekäs, R. Contribution of the o-carboranyl fragment to the chemical stability and the 31P-NMR chemical shift in closo-carboranylphosphines. Crystal structure of bis(1-yl-2-methyl-1,2-dicarba-closo-dodecaborane)phenylphosphine. J. Organometal. Chem. 1999, 592, 22–28. [Google Scholar] [CrossRef]
- Fey, N.; Haddow, M.F.; Mistry, R.; Norman, N.C.; Orpen, A.G.; Reynolds, T.J.; Pringle, P.G. Regioselective B-Cyclometalation of a bulky o-carboranyl phosphine and the unexpected formation of a dirhodium(ii) complex. Organometallics 2012, 31, 2907–2913. [Google Scholar] [CrossRef]
- Fein, M.M.; Grafstein, D.; Paustian, J.E.; Bobinski, J.; Lichstein, B.M.; Mayes, N.; Schwartz, N.N.; Cohen, M.S. Carboranes. II. The preparation of 1- and 1,2-substituted carboranes. Inorg. Chem. 1963, 2, 1115–1119. [Google Scholar] [CrossRef]
- Brain, P.T.; Cowie, J.; Donohue, D.J.; Hnyk, D.; Rankin, D.W.H.; Reed, D.; Reid, B.D.; Robertson, H.E.; Welch, A.J.; Hofmann, M.; et al. 1-Phenyl-1, 2-dicarba-closo-dodecaborane, 1-Ph-1, 2-closo-C2B10H11. Synthesis, characterization, and structure as determined in the gas phase by electron diffraction, in the crystalline phase at 199 K by X-ray diffraction, and by ab initio computations. Inorg. Chem. 1996, 35, 1701–1708. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- McAnaw, A.; Scott, G.; Elrick, L.; Rosair, G.M.; Welch, A.J. The VCD method‚A simple and reliable way to distinguish cage C and B atoms in (hetero)carborane structures determined crystallographically. Dalton Trans. 2013, 42, 645–664. [Google Scholar] [CrossRef]
- McAnaw, A.; Lopez, M.E.; Ellis, D.; Rosair, G.M.; Welch, A.J. Asymmetric 1,8/13,2,x-M2C2B10 14-vertex metallacarboranes by direct electrophilic insertion reactions; the VCD and BHD methods in critical analysis of cage C atom positions. Dalton Trans. 2014, 43, 5095–5105. [Google Scholar] [CrossRef]
- Welch, A.J. What can we learn from the crystal structures of metallacarboranes? Crystals 2017, 7, 234. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds not available. |







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).