Mepazine Inhibits RANK-Induced Osteoclastogenesis Independent of Its MALT1 Inhibitory Function


 ,
,
Abstract
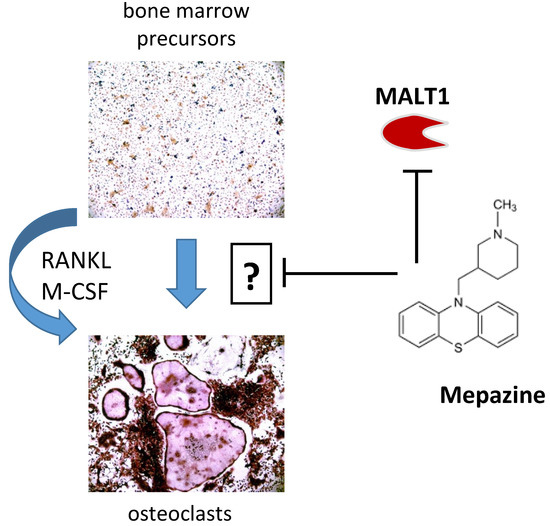
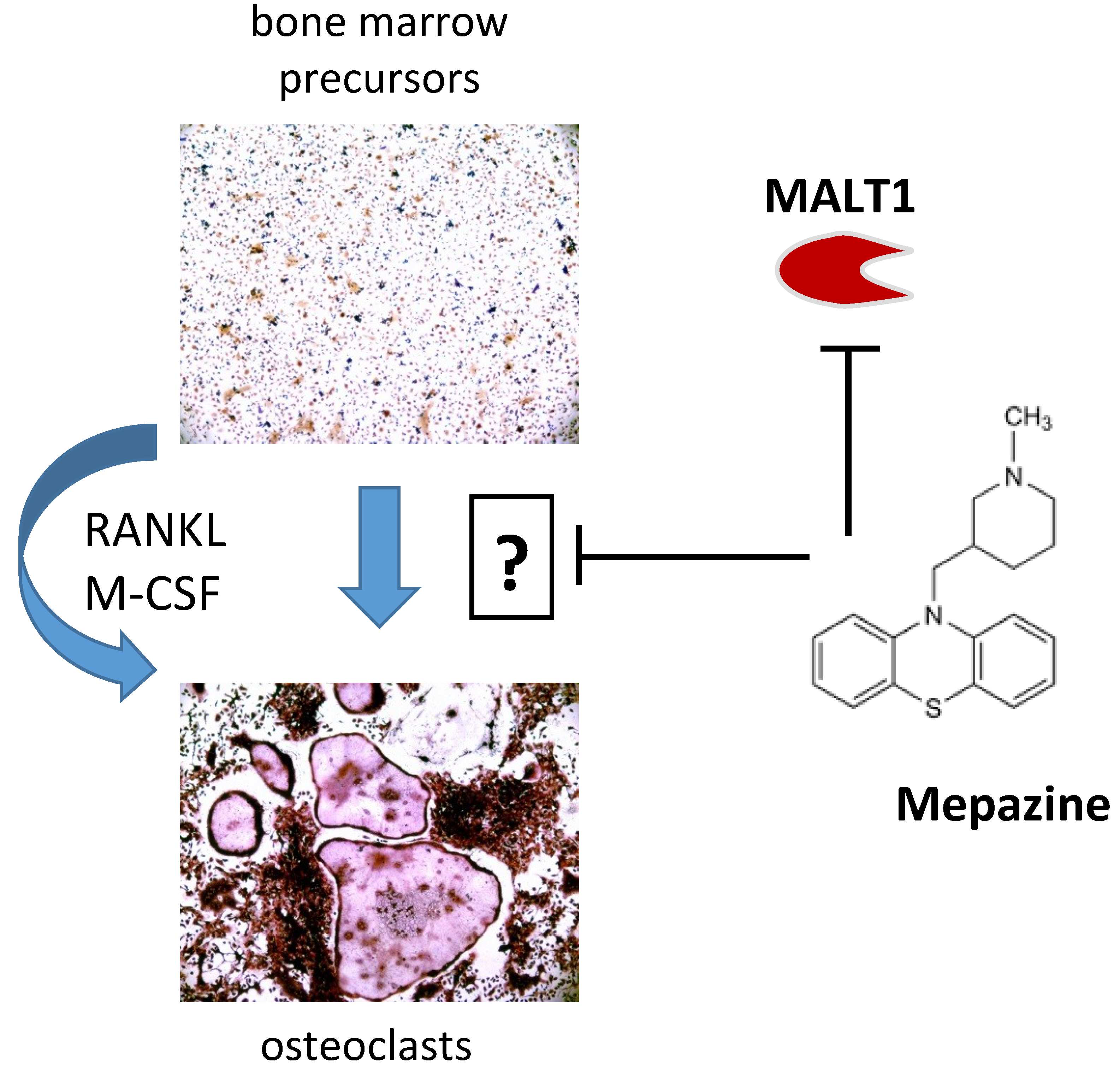
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
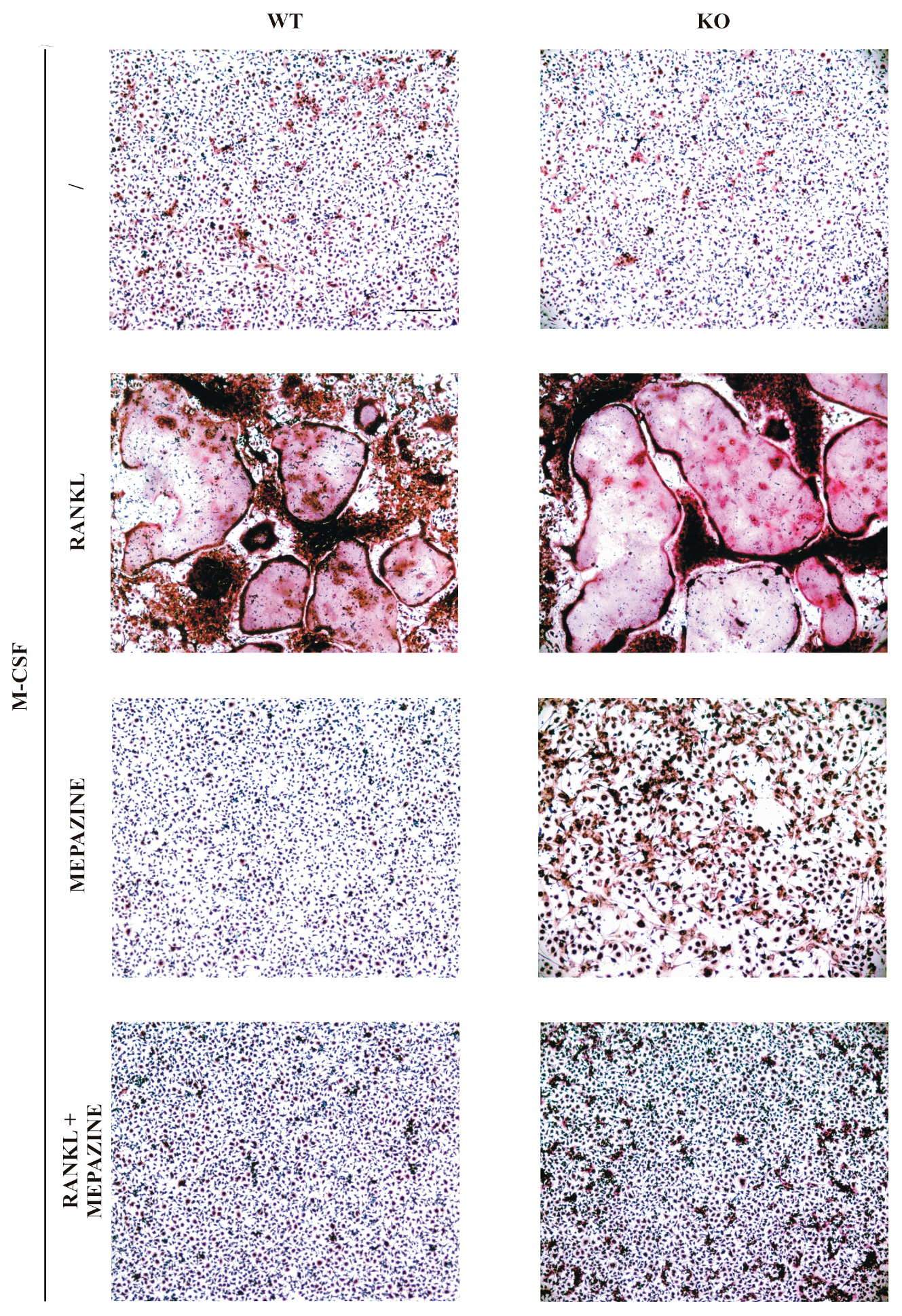
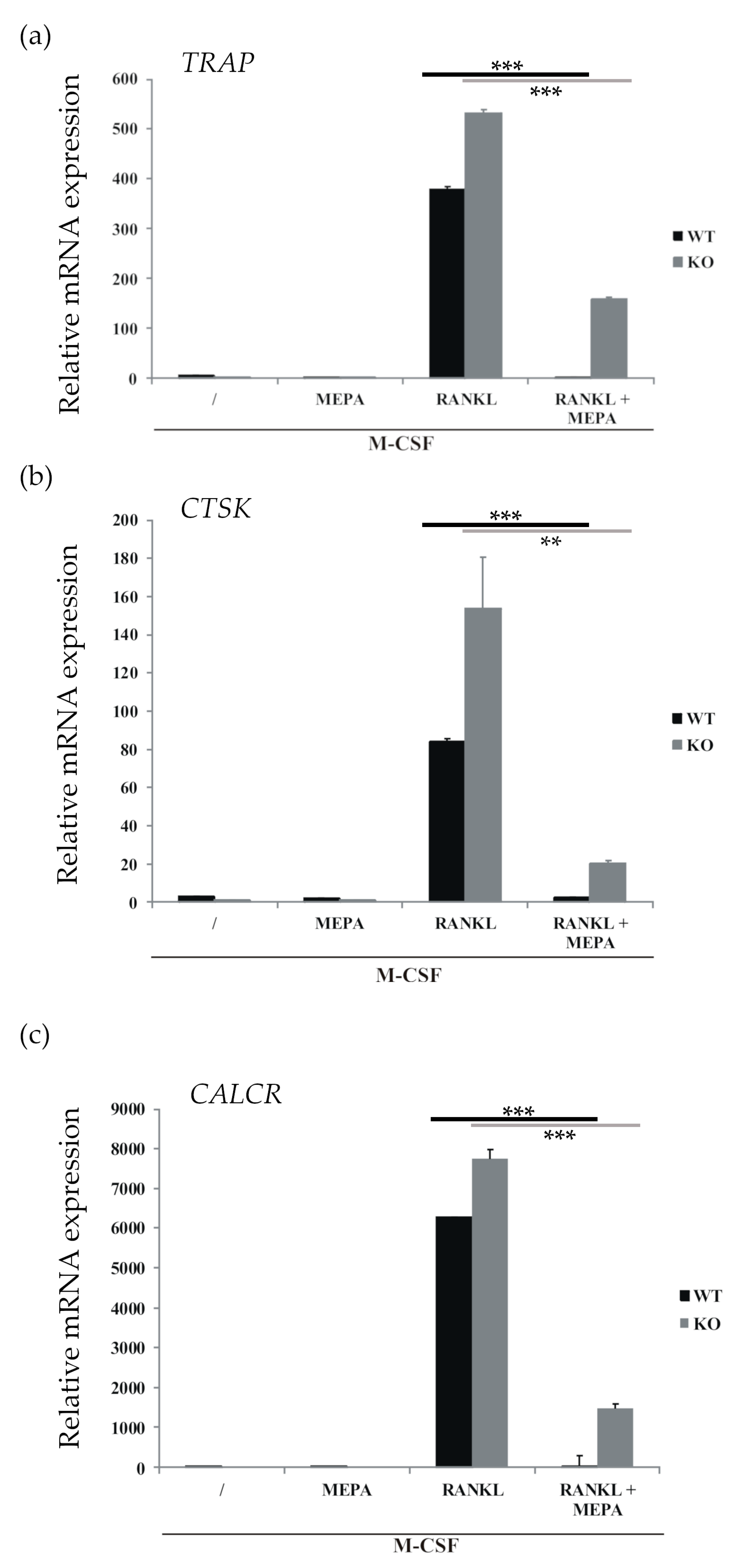
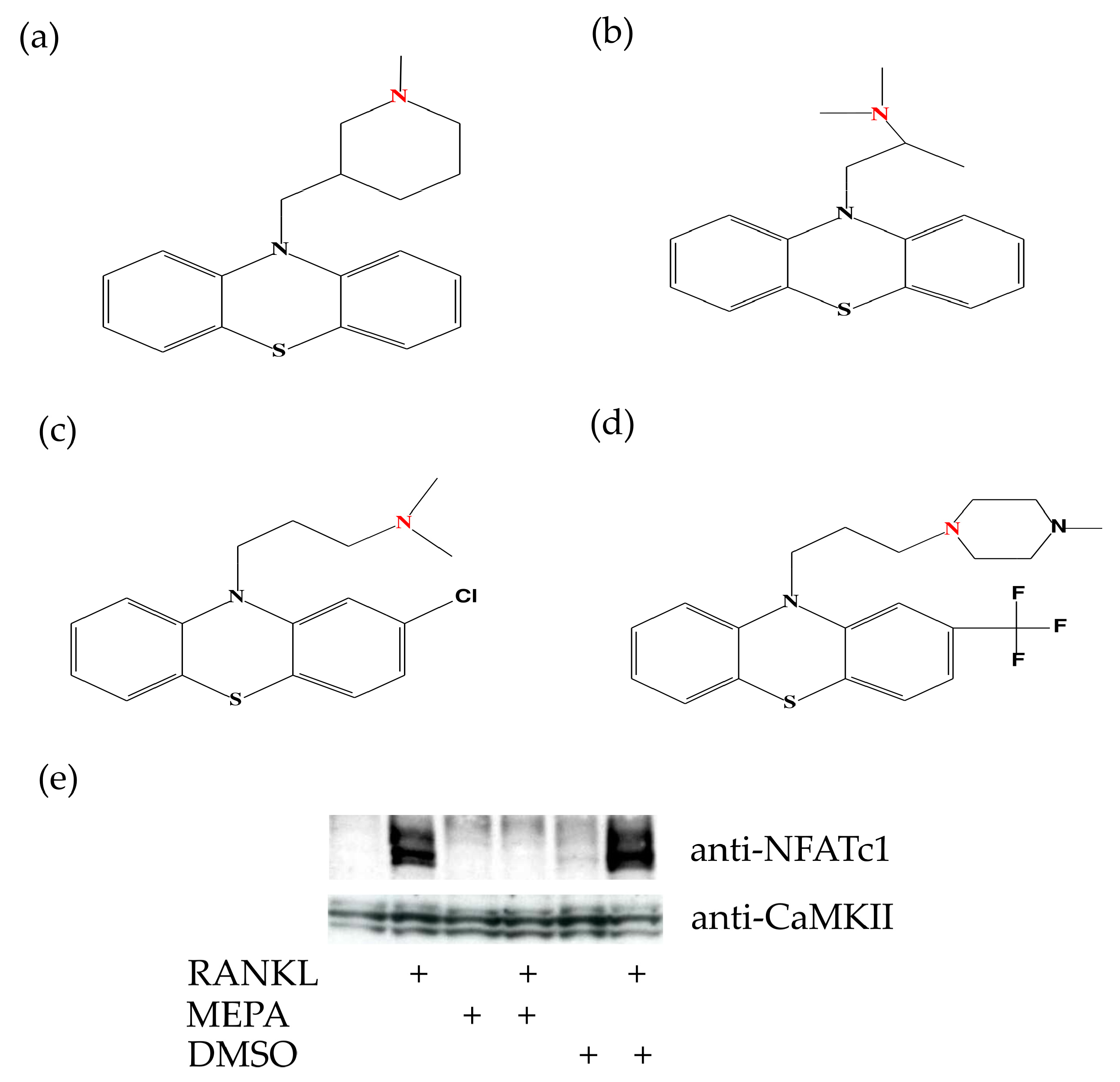
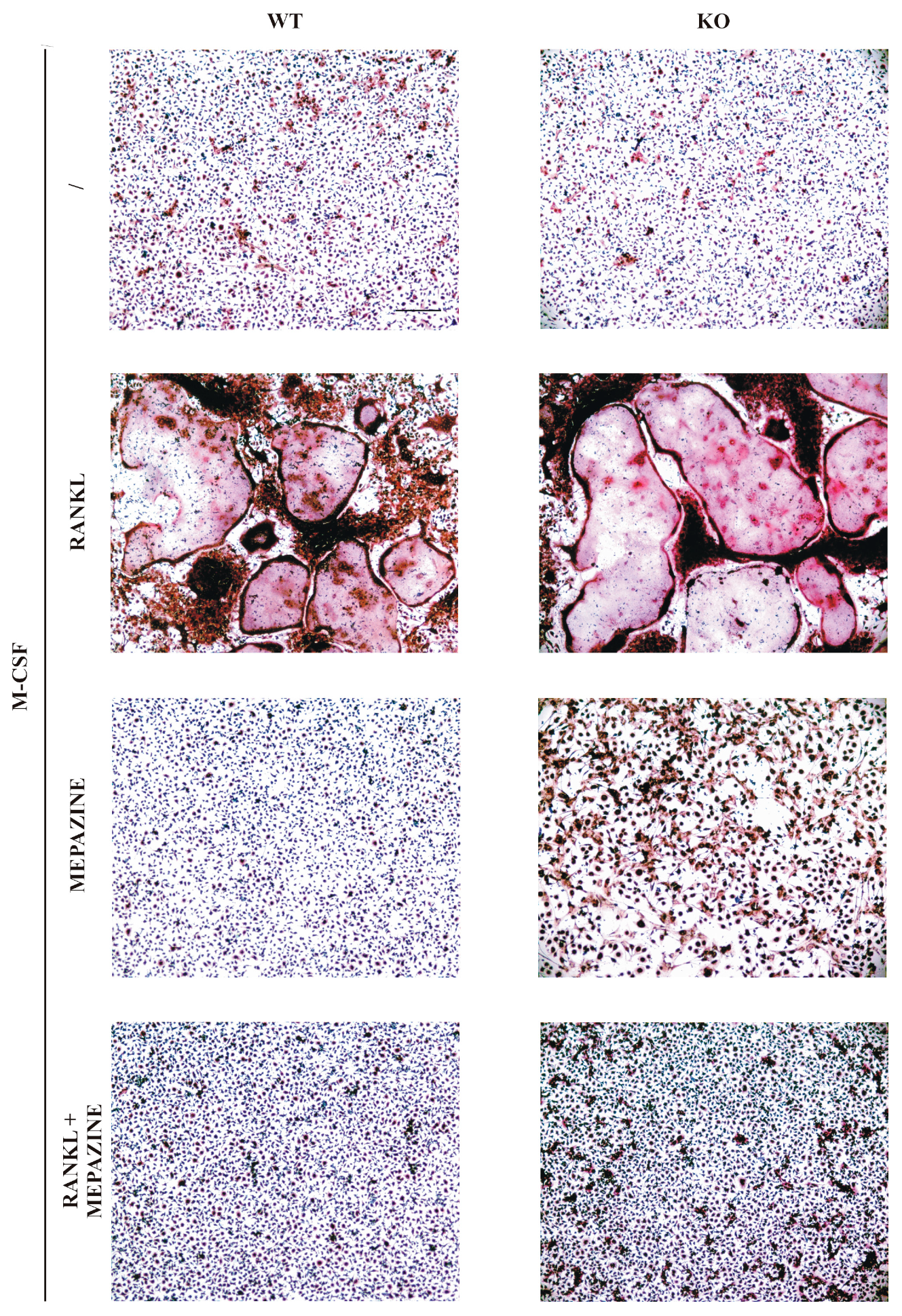
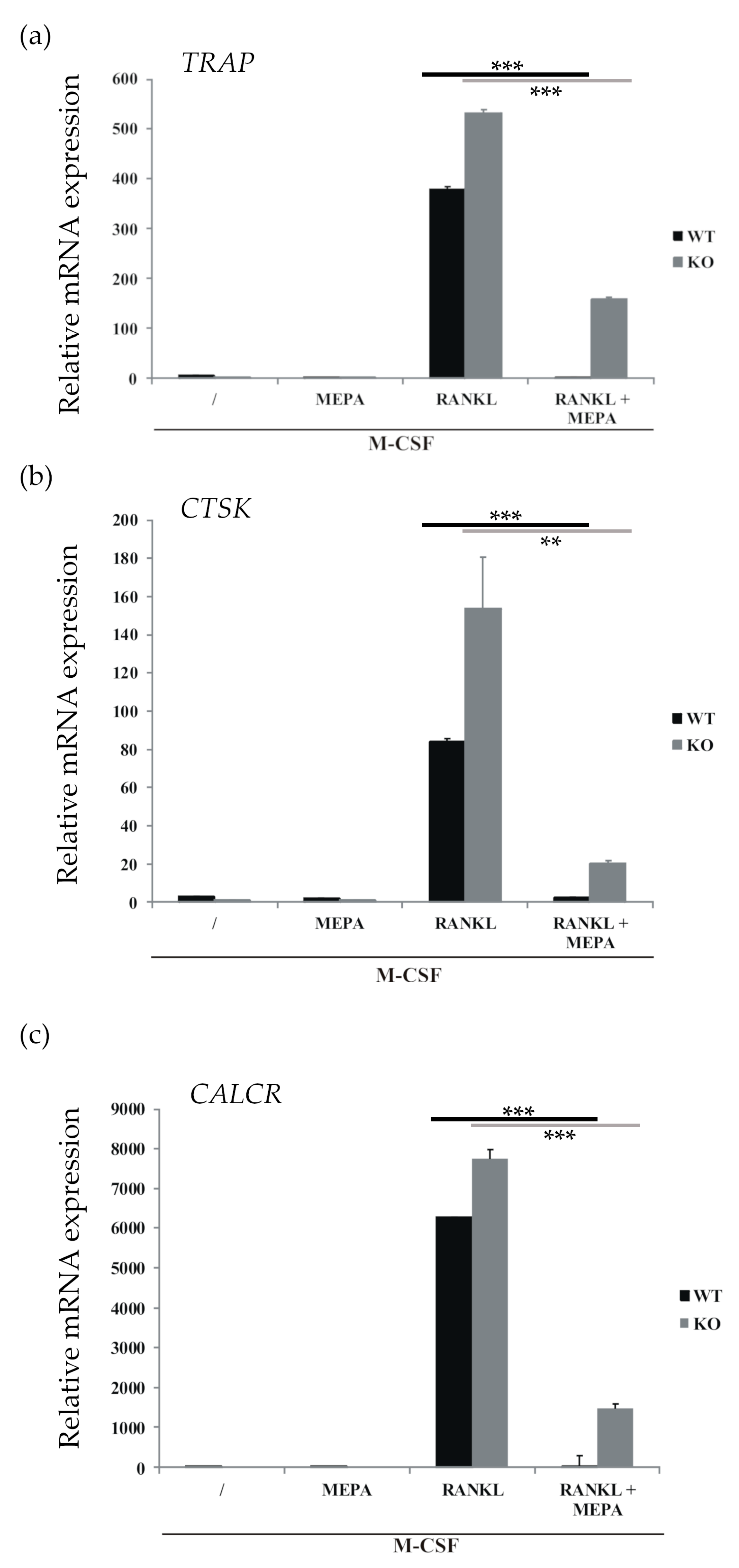
2.1. Mepazine Inhibits RANK-Induced Osteoclastogenesis Independent of MALT1
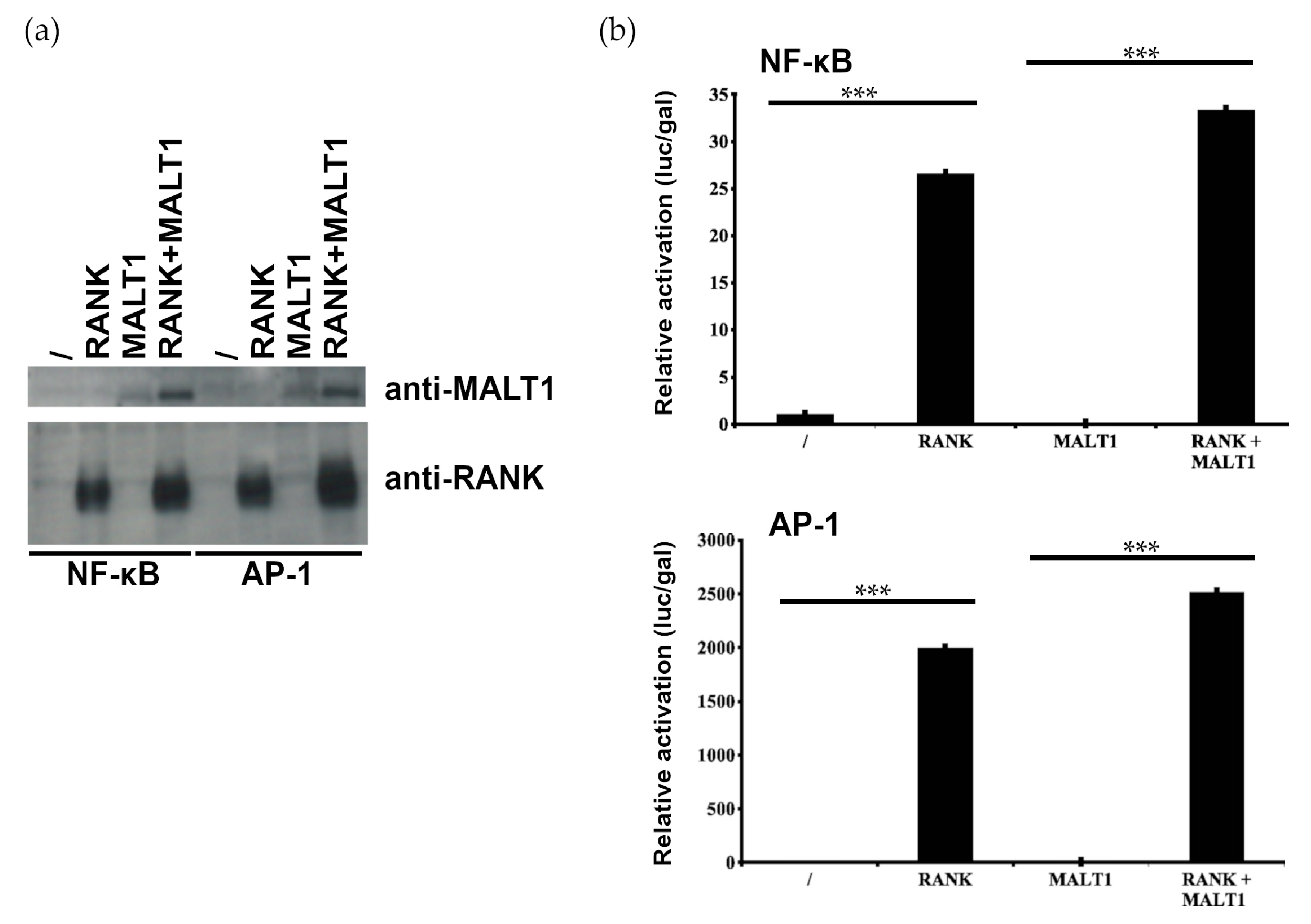
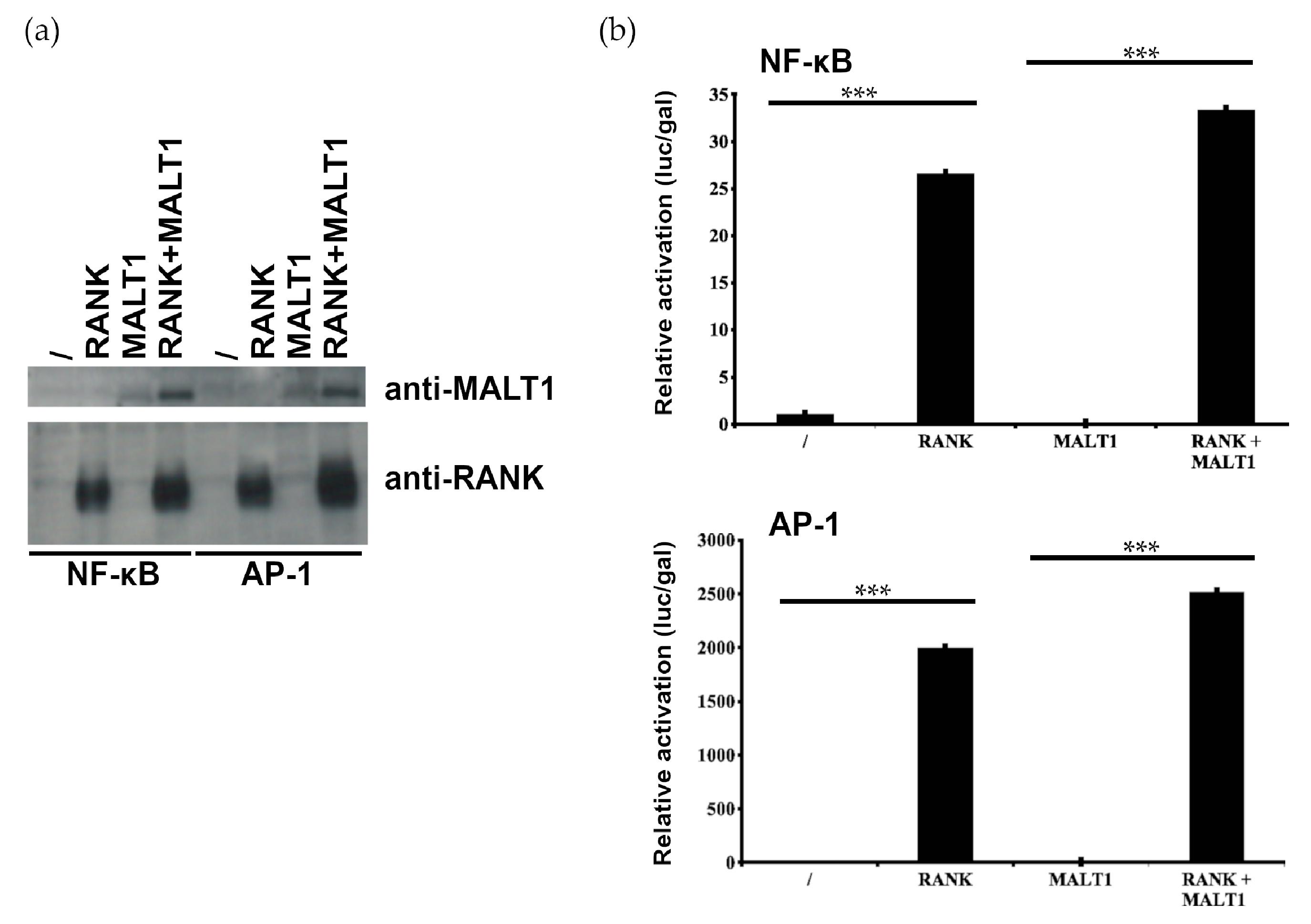
2.2. MALT1 Is not Involved in RANK-Induced NF-κB and AP1 Signaling
2.3. Mepazine Inhibits CaMKII Phosphorylation and NFATc1 Expression Independently of MALT1
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Osteoclast Cell Culture and TRAP Staining
4.3. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
| ACTIN FW | GCTTCTAGGCGGACTGTTACTGA | MALT1 FW | GGACAAAGTCGCCCTTTTGAT |
| ACTIN REV | GCCATGCCAATGTTGTCTCTTAT | MALT1 REV | TCCACAGCGTTACACATCTCA |
| GADPH FW | TGAAGCAGGCATCTGAGGG | TRAP FW | TGGTCCAGGAGCTTAACTGC |
| GADPH REV | CGAAGGTGGAAGAGTGGGAG | TRAP REV | GTCAGGAGTGGGAGCCATATG |
| CATHEPSIN K FW | GTGGGTGTTCAAGTTTCTGC | CALCITONIN receptor FW | CTCCAACAAGGTGCTTGGGA |
| CATHEPSIN K REV | GGTGAGTCTTCTTCCATAGC | CALCITONIN receptor REV | GAAGCAGTAGATAGTCGCCA |
4.4. Luciferase Assay and Western Blot Analysis
4.5. Chemical Drawings
Author Contributions
Funding
Conflicts of Interest
References
- Jaszczyszyn, A.; Gąsiorowski, K.; Świątek, P.; Malinka, W.; Cieślik-Boczula, K.; Petrus, J.; Czarnik-Matusewicz, B. Chemical structure of phenothiazines and their biological activity. Pharmacol. Rep. 2012, 64, 16–23. [Google Scholar] [CrossRef]
- Pluta, K.; Morak-Młodawska, B.; Jeleń, M. Recent progress in biological activities of synthesized phenothiazines. Eur. J. Med. Chem. 2011, 46, 3179–3189. [Google Scholar] [CrossRef] [PubMed]
- Nagel, D.; Spranger, S.; Vincendeau, M.; Grau, M.; Raffegerst, S.; Kloo, B.; Hlahla, D.; Neuenschwander, M.; Peter von Kries, J.; Hadian, K.; et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell. 2012, 22, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Schlauderer, F.; Lammens, K.; Nagel, D.; Vincendeau, M.; Eitelhuber, A.C.; Verhelst, S.H.L.; Kling, D.; Chrusciel, A.; Ruland, J.; Krappmann, D.; et al. Structural Analysis of Phenothiazine Derivatives as Allosteric Inhibitors of the MALT1 Paracaspase. Angew. Chem. Int. Ed. 2013, 52, 10384–10387. [Google Scholar] [CrossRef] [PubMed]
- Mc Guire, C.; Elton, L.; Wieghofer, P.; Staal, J.; Voet, S.; Demeyer, A.; Nagel, D.; Krappmann, D.; Prinz, M.; Beyaert, R.; et al. Pharmacological inhibition of MALT1 protease activity protects mice in a mouse model of multiple sclerosis. J. Neuroinflamm. 2014, 11, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kip, E.; Staal, J.; Verstrepen, L.; Tima, H.G.; Terryn, S.; Romano, M.; Lemeire, K.; Suin, V.; Hamouda, A.; Kalai, M.; et al. MALT1 controls attenuated rabies virus by inducing early inflammation and T cell activation in the brain. J. Virol. 2018, 92, e02029-17. [Google Scholar] [CrossRef] [PubMed]
- Kip, E.; Staal, J.; Tima, H.G.; Verstrepen, L.; Romano, M.; Lemeire, K.; Suin, V.; Hamouda, A.; Baens, M.; Libert, C.; et al. Inhibition of MALT1 decreases neuroinflammation and pathogenicity of virulent rabies virus in mice. J. Virol. 2018, 92, e00720-18. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Guo, W.; Hang, N.; Yang, Y.; Wu, X.; Shen, Y.; Cao, J.; Sun, Y.; Xu, Q. MALT1 inhibitors prevent the development of DSS-induced experimental colitis in mice via inhibiting NF-κB and NLRP3 inflammasome activation. Oncotarget 2016, 7, 30536–30549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulpiau, P.; Driege, Y.; Staal, J.; Beyaert, R. MALT1 is not alone after all: Identification of novel paracaspases. Cell. Mol. Life Sci. 2016, 73, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Elton, L.; Carpentier, I.; Beyaert, R. MALT1--a universal soldier: Multiple strategies to ensure NF-κB activation and target gene expression. FEBS J. 2015, 282, 3286–3297. [Google Scholar] [CrossRef] [PubMed]
- Demeyer, A.; Staal, J.; Beyaert, R. Targeting MALT1 Proteolytic Activity in Immunity, Inflammation and Disease: Good or Bad? Trends Mol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Komoda, T.; Ikeda, E.; Nakatani, Y.; Sakagishi, Y.; Maeda, N.; Kato, T.; Kumegawa, M. Inhibitory effect of phenothiazine derivatives on bone in vivo and osteoblastic cells in vitro. Biochem. Pharmacol. 1985, 34, 3885–3889. [Google Scholar] [CrossRef]
- Chikuma, T.; Ishii, Y.; Kato, T.; Kurihara, N.; Hakeda, Y.; Kumegawa, M. Effect of chlorpromazine on PZ-peptidase and several other peptidase activities in cloned osteoblastic cells (MC3T3-E1). Biochem. Pharmacol. 1987, 36, 4319–4324. [Google Scholar] [CrossRef]
- Hall, T.J.; Schaueblin, M. Promethazine inhibits osteoclastic bone resorption in vitro. Calcif. Tissue Int. 1994, 55, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Goldhaber, P.; Rabadjija, L. Effect of promethazine hydrochloride on bone resorption in tissue culture. Exp. Biol. Med. 1982, 169, 105–109. [Google Scholar] [CrossRef]
- Zhang, L.; Feng, X.; McDonald, J.M. The role of calmodulin in the regulation of osteoclastogenesis. Endocrinology 2003, 144, 4536–4543. [Google Scholar] [CrossRef] [PubMed]
- de Lima, V.; Bezerra, M.M.; de Menezes Alencar, V.B.; Vidal, F.D.; da Rocha, F.A.; de Castro Brito, G.A.; de Albuquerque Ribeiro, R. Effects of chlorpromazine on alveolar bone loss in experimental periodontal disease in rats. Eur. J. Oral Sci. 2000, 108, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.J.; Nyugen, H.; Schaeublin, M.; Michalsky, M.; Missbach, M. Phenothiazines are potent inhibitors of osteoclastic bone resorption. Gen. Pharmacol. 1996, 27, 845–848. [Google Scholar] [CrossRef]
- Kawamura, H.; Arai, M.; Togari, A. Inhibitory effect of chlorpromazine on RANKL-induced osteoclastogenesis in mouse bone marrow cells. J. Pharmacol. Sci. 2011, 117, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; Bollerslev, J.; Everts, V.; Karsdal, M.A. Osteoclast activity and subtypes as a function of physiology and pathology--implications for future treatments of osteoporosis. Endocr. Rev. 2011, 32, 31–63. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, T.; Hayashi, M.; Takayanagi, H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol. Metab. 2012, 23, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Eferl, R. Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 2005, 208, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev. Cell. 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Coornaert, B.; Baens, M.; Heyninck, K.; Bekaert, T.; Haegman, M.; Staal, J.; Sun, L.; Chen, Z.J.; Marynen, P.; Beyaert, R. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-κB inhibitor A20. Nat. Immunol 2008, 9, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Staal, J.; Driege, Y.; Bekaert, T.; Demeyer, A.; Muyllaert, D.; Damme, P.V.; Gevaert, K.; Beyaert, R. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. 2011, 30, 1742–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailfinger, S.; Nogai, H.; Pelzer, C.; Jaworski, M.; Cabalzar, K.; Charton, J.E.; Guzzardi, M.; Décaillet, C.; Grau, M.; Dörken, B.; et al. Malt1-Dependent RelB Cleavage Promotes Canonical NF-κB Activation in Lymphocytes and Lymphoma Cell Lines. Proc. Natl. Acad. Sci. 2011, 108, 14596–14601. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Chang, M.; Paul, E.M.; Babu, G.; Lee, A.J.; Reiley, W.; Wright, A.; Zhang, M.; You, J.; Sun, S.C. Deubiquitinating enzyme CYLD negatively regulates RANK signaling and osteoclastogenesis in mice. J. Clin. Investig. 2008, 118, 1858–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matmati, M.; Jacques, P.; Maelfait, J.; Verheugen, E.; Kool, M.; Sze, M.; Geboes, L.; Louagie, E.; Mc Guire, C.; Vereecke, L.; et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat. Genet. 2011, 43, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Vaira, S.; Johnson, T.; Hirbe, A.C.; Alhawagri, M.; Anwisye, I.; Sammut, B.; O’Neal, J.; Zou, W.; Weilbaecher, K.N.; Faccio, R.; et al. RelB is the NF-κB subunit downstream of NIK responsible for osteoclast differentiation. Proc. Natl. Acad. Sci. USA. 2008, 105, 3897–3902. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xiu, Y.; Li, J.; Xing, L.; Yao, Z. NF-κB-Mediated Regulation of Osteoclastogenesis. Endocrinol. Metab. 2015, 30, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Matsuo, K. Signalling in osteoclasts and the role of Fos/AP1 proteins. Ann. Rheum. Dis. 2003, 62, ii83–ii85. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, K.; Kudo, A. Self-assembled RANK induces osteoclastogenesis ligand-independently. J. Bone Miner. Res. 2005, 20, 2053–2060. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Weber, G.F. Synergistic activation of the CMV promoter by NF-κB P50 and PKG. Biochem. Biophys. Res. Commun. 2004, 321, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Rodova, M.; Jayini, R.; Singasani, R.; Chipps, E.; Islam, M.R. CMV promoter is repressed by p53 and activated by JNK pathway. Plasmid 2013, 69, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, B.; Prozialeck, W.C.; Wallace, T.L. Interaction of drugs with calmodulin. Biochemical, pharmacological and clinical implications. Biochem. Pharmacol. 1982, 31, 2217–2226. [Google Scholar] [CrossRef]
- Nagel, D.; Bognar, M.; Eitelhuber, A.C.; Kutzner, K.; Vincendeau, M.; Krappmann, D.; Nagel, D.; Bognar, M.; Eitelhuber, A.C.; Kutzner, K.; et al. Combinatorial BTK and MALT1 inhibition augments killing of CD79 mutant diffuse large B cell lymphoma. Oncotarget 2015, 6, 42232–42242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howes, A.; O’Sullivan, P.A.; Breyer, F.; Ghose, A.; Cao, L.; Krappmann, D.; Bowcock, A.M.; Ley, S.C. Psoriasis mutations disrupt CARD14 autoinhibition promoting BCL10-MALT1-dependent NF-κB activation. Biochem. J. 2016, BCJ20160270. [Google Scholar] [CrossRef]
- Afonina, I.S.; Van Nuffel, E.; Baudelet, G.; Driege, Y.; Kreike, M.; Staal, J.; Beyaert, R. The paracaspase MALT1 mediates CARD14-induced signaling in keratinocytes. EMBO Rep. 2016, 17, 914–927. [Google Scholar] [CrossRef] [PubMed]
- Douanne, T.; Gavard, J.; Bidère, N. The paracaspase MALT1 cleaves the LUBAC subunit HOIL1 during antigen receptor signaling. J. Cell. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Elton, L.; Carpentier, I.; Staal, J.; Driege, Y.; Haegman, M.; Beyaert, R. MALT1 cleaves the E3 ubiquitin ligase HOIL-1 in activated T cells, generating a dominant negative inhibitor of LUBAC-induced NF-κB signaling. FEBS J. 2016, 283, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Bonsignore, L.; Passelli, K.; Pelzer, C.; Perroud, M.; Konrad, A.; Thurau, M.; Stürzl, M.; Dai, L.; Trillo-Tinoco, J.; Del Valle, L.; et al. A role for MALT1 activity in Kaposi’s sarcoma-associated herpes virus latency and growth of primary effusion lymphoma. Leukemia 2017, 31, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Bae, S.J.; Kim, M. Mucosa-associated lymphoid tissue lymphoma translocation 1 as a novel therapeutic target for rheumatoid arthritis. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Bardet, M.; Unterreiner, A.; Malinverni, C.; Lafossas, F.; Vedrine, C.; Boesch, D.; Kolb, Y.; Kaiser, D.; Glück, A.; Schneider, M.A.; et al. The T-cell fingerprint of MALT1 paracaspase revealed by selective inhibition. Immunol. Cell. Biol. 2018, 96, 81–99. [Google Scholar] [CrossRef] [PubMed]
- Lork, M.; Kreike, M.; Staal, J.; Beyaert, R. Importance of Validating Antibodies and Small Compound Inhibitors Using Genetic Knockout Studies-T Cell Receptor-Induced CYLD Phosphorylation by IKKε/TBK1 as a Case Study. Front. Cell. Dev. Biol. 2018, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Bowes, H.A. The ataractic drugs: The present position of chlorpromazine, frenquel, pacatal, and reserpine in the psychiatric hospital. Am. J. Psychiatry 1956, 113, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Whittier, J.R.; Klein, D.F.; Levine, G.; Weiss, D. Mepazine (pacatal): Clinical trial with placebo control and psychological study. Psychopharmacologia 1960, 1, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Federal Register, 1998, 63:195, 54085. Available online: https://www.gpo.gov/fdsys/pkg/FR-1998-10-08/html/98-26923.htm (accessed on 30 November 2018).
- Krappmann, D.; Nagel, D.; Schlauderer, F.; Lammens, K.; Hopfner, K.P.; Chrusciel, R.A.; Kling, D.L. (S)-Enantiomer of Mepazine. U.S. Patent 20160137635A1, 19 May 2016. [Google Scholar]
- Hamerman, J.A.; Ni, M.; Killebrew, J.R.; Chu, C.L.; Lowell, C.A. The expanding roles of ITAM adapters FcRgamma and DAP12 in myeloid cells. Immunol. Rev. 2009, 232, 42–58. [Google Scholar] [CrossRef] [PubMed]
- Barrow, A.D.; Raynal, N.; Andersen, T.L.; Slatter, D.A.; Bihan, D.; Pugh, N.; Cella, M.; Kim, T.; Rho, J.; Negishi-Koga, T.; et al. OSCAR is a collagen receptor that costimulates osteoclastogenesis in DAP12-deficient humans and mice. J. Clin. Investig. 2011, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbull, I.R.; Gilfillan, S.; Cella, M.; Aoshi, T.; Miller, M.; Piccio, L.; Hernandez, M.; Colonna, M. Cutting Edge: TREM-2 Attenuates Macrophage Activation. J. Immunol. 2006, 177, 3520–3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.S.; Sarin, R.; Dixit, N.; Wu, J.; Gershwin, E.; Bowman, E.P.; Adamopoulos, I.E. Crosstalk among IL-23 and DNAX Activating Protein of 12 kDa–Dependent Pathways Promotes Osteoclastogenesis. J. Immunol. 2015, 194, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Harre, U.; Lang, S.C.; Pfeifle, R.; Rombouts, Y.; Frühbeißer, S.; Amara, K.; Bang, H.; Lux, A.; Koeleman, C.A.; Baum, W.; et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat. Commun. 2015, 6, 6651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaiss, M.M.; Sarter, K.; Hess, A.; Engelke, K.; Böhm, C.; Nimmerjahn, F.; Voll, R.; Schett, G.; David, J.P. Increased bone density and resistance to ovariectomy-induced bone loss in FoxP3-transgenic mice based on impaired osteoclast differentiation. Arthritis Rheum. 2010, 62, 2328–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinnon, M.L.; Rozmus, J.; Fung, S.Y.; Hirschfeld, A.F.; Del Bel, K.L.; Thomas, L.; Marr, N.; Martin, S.D.; Marwaha, A.K.; Priatel, J.J.; et al. Combined immunodeficiency associated with homozygous MALT1 mutations. J. Allergy Clin. Immunol. 2014, 133, 1458–1462. [Google Scholar] [CrossRef] [PubMed]
- Rozmus, J.; McDonald, R.; Fung, S.Y.; Del Bel, K.L.; Roden, J.; Senger, C.; Schultz, K.R.; McKinnon, M.L.; Davis, J.; Turvey, S.E. Successful clinical treatment and functional immunological normalization of human MALT1 deficiency following hematopoietic stem cell transplantation. Clin. Immunol. Orlando Fla 2016, 168, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, K.; Ando, T.; Goto, H.; Xavier, R. Bcl10 is phosphorylated on Ser138 by Ca2+/calmodulin-dependent protein kinase II. Mol. Immunol. 2007, 44, 2095–2100. [Google Scholar] [CrossRef] [PubMed]
- Oruganti, S.R.; Edin, S.; Grundström, C.; Grundström, T. CaMKII targets Bcl10 in T-cell receptor induced activation of NF-κB. Mol. Immunol. 2011, 48, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, K.; Green, T.; Rapley, J.; Wachtel, H.; Giallourakis, C.; Landry, A.; Cao, Z.; Lu, N.; Takafumi, A.; Goto, H.; et al. Ca2+/calmodulin-dependent protein kinase II is a modulator of CARMA1-mediated NF-κB activation. Mol. Cell. Biol. 2006, 26, 5497–5508. [Google Scholar] [CrossRef] [PubMed]
- Frischbutter, S.; Gabriel, C.; Bendfeldt, H.; Radbruch, A.; Baumgrass, R. Dephosphorylation of Bcl-10 by calcineurin is essential for canonical NF-κB activation in Th cells. Eur. J. Immunol. 2011, 41, 2349–2357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraccioli, G.F.; Tomietto, P.; De Santis, M. Rationale for T cell inhibition by cyclosporin A in major autoimmune diseases. Ann. N. Y. Acad. Sci. 2005, 1051, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Ruland, J.; Duncan, G.S.; Wakeham, A.; Mak, T.W. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity 2003, 19, 749–758. [Google Scholar] [CrossRef]
- Takahashi, N.; Yamana, H.; Yoshiki, S.; Roodman, G.D.; Mundy, G.R.; Jones, S.J.; Boyde, A.; Suda, T. Osteoclast-like cell formation and its regulation by osteotropic hormones in mouse bone marrow cultures. Endocrinology 1988, 122, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Staal, J.; Driege, Y.; Haegman, M.; Borghi, A.; Hulpiau, P.; Lievens, L.; Gul, I.S.; Sundararaman, S.; Gonçalves, A.; Dhondt, I.; et al. Ancient Origin of the CARD–Coiled Coil/Bcl10/MALT1-Like Paracaspase Signaling Complex Indicates Unknown Critical Functions. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüstle, M. Chemtool—Moleküle zeichnen mit dem Pinguin. Nachrichten Aus Chem. 2010, 49, 1310–1313. [Google Scholar] [CrossRef]
Sample Availability: Mepazine and other products used in this study are commercially available from multiple sources, and the plasmids used are available via the BCCM/GeneCorner plasmid collection. MALT1 KO HEK293T cells are available from the authors upon request. |

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meloni, L.; Verstrepen, L.; Kreike, M.; Staal, J.; Driege, Y.; Afonina, I.S.; Beyaert, R. Mepazine Inhibits RANK-Induced Osteoclastogenesis Independent of Its MALT1 Inhibitory Function. Molecules 2018, 23, 3144. https://doi.org/10.3390/molecules23123144
Meloni L, Verstrepen L, Kreike M, Staal J, Driege Y, Afonina IS, Beyaert R. Mepazine Inhibits RANK-Induced Osteoclastogenesis Independent of Its MALT1 Inhibitory Function. Molecules. 2018; 23(12):3144. https://doi.org/10.3390/molecules23123144
Chicago/Turabian StyleMeloni, Laura, Lynn Verstrepen, Marja Kreike, Jens Staal, Yasmine Driege, Inna S. Afonina, and Rudi Beyaert. 2018. "Mepazine Inhibits RANK-Induced Osteoclastogenesis Independent of Its MALT1 Inhibitory Function" Molecules 23, no. 12: 3144. https://doi.org/10.3390/molecules23123144
APA StyleMeloni, L., Verstrepen, L., Kreike, M., Staal, J., Driege, Y., Afonina, I. S., & Beyaert, R. (2018). Mepazine Inhibits RANK-Induced Osteoclastogenesis Independent of Its MALT1 Inhibitory Function. Molecules, 23(12), 3144. https://doi.org/10.3390/molecules23123144