Pd-Catalyzed Suzuki-Miyaura Cross-Coupling of Pentafluorophenyl Esters



Abstract
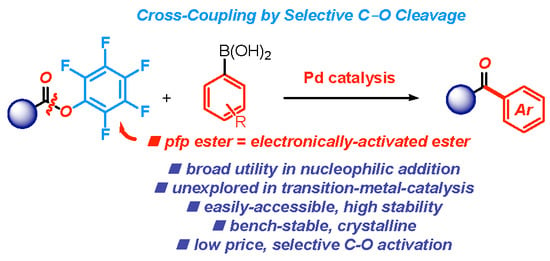
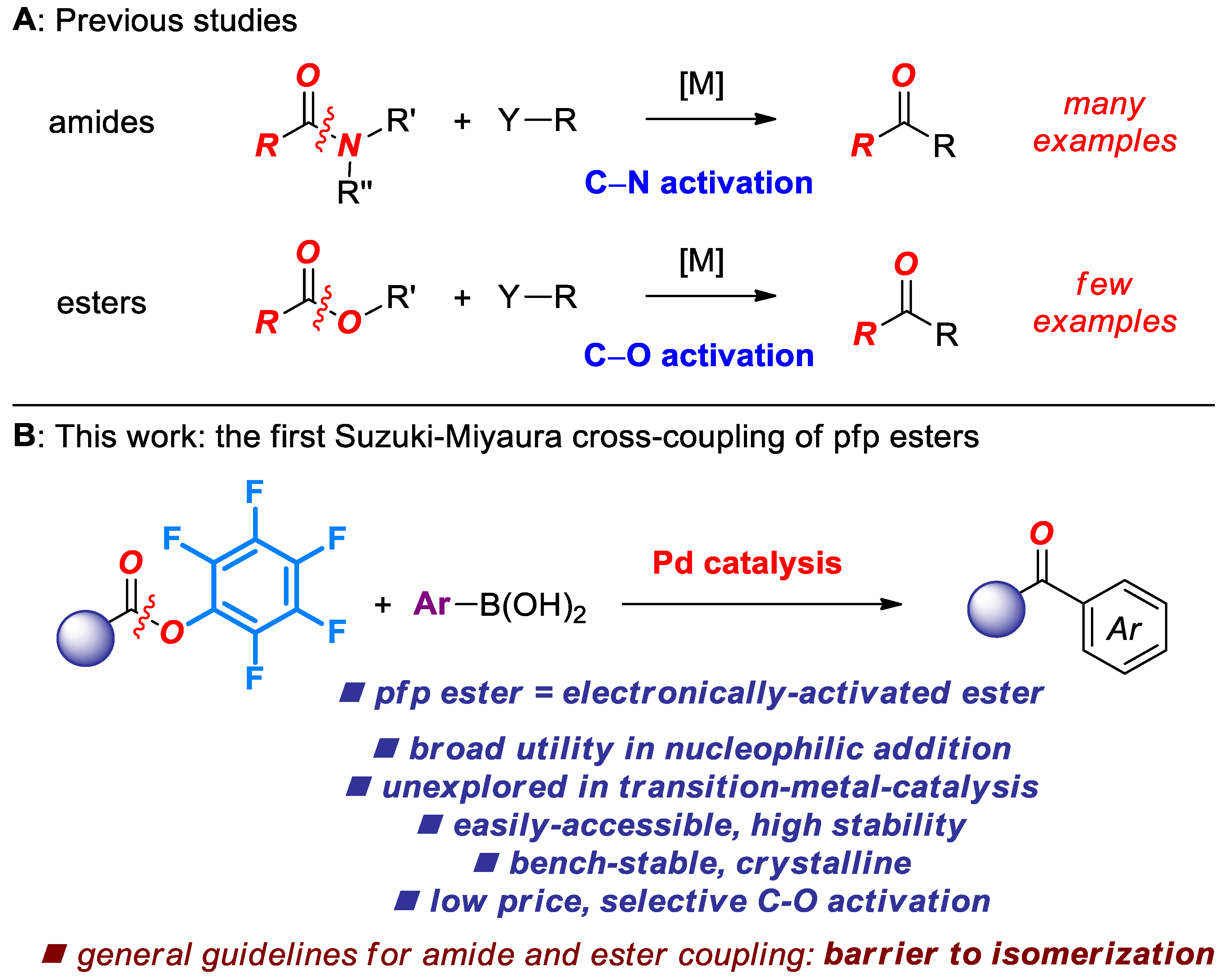
1. Introduction
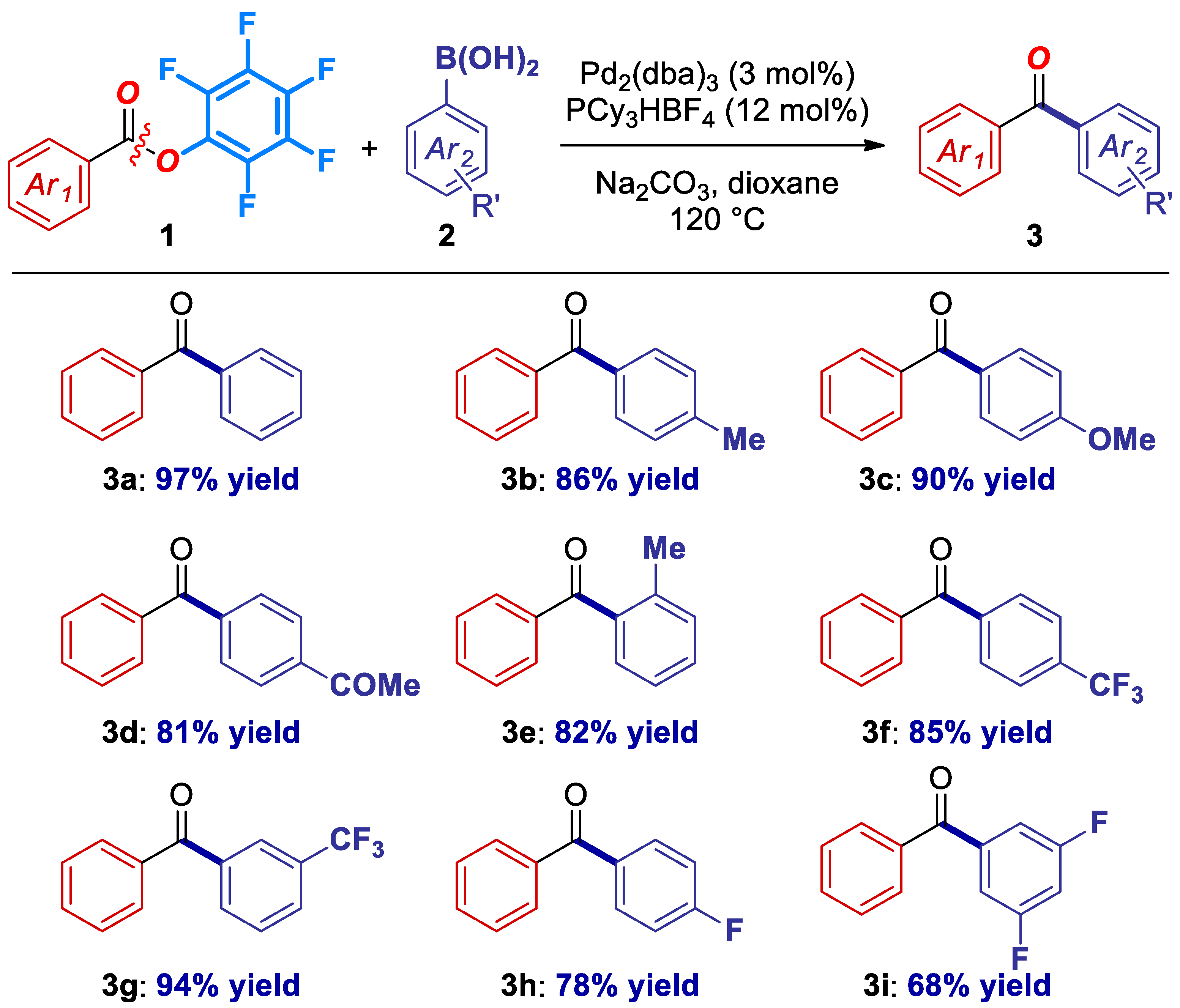
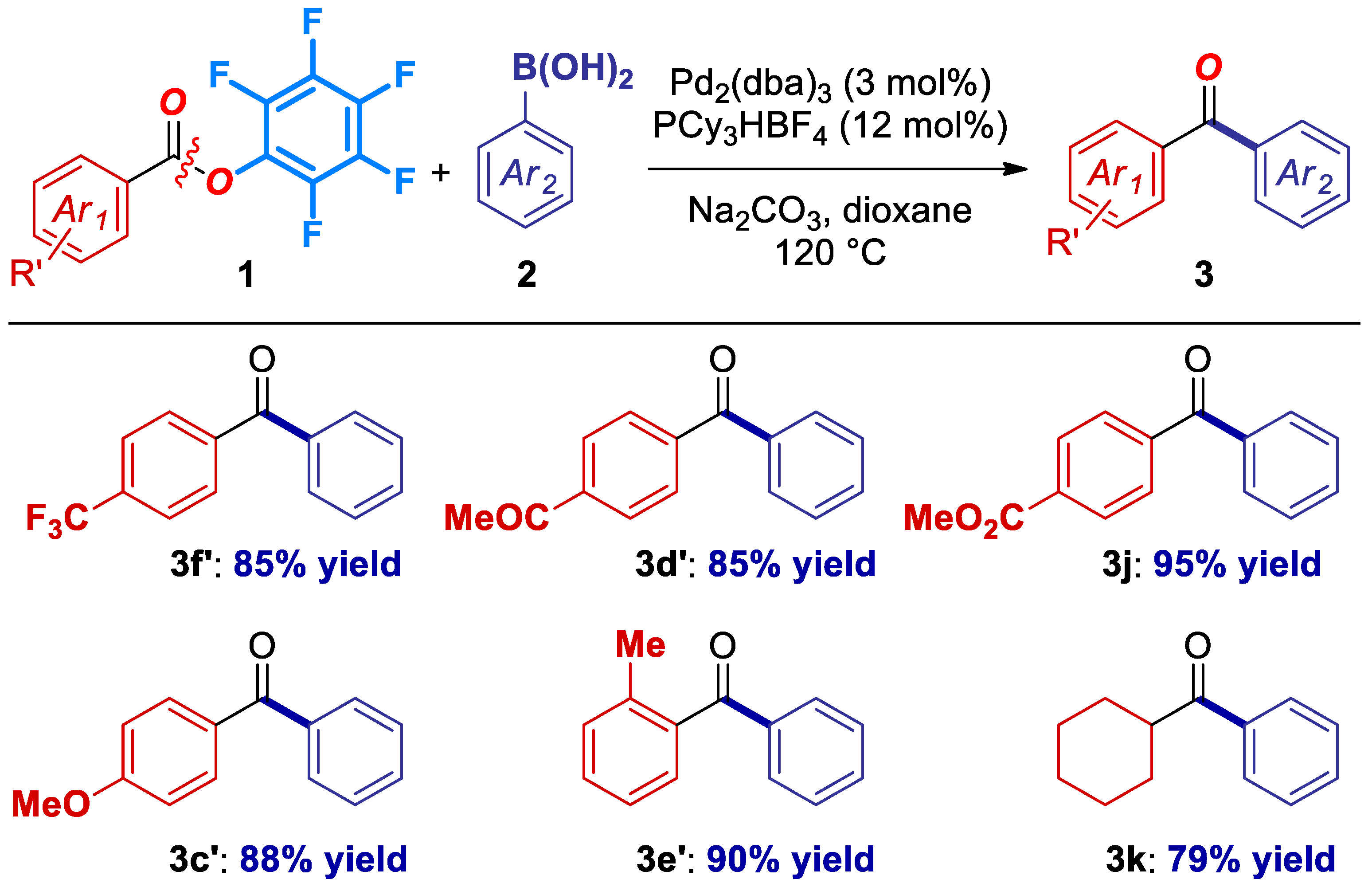
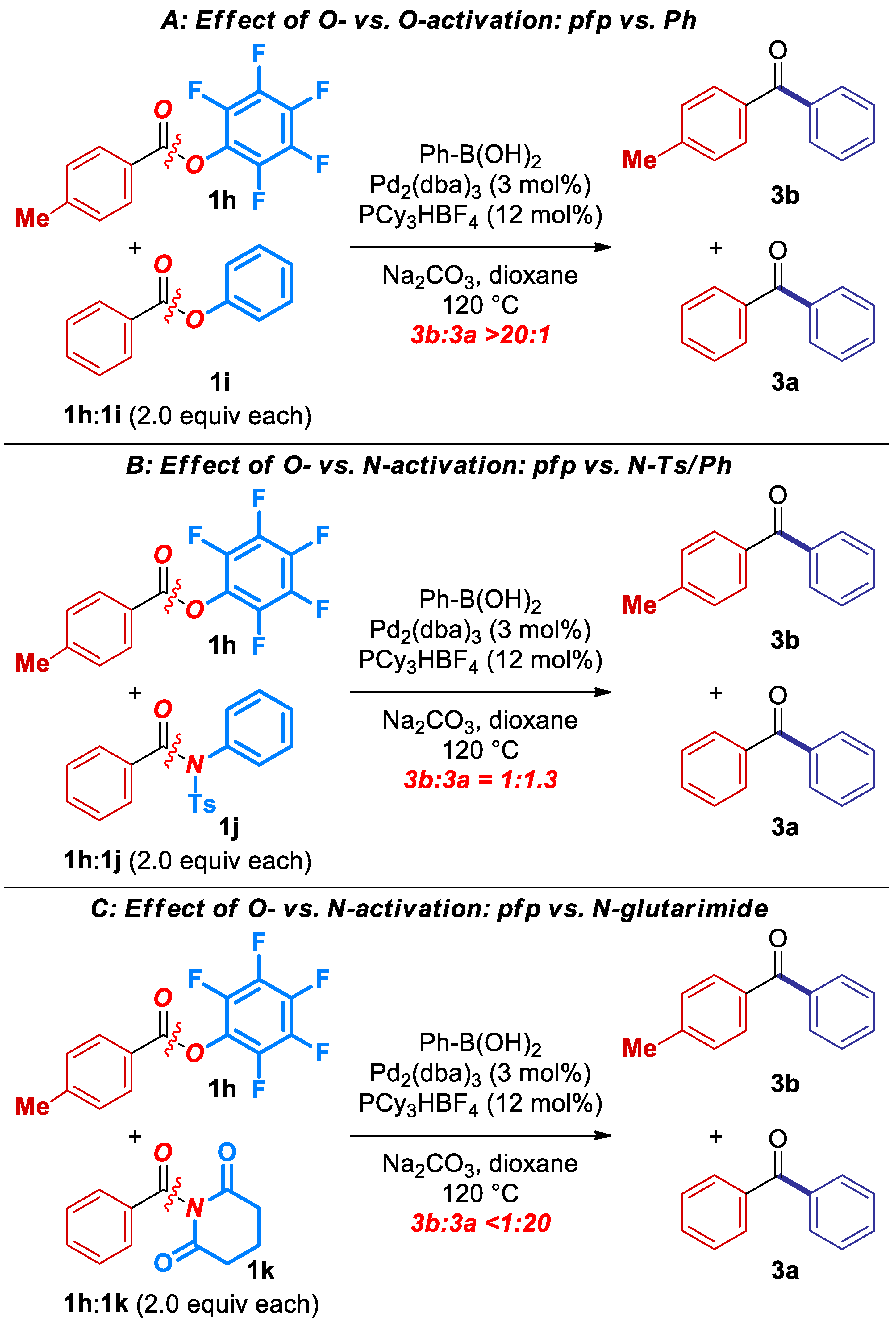
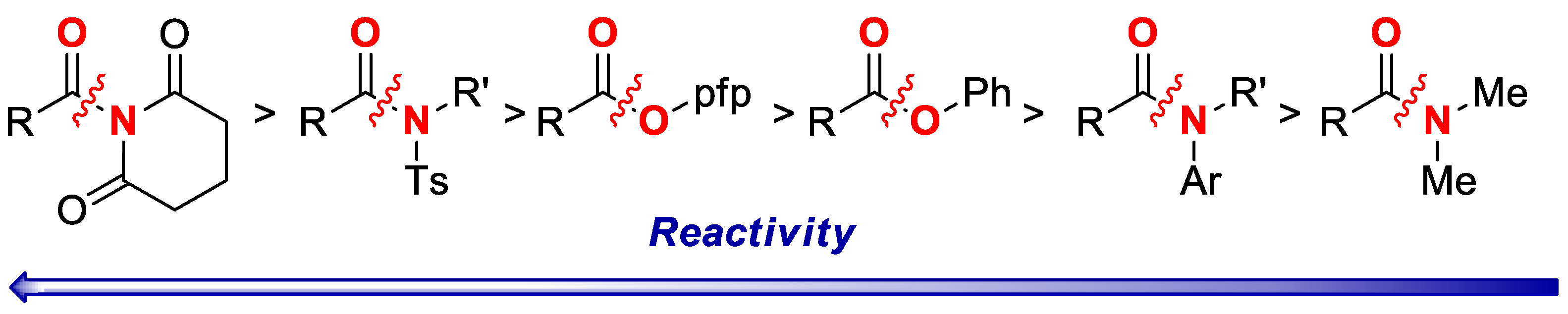
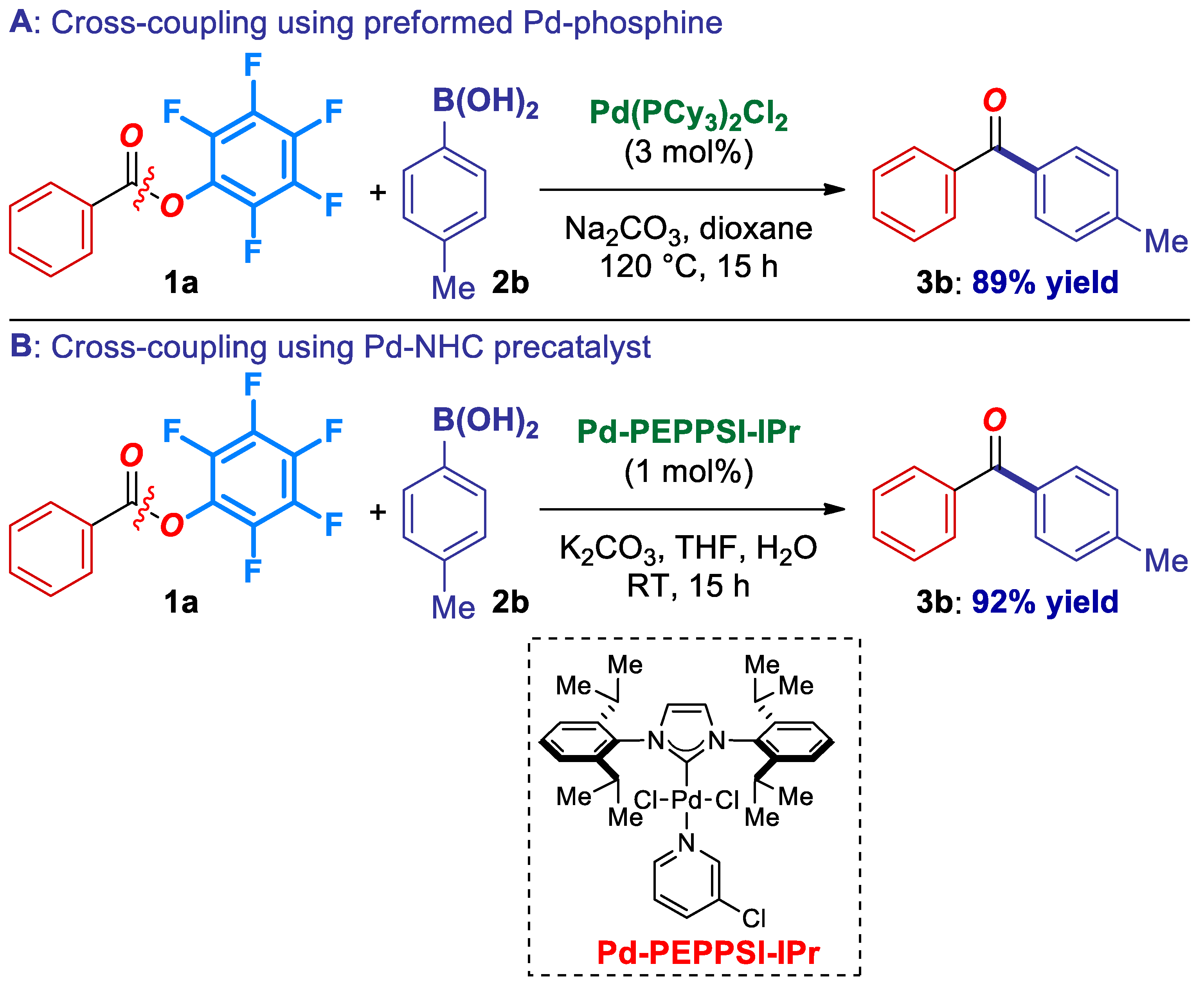
2. Results
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. General Procedure for Cross-Coupling of Pentafluorophenyl Esters
4.3. Representative Procedure for Cross-Coupling of Pentafluorophenyl Esters
4.4. Characterization Data for Products 3a–3k (Figures 1 and 2)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shi, S.; Nolan, S.P.; Szostak, M. Well-Defined Palladium(II)-NHC (NHC = N-Heterocyclic Carbene) Precatalysts for Cross-Coupling Reactions of Amides and Esters by Selective Acyl CO–X (X = N, O) Cleavage. Acc. Chem. Res. 2018, 51, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.; Bauer, A.; Lemmerer, M.; Maulide, N. Amide Activation: An Emerging Tool for Chemoselective Synthesis. Chem. Soc. Rev. 2018, 47, 7899–7925. [Google Scholar] [CrossRef] [PubMed]
- Takise, R.; Muto, K.; Yamaguchi, J. Cross-Coupling of Aromatic Esters and Amides. Chem. Soc. Rev. 2017, 46, 5864–5888. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Szostak, M. Twisted Amides: From Obscurity to Broadly Useful Transition-Metal-Catalyzed Reactions by N–C Amide Bond Activation. Chem. Eur. J. 2017, 23, 7157–7173. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Shi, S.; Szostak, M. Cross-Coupling of Amides by N–C Bond Activation. Synlett 2016, 27, 2530–2540. [Google Scholar] [CrossRef]
- Meng, G.; Szostak, M. N-Acyl-Glutarimides: Privileged Scaffolds in Amide N–C Bond Cross-Coupling. Eur. J. Org. Chem. 2018, 20–21, 2352–2365. [Google Scholar] [CrossRef]
- Liu, C.; Szostak, M. Decarbonylative Cross-Coupling of Amides. Org. Biomol. Chem. 2018, 16. in press. [Google Scholar] [CrossRef] [PubMed]
- Dander, J.E.; Garg, N.K. Breaking Amides using Nickel Catalysis. ACS Catal. 2017, 7, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Shi, S.; Lalancette, R.; Szostak, R.; Szostak, M. Reversible Twisting of Primary Amides via Ground State N–C(O) Destabilization: Highly Twisted Rotationally Inverted Acyclic Amides. J. Am. Chem. Soc. 2018, 140, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Szostak, R.; Shi, S.; Meng, G.; Lalancette, R.; Szostak, M. Ground-State Distortion in N-Acyl-tert-butyl-carbamates (Boc) and N-Acyl-tosylamides (Ts): Twisted Amides of Relevance to Amide N–C Cross-Coupling. J. Org. Chem. 2016, 81, 8091–8094. [Google Scholar] [CrossRef] [PubMed]
- Pace, V.; Holzer, W.; Meng, G.; Shi, S.; Lalancette, R.; Szostak, R.; Szostak, M. Structures of Highly Twisted Amides Relevant to Amide N–C Cross-Coupling: Evidence for Ground-State Amide Destabilization. Chem. Eur. J. 2016, 22, 14494–14498. [Google Scholar] [CrossRef] [PubMed]
- Szostak, R.; Meng, G.; Szostak, M. Resonance Destabilization in N-Acylanilines (Anilides): Electronically-Activated Planar Amides of Relevance in N–C(O) Cross-Coupling. J. Org. Chem. 2017, 82, 6373–6378. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, G.; Shi, S.; Meng, G.; Lalancette, R.; Szostak, R.; Szostak, M. Acyl and Decarbonylative Suzuki Coupling of N-Acetyl Amides: Electronic Tuning of Twisted, Acyclic Amides in Catalytic Carbon−Nitrogen Bond Cleavage. ACS Catal. 2018, 8, 9131–9139. [Google Scholar] [CrossRef]
- Halima, T.B.; Zhang, W.; Yalaoui, I.; Hong, X.; Yang, Y.-F.; Houk, K.N.; Newman, S.G. Palladium-Catalyzed Suzuki-Miyaura Coupling of Aryl Esters. J. Am. Chem. Soc. 2017, 139, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Meng, G.; Shi, S.; Ling, Y.; An, J.; Szostak, R.; Szostak, M. Suzuki-Miyaura Cross-Coupling of Amides and Esters at Room Temperature: Correlation with Barriers to Rotation around C–N and C–O Bonds. Chem. Sci. 2017, 8, 6525–6530. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Lei, P.; Szostak, M. Pd-PEPPSI: A General Pd-NHC Precatalyst for Suzuki-Miyaura Cross- Coupling of Esters by C–O Cleavage. Organometallics 2017, 36, 3784–3789. [Google Scholar] [CrossRef]
- Li, G.; Shi, S.; Szostak, M. Pd-PEPPSI: Water-Assisted Suzuki-Miyaura Cross-Coupling of Aryl Esters at Room Temperature using a Practical Palladium-NHC (NHC = N-Heterocyclic Carbene) Precatalyst. Adv. Synth. Catal. 2018, 360, 1538–1543. [Google Scholar] [CrossRef]
- Dardir, A.H.; Melvin, P.R.; Davis, R.M.; Hazari, N.; Beromi, M.M. Rapidly Activating Pd-Precatalyst for Suzuki-Miyaura and Buchwald-Hartwig Couplings of Aryl Esters. J. Org. Chem. 2017, 83, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Liebman, J.; Greenberg, A. The Origin of Rotational Barriers in Amides and Esters. Biophys. Chem. 1974, 1, 222–226. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Natarajan, S.; Li, H.; Jain, N.F.; Hughes, R.; Solomon, M.E.; Ramanjulu, J.M.; Boddy, C.N.C.; Takayanagi, M. Total Synthesis of Vancomycin Aglycon. Part 1: Synthesis of Amino Acids 4–7 and Construction of the AB-COD Ring Skeleton. Angew. Chem. Int. Ed. 1998, 37, 2708–2714. [Google Scholar] [CrossRef]
- Al-Warhi, T.I.; Al-Hazimi, H.M.A.; El-Faham, A. Recent development in peptide coupling reagents. J. Saudi Chem. Soc. 2012, 16, 97–116. [Google Scholar] [CrossRef]
- Specklin, S.; Cossy, J. Chemoselective Synthesis of β-Ketophosphonates Using Lithiated α-(Trimethylsilyl)methylphosphonate. J. Org. Chem. 2015, 80, 3302–3308. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Rueping, M. Decarbonylative Cross-Couplings: Nickel Catalyzed Functional Group Interconversion Strategies for the Construction of Complex Organic Molecules. Acc. Chem. Res. 2018, 51, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Rueping, M. Transition-Metal-Catalyzed Decarbonylative Coupling Reactions: Concepts, Classifications, and Applications. Chem. Eur. J. 2018, 24, 7794–7809. [Google Scholar] [CrossRef] [PubMed]
- Gildner, P.G.; Colacot, T.J. Reactions of the 21st Century: Two Decades of Innovative Catalyst Design for Palladium-Catalyzed Cross-Couplings. Organometallics 2015, 34, 5497–5508. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Ligand | Base | [Pd]:L | Yield (%) |
|---|---|---|---|---|---|
| 1 | Pd(OAc)2 | PCy3HBF4 | Na2CO3 | 1:4 | 52 |
| 2 | Pd(OAc)2 | PCy3HBF4 | KHCO3 | 1:4 | 53 |
| 3 | Pd(OAc)2 | PCy3HBF4 | NaHCO3 | 1:4 | 30 |
| 4 | Pd(OAc)2 | PCy3HBF4 | K2CO3 | 1:4 | 12 |
| 5 | Pd(OAc)2 | PCy3HBF4 | K3PO4 | 1:4 | 47 |
| 6 | Pd(OAc)2 | PCy3HBF4 | KF | 1:4 | 56 |
| 7 | Pd(OAc)2 | PPhCy2 | Na2CO3 | 1:4 | 60 |
| 8 | Pd(OAc)2 | PPh2Cy | Na2CO3 | 1:4 | 5 |
| 9 | Pd(OAc)2 | PPh3 | Na2CO3 | 1:4 | 5 |
| 10 | Pd(OAc)2 | DPPB | Na2CO3 | 1:4 | 13 |
| 11 | Pd(OAc)2 | Xantphos | Na2CO3 | 1:4 | <5 |
| 12 | Pd(OAc)2 | Pt-Bu3HBF4 | Na2CO3 | 1:4 | <5 |
| 13 | Pd(dba)2 | PCy3HBF4 | Na2CO3 | 1:4 | 23 |
| 14 | PdCl2 | PCy3HBF4 | Na2CO3 | 1:4 | 25 |
| 15 | Pd2(dba)3 | PCy3HBF4 | Na2CO3 | 1:2 | 74 |
| 16 2 | Pd2(dba)3 | PCy3HBF4 | Na2CO3 | 1:2 | 85 |
| 17 3 | Pd2(dba)3 | PCy3HBF4 | Na2CO3 | 1:1 | 44 |
| 18 4 | Pd2(dba)3 | PCy3HBF4 | Na2CO3 | 2:1 | 30 |
| 19 5 | Pd(OAc)2 | PCy3HBF4 | Na2CO3 | 1:4 | 83 |
| 20 5 | Pd2(dba)3 | PCy3HBF4 | Na2CO3 | 1:2 | 92 |
| 21 6 | Pd2(dba)3 | PCy3HBF4 | Na2CO3 | 1:2 | 75 |
| 22 7 | Pd2(dba)3 | PCy3HBF4 | Na2CO3 | 1:2 | 89 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buchspies, J.; J. Pyle, D.; He, H.; Szostak, M. Pd-Catalyzed Suzuki-Miyaura Cross-Coupling of Pentafluorophenyl Esters. Molecules 2018, 23, 3134. https://doi.org/10.3390/molecules23123134
Buchspies J, J. Pyle D, He H, Szostak M. Pd-Catalyzed Suzuki-Miyaura Cross-Coupling of Pentafluorophenyl Esters. Molecules. 2018; 23(12):3134. https://doi.org/10.3390/molecules23123134
Chicago/Turabian StyleBuchspies, Jonathan, Daniel J. Pyle, Huixin He, and Michal Szostak. 2018. "Pd-Catalyzed Suzuki-Miyaura Cross-Coupling of Pentafluorophenyl Esters" Molecules 23, no. 12: 3134. https://doi.org/10.3390/molecules23123134
APA StyleBuchspies, J., J. Pyle, D., He, H., & Szostak, M. (2018). Pd-Catalyzed Suzuki-Miyaura Cross-Coupling of Pentafluorophenyl Esters. Molecules, 23(12), 3134. https://doi.org/10.3390/molecules23123134