Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives


Abstract
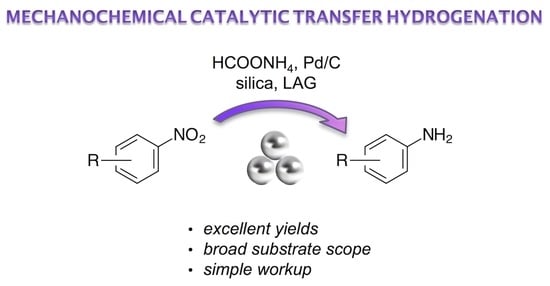
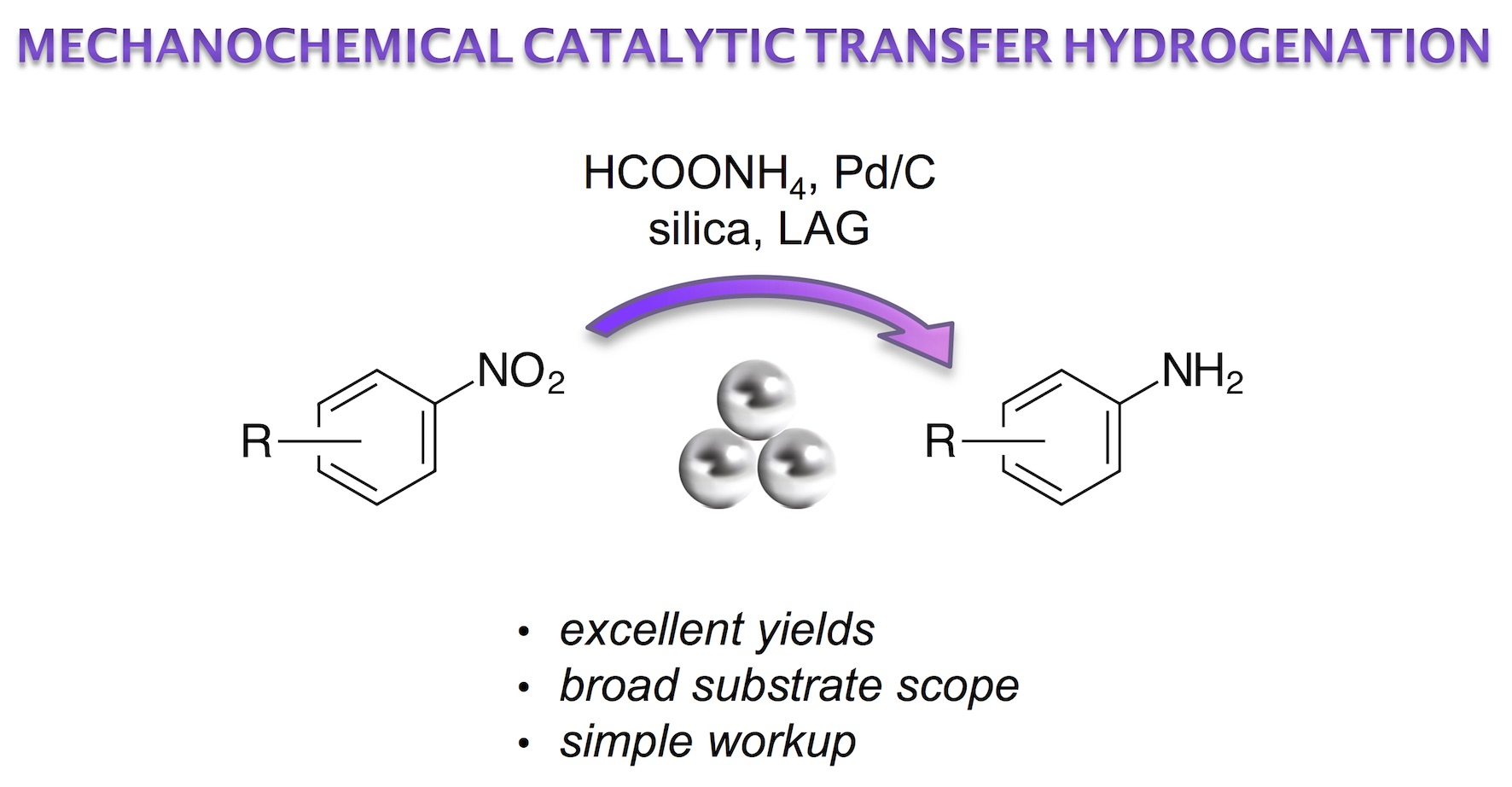
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Brieger, G.; Nestrick, T.J. Catalytic Transfer Hydrogenation. Chem. Rev. 1974, 74, 567–580. [Google Scholar] [CrossRef]
- Wang, D.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef] [PubMed]
- Hagen, J. Heterogeneously Catalyzed Processes in Industry. In Industrial Catalysis: A Practical Approach, 2nd ed.; Hagen, J., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 261–293. ISBN 978-3-527-31144-6. [Google Scholar]
- Ram, S.; Ehrekaufer, R.E. Ammonium Formate in Organic Synthesis: A Versatile Agent in Catalytic Hydrogen Transfer Reductions. Synthesis 1988, 1988, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Zoran, A.; Sasson, Y. Catalytic transfer hydrogenation of unsaturated compounds by solid sodium formate in the presence of palladium on carbon. J. Mol. Catal. 1984, 26, 321–326. [Google Scholar] [CrossRef]
- Paryzek, Z.; Koenig, H.; Tabaczka, B. Ammonium Formate/Palladium on Carbon: A Versatile System for Catalytic Hydrogen Transfer Reductions of Carbon-Carbon Double Bonds. Synthesis 2003, 2023–2026. [Google Scholar] [CrossRef]
- Dobrovolná, Z.; Červeny, L. Ammonium formate decomposition using palladium catalyst. Res. Chem. Intermed. 2000, 26, 489–497. [Google Scholar] [CrossRef]
- Banik, B.K.; Barakat, K.J.; Wagle, D.R.; Manhas, M.S.; Bose, A.K. Microwave-Assisted Rapid and Simplified Hydrogenation. J. Org. Chem. 1999, 64, 5746–5753. [Google Scholar] [CrossRef]
- Vass, A.; Dudás, J.; Tóth, J.; Varma, R.S. Solvent-free reduction of aromatic nitro compounds with alumina-supported hydrazine under microwave irradiation. Tetrahedron Lett. 2001, 42, 5347–5349. [Google Scholar] [CrossRef]
- Berthold, H.; Schotten, T.; Hönig, H. Transfer Hydrogenation in Ionic Liquids under Microwave Irradiation. Synthesis 2002, 1607–1610. [Google Scholar] [CrossRef]
- Do, J.-L.; Friščić, T. Mechanochemistry: A Force of Synthesis. ACS Cent. Sci. 2017, 3, 13–19. [Google Scholar] [CrossRef]
- Howard, J.L.; Cao, Q.; Browne, D.L. Mechanochemistry as an emerging tool for molecular synthesis: What can it offer? Chem. Sci. 2018, 9, 3080–3094. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; De, S.; Balu, A.M.; Ojeda, M.; Luque, R. Mechanochemical synthesis of advanced nanomaterials for catalytic applications. Chem. Commun. 2015, 51, 6698–6713. [Google Scholar] [CrossRef]
- Gečiauskaite, A.A.; García, F. Main group mechanochemistry. Beilstein J. Org. Chem. 2017, 13, 2068–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, V.; Quaresma, S.; da Silva, J.L.F.; Duarte, M.T. Exploring mechanochemistry to turn organic bio-relevant molecules into metal-organic frameworks: A short review. Beilstein J. Org. Chem. 2017, 13, 2416–2427. [Google Scholar] [CrossRef] [PubMed]
- Wixtrom, A.; Buhler, J.; Abdel-Fattah, T. Mechanochemical Synthesis of Two Polymorphs of the Tetrathiafulvalene-Chloranil Charge Transfer Salt: An Experiment for Organic Chemistry. J. Chem. Educ. 2014, 91, 1232–1235. [Google Scholar] [CrossRef]
- Crawford, D.E.; Miskimmin, C.K.G.; Albadarin, A.B.; Walker, G.; James, S.L. Organic synthesis by Twin Screw Extrusion (TSE): Continuous, scalable and solvent-free. Green Chem. 2017, 19, 1507–1518. [Google Scholar] [CrossRef]
- Crawford, D.E.; Wright, L.A.; James, S.L.; Abbott, A.P. Efficient continuous synthesis of high purity deep eutectic solvents by twin screw extrusion. Chem. Commun. 2016, 52, 4215–4218. [Google Scholar] [CrossRef]
- Crawford, D.; Casaban, J.; Haydon, R.; Giri, N.; McNally, T.; James, S.L. Synthesis by extrusion: Continuous, large-scale preparation of MOFs using little or no solvent. Chem. Sci. 2015, 6, 1645–1649. [Google Scholar] [CrossRef]
- Michalchuk, A.A.L.; Hope, K.S.; Kennedy, S.R.; Blanco, M.V.; Boldyreva, E.V.; Pulham, C.R. Ball-free mechanochemistry: In situ real-time monitoring of pharmaceutical co-crystal formation by resonant acoustic mixing. Chem. Commun. 2018, 54, 4033–4036. [Google Scholar] [CrossRef]
- Martina, K.; Rotolo, L.; Porcheddu, A.; Delogu, F.; Bysouth, S.R.; Cravotto, G.; Colacino, E. High throughput mechanochemistry: Application to parallel synthesis of benzoxazines. Chem. Commun. 2018, 54, 551–554. [Google Scholar] [CrossRef]
- Štrukil, V.; Sajko, I. Mechanochemically-assisted solid-state photocatalysis (MASSPC). Chem. Commun. 2017, 53, 9101–9104. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. [Google Scholar] [CrossRef]
- Štrukil, V. Mechanochemical Organic Synthesis: The Art of Making Chemistry Green. Synlett 2018, 29, 1281–1288. [Google Scholar] [CrossRef]
- Tan, D.; Friščić, T. Mechanochemistry for Organic Chemists: An Update. Eur. J. Org. Chem. 2018, 18–33. [Google Scholar] [CrossRef]
- Colacino, E.; Porcheddu, A.; Halasz, I.; Charnay, C.; Delogu, F.; Guerra, R.; Fullenwarth, J. Mechanochemistry for “no solvent, no base” preparation of hydantoin-based active pharmaceutical ingredients: Nitrofurantoin and dantrolene. Green Chem. 2018, 20, 2973–2977. [Google Scholar] [CrossRef]
- Andersen, J.; Mack, J. Mechanochemistry and organic synthesis: From mystical to practical. Green Chem. 2018, 20, 1435–1443. [Google Scholar] [CrossRef]
- Chauhan, P.; Chimni, S.S. Mechanochemistry assisted asymmetric organocatalysis: A sustainable approach. Beilstein J. Org. Chem. 2012, 8, 2132–2141. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, B.; Rantanen, T.; Bolm, C. Solvent-Free Asymmetric Organocatalysis in a Ball Mill. Angew. Chem. Int. Ed. 2006, 45, 6924–6926. [Google Scholar] [CrossRef]
- Rantanen, T.; Schiffers, I.; Bolm, C. Solvent-Free Asymmetric Anhydride Opening in a Ball Mill. Org. Process Res. Dev. 2007, 11, 592–597. [Google Scholar] [CrossRef]
- Hernández, J.G.; Friščić, T. Metal-catalyzed organic reactions using mechanochemistry. Tetrahedron Lett. 2015, 56, 4253–4265. [Google Scholar] [CrossRef]
- Hernández, J.G. C−H Bond Functionalization by Mechanochemistry. Chem. Eur. J. 2017, 23, 17157–17165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Li, Y.; Liu, C.; Zhao, Y. Recent advances in mechanochemical C–H functionalization reactions. Tetrahedron Lett. 2018, 59, 317–324. [Google Scholar] [CrossRef]
- Leonardi, M.; Villacampa, M.; Menéndez, J.C. Multicomponent mechanochemical synthesis. Chem. Sci. 2018, 9, 2042–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delogu, F.; Mulas, G.; Garroni, S. Hydrogenation of carbon monoxide under mechanical activation conditions. Appl. Catal. A Gen. 2009, 366, 201–205. [Google Scholar] [CrossRef]
- Mack, J.; Fulmer, D.; Stofel, S.; Santos, N. The first solvent-free method for the reduction of esters. Green Chem. 2007, 9, 1041–1043. [Google Scholar] [CrossRef]
- Kaupp, G. Organic Solid-State Reactions with 100% Yield. Top. Curr. Chem. 2005, 254, 95–183, (pp. 117, unpublished results). [Google Scholar] [CrossRef]
- Birke, V.; Mattik, J.; Runne, D. Mechanochemical reductive dehalogenation of hazardous polyhalogenated contaminants. J. Mat. Sci. 2004, 39, 5111–5116. [Google Scholar] [CrossRef]
- Lu, S.; Huang, J.; Peng, Z.; Li, X.; Yan, J. Ball milling 2,4,6-trichlorophenol with calcium oxide: Dechlorination experiment and mechanism considerations. Chem. Eng. J. 2012, 195, 62–68. [Google Scholar] [CrossRef]
- Loiselle, S.; Branca, M.; Mulas, G.; Cocco, G. Selective Mechanochemical Dehalogenation of Chlorobenzenes over Calcium Hydride. Environ. Sci. Technol. 1996, 31, 261–265. [Google Scholar] [CrossRef]
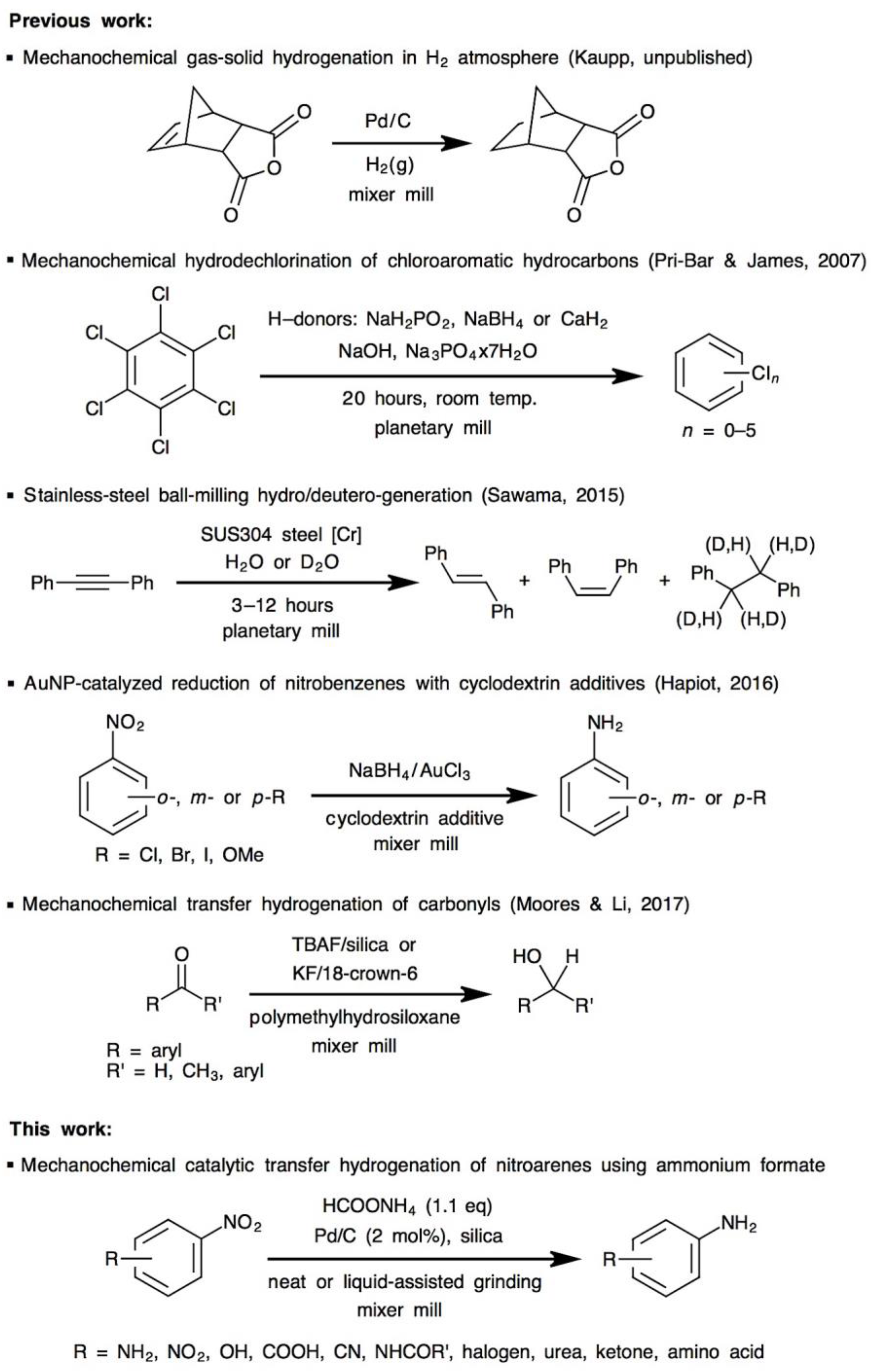
- Pri-Bar, I.; James, B.R. Mechanochemical, solvent free, palladium-catalyzed hydrodechlorination of chloroaromatic hydrocarbons. J. Mol. Catal. A Chem. 2007, 264, 135–139. [Google Scholar] [CrossRef]
- Sawama, Y.; Kawajiri, T.; Niikawa, M.; Goto, R.; Yabe, Y.; Takahashi, T.; Marumoto, T.; Itoh, M.; Kimura, Y.; Monguchi, Y.; et al. Stainless-Steel Ball-Milling Method for Hydro-/Deutero-genation using H2O/D2O as a Hydrogen/Deuterium Source. ChemSusChem 2015, 8, 3773–3776. [Google Scholar] [CrossRef] [PubMed]
- Sawama, Y.; Niikawa, M.; Yabe, Y.; Goto, R.; Kawajiri, T.; Marumoto, T.; Takahashi, T.; Itoh, M.; Kimura, Y.; Sasai, Y.; et al. Stainless-Steel-Mediated Quantitative Hydrogen Generation from Water under Ball Milling Conditions. ACS Sustain. Chem. Eng. 2015, 3, 683–689. [Google Scholar] [CrossRef]
- Sawama, Y.; Yasukawa, N.; Ban, K.; Goto, R.; Niikawa, M.; Monguchi, Y.; Itoh, M.; Sajiki, H. Stainless Steel-Mediated Hydrogen Generation from Alkanes and Diethyl Ether and Its Application for Arene Reduction. Org. Lett. 2018, 20, 2892–2896. [Google Scholar] [CrossRef] [PubMed]
- Menuel, S.; Léger, B.; Addad, A.; Monflier, E.; Hapiot, F. Cyclodextrins as effective additives in AuNP-catalyzed reduction of nitrobenzene derivatives in a ball-mill. Green Chem. 2016, 18, 5500–5509. [Google Scholar] [CrossRef]
- Li, A.Y.; Segalla, A.; Li, C.-J.; Moores, A. Mechanochemical Metal-Free Transfer Hydrogenation of Carbonyls Using Polymethylhydrosiloxane as the Hydrogen Source. ACS Sustain. Chem. Eng. 2017, 5, 11752–11760. [Google Scholar] [CrossRef]
- Kadam, H.K.; Tilve, S.G. Advancement in methodologies for reduction of nitroarenes. RSC Adv. 2015, 5, 83391–83407. [Google Scholar] [CrossRef]
- Zhao, Z.; Yang, H.; Li, Y.; Guo, X. Cobalt-modified molybdenum carbide as an efficient catalyst for chemoselective reduction of aromatic nitro compounds. Green Chem. 2014, 16, 1274–1281. [Google Scholar] [CrossRef]
- Yang, X.-J.; Chen, B.; Zheng, L.-Q.; Wu, L.-Z.; Tung, C.-H. Highly efficient and selective photocatalytic hydrogenation of functionalized nitrobenzenes. Green Chem. 2014, 16, 1082–1086. [Google Scholar] [CrossRef]
- Wang, L.; Li, P.; Wu, Z.; Yan, J.; Wang, M.; Ding, Y. Reduction of Nitroarenes to Aromatic Amines with Nanosized Activated Metallic Iron Powder in Water. Synthesis 2003, 2001–2004. [Google Scholar] [CrossRef]
- Wienhöfer, G.; Baseda-Krüger, M.; Ziebart, C.; Westerhaus, F.A.; Baumann, W.; Jackstell, R.; Junge, K.; Beller, M. Hydrogenation of nitroarenes using defined iron–phosphine catalysts. Chem. Commun. 2013, 49, 9089–9091. [Google Scholar] [CrossRef]
- Vaidya, M.J.; Kulkarni, S.M.; Chaudhari, R.V. Synthesis of p-Aminophenol by Catalytic Hydrogenation of p-Nitrophenol. Org. Process Res. Dev. 2003, 7, 202–208. [Google Scholar] [CrossRef]
- Baron, M.; Métay, E.; Lemaire, M.; Popowycz, F. Reduction of aromatic and aliphatic nitro groups to anilines and amines with hypophosphites associated with Pd/C. Green Chem. 2013, 15, 1006–1015. [Google Scholar] [CrossRef]
- Orlandi, M.; Brenna, D.; Harms, R.; Jost, S.; Benaglia, M. Recent Developments in the Reduction of Aromatic and Aliphatic Nitro Compounds to Amines. Org. Process Res. Dev. 2018, 22, 430–445. [Google Scholar] [CrossRef]
- Ram, S.; Ehrekaufer, R.E. A general procedure for mild and rapid reduction of aliphatic and aromatic nitro compounds using ammonium formate as a catalytic hydrogen transfer agent. Tetrahedron Lett. 1984, 25, 3415–3418. [Google Scholar] [CrossRef]
- Ram, S.; Ehrekaufer, R.E. A Facile Synthesis of α-Amino Esters via Reduction of α-Nitro Esters Using Ammonium Formate as a Catalytic Hydrogen Transfer Agent. Synthesis 1986, 133–135. [Google Scholar] [CrossRef]
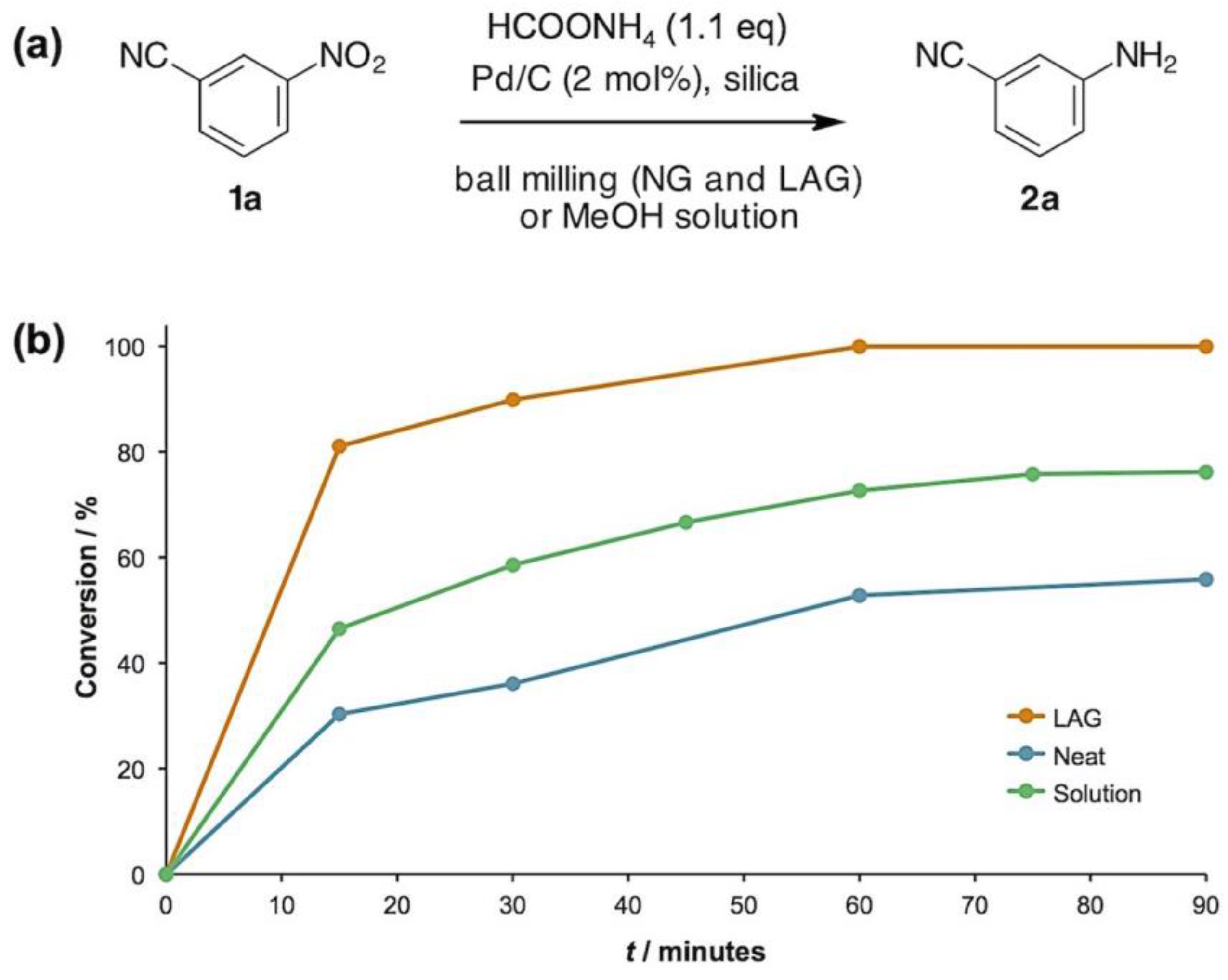
- HPLC analysis showed that the CTH of 3-nitrobenzonitrile (1a) (1.0 mmol) in methanol solution (10 mL) with 3 eq of ammonium formate (9.9 mmol) reached quantitative yield in 30 minutes.
- Đud, M.; Magdysyuk, O.V.; Margetić, D.; Štrukil, V. Synthesis of monosubstituted thioureas by vapour digestion and mechanochemical amination of thiocarbamoyl benzotriazoles. Green Chem. 2016, 18, 2666–2674. [Google Scholar] [CrossRef] [Green Version]
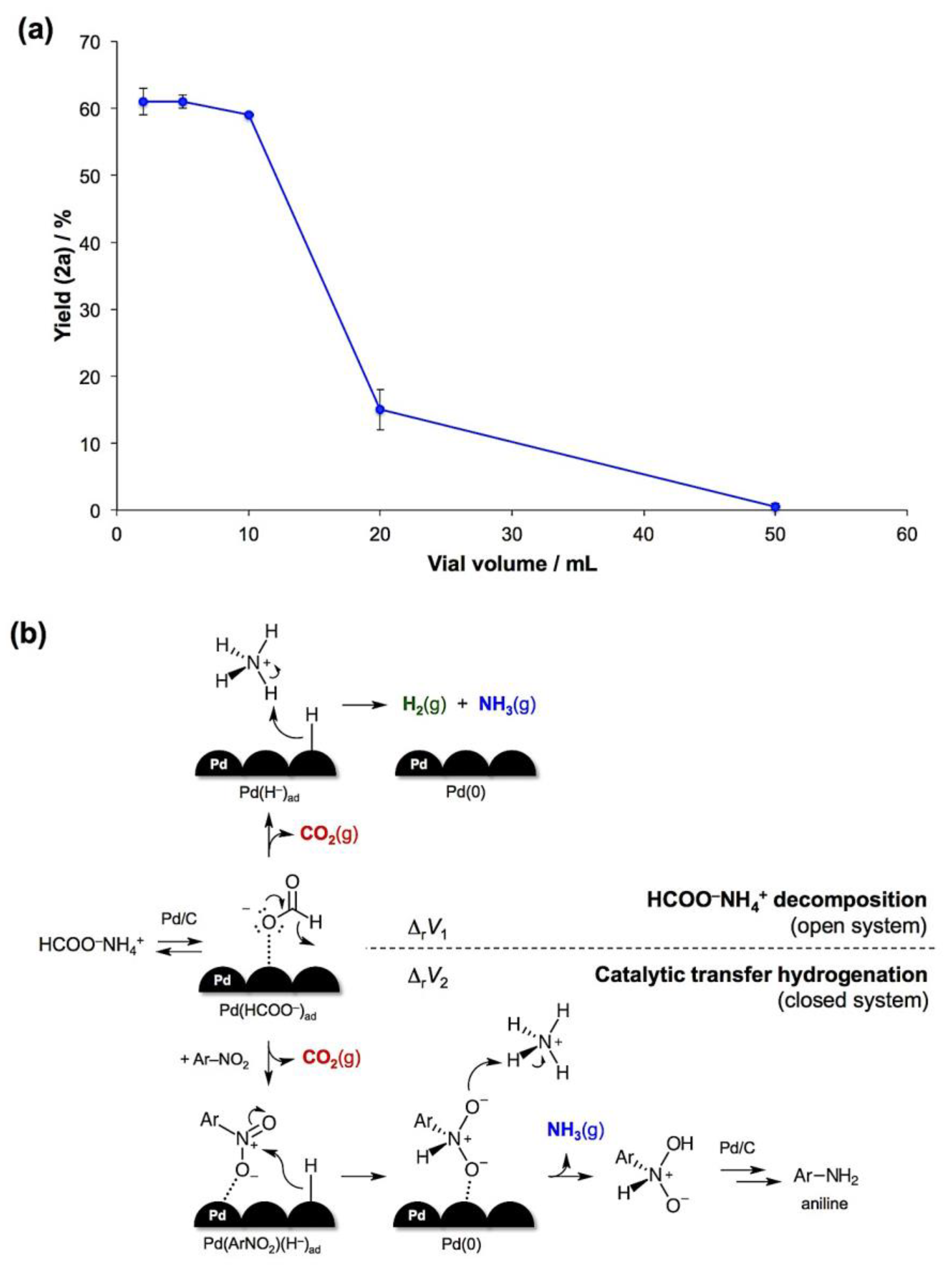
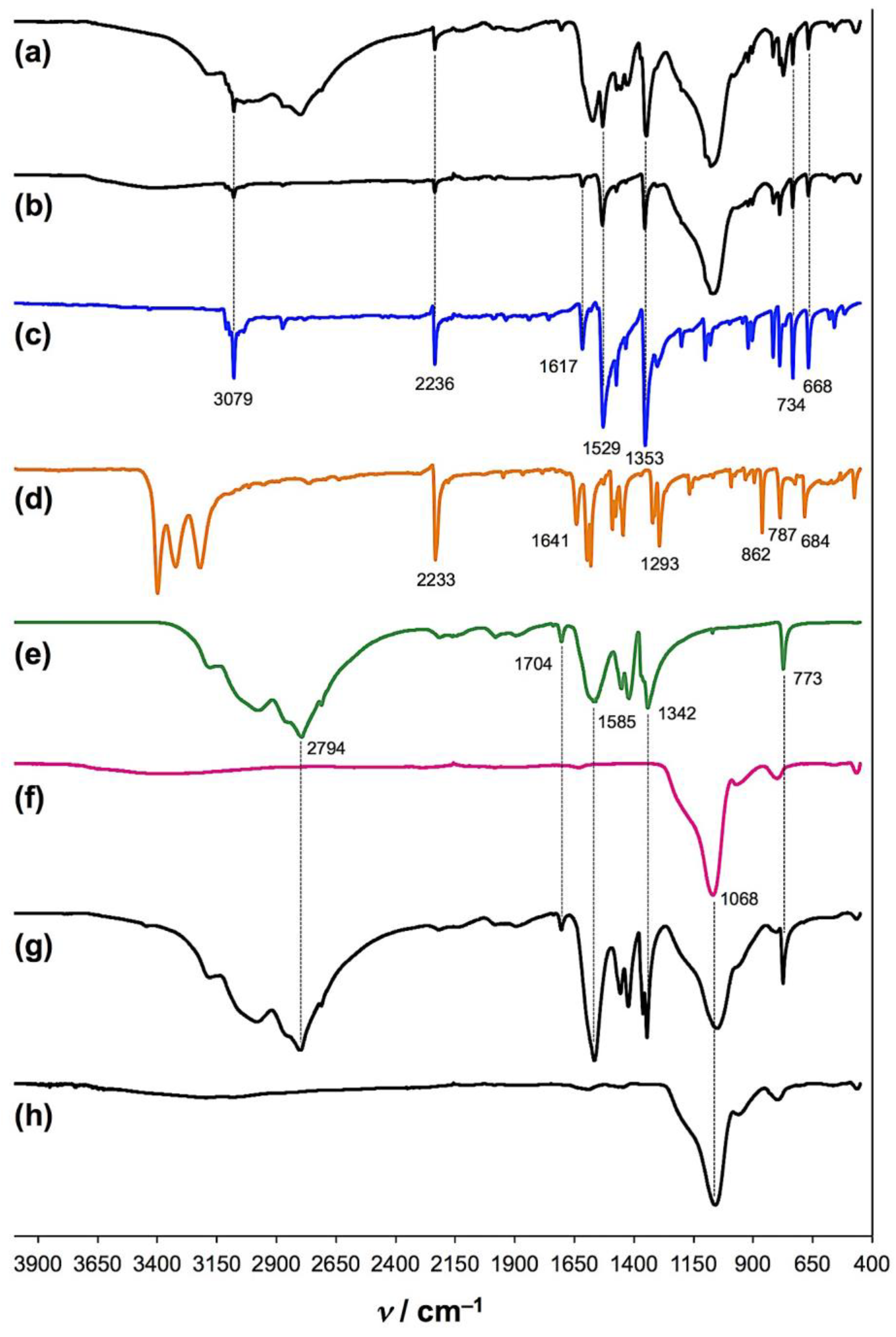
- The reaction mass loss upon opening the jars indicated ca. 60% conversion after 1 hour and quantitative decomposition after 2 hours in air.
- Rajagopal, S.; Anwer, M.K.; Spatola, A.F. Catalytic transfer hydrogenation and hydrogenolysis by formic acid and its salts. In Peptides Design, Synthesis, and Biological Activity, 1st ed.; Basava, C., Anantharamaiah, G.M., Eds.; Birkhäuser: Boston, MA, USA, 1994; Chapter 2; pp. 11–26. ISBN 978-1-4615-8178-9. [Google Scholar]
- Byun, E.; Hong, B.; De Castro, K.A.; Lim, M.; Rhee, H. One-Pot Reductive Mono-N-alkylation of Aniline and Nitroarene Derivatives Using Aldehydes. J. Org. Chem. 2007, 72, 9815–9817. [Google Scholar] [CrossRef]
- Tuteja, J.; Nishimura, S.; Ebitani, K. Base-free chemoselective transfer hydrogenation of nitroarenes to anilines with formic acid as hydrogen source by a reusable heterogeneous Pd/ ZrP catalyst. RSC Adv. 2014, 4, 38241–38249. [Google Scholar] [CrossRef]
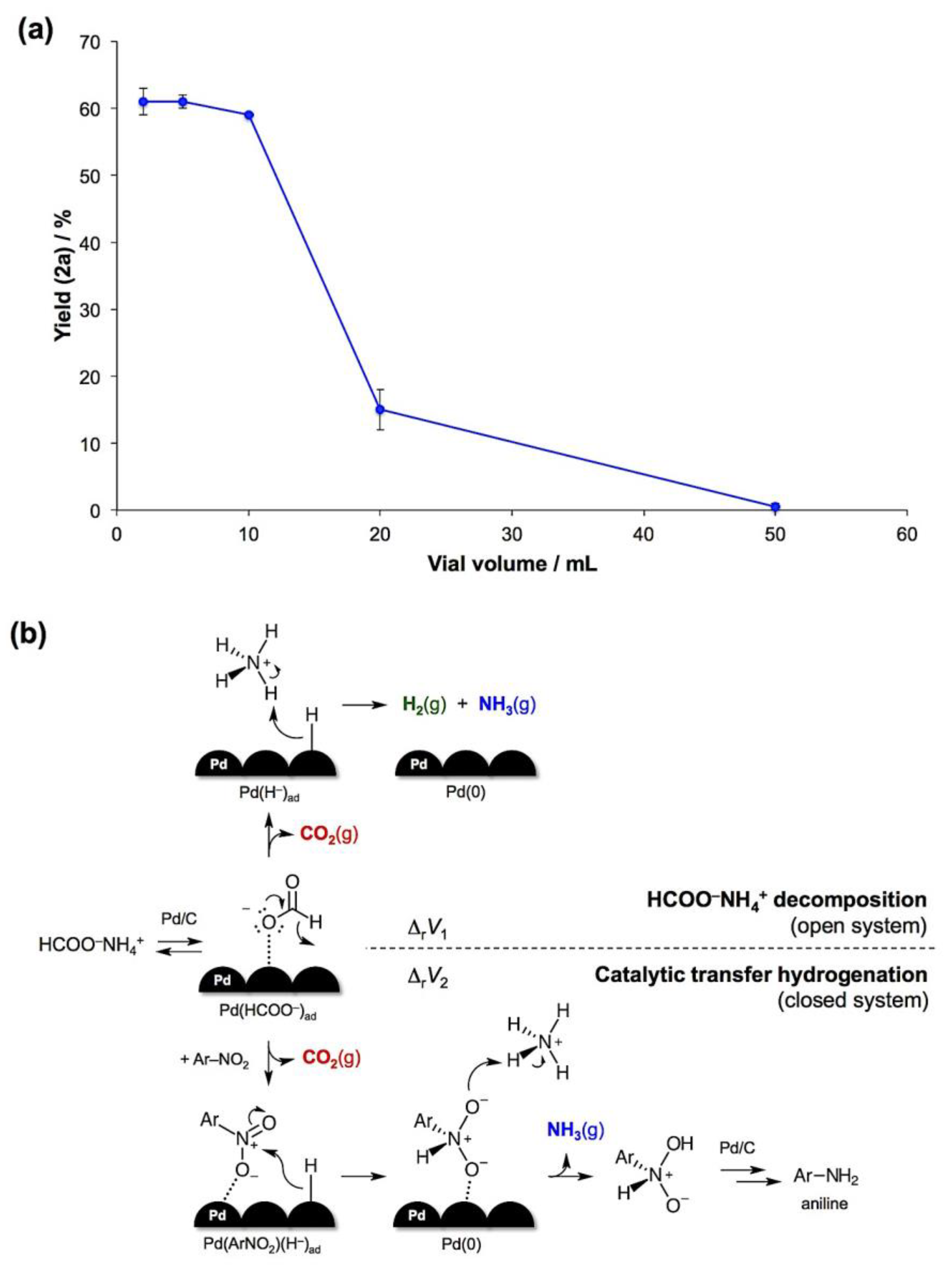
- The reaction volume change is defined as the difference between the sums of molar volumes of products and reactants, ΔrV = ∑Vm(products) − ∑Vm(reactants). For solids, the difference in molar volumes is typically small and can be neglected. With this assumption, the CTH will generate 2 moles of gaseous products per 1 mole of the formate salt, while the complete decomposition of HCOONH4 will result in 3 moles of gases per 1 mole of the salt. Therefore, ΔrV1 (decomposition) > ΔrV2 (CTH).
- Maegawa, T.; Fujiwara, Y.; Ikawa, T.; Hisashi, H.; Monguchi, Y.; Sajiki, H. Novel deprotection method of Fmoc group under neutral hydrogenation conditions. Amino Acids 2009, 36, 493–499. [Google Scholar] [CrossRef]
- Albers, P.; Pietsch, J.; Parker, S.F. Poisoning and deactivation of palladium catalysts. J. Mol. Catal. A Chem. 2001, 173, 275–286. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Sane, M.G. Poisoning of Pd–carbon catalysts by sulphur, chloro and heavy metal compounds in liquid phase hydrogenation of o-nitrophenol to O-aminophenol. J. Chem. Technol. Biotechnol. 1998, 73, 336–340. [Google Scholar] [CrossRef]
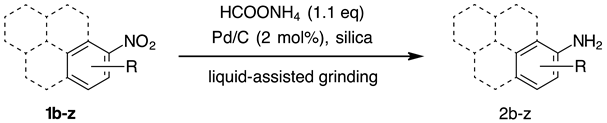
- Kulla, H.; Fischer, F.; Benemann, S.; Rademann, K.; Emmerling, F. The effect of the ball to reactant ratio on mechanochemical reaction times studied by in situ PXRD. Cryst. Eng. Comm. 2017, 19, 3902–3907. [Google Scholar] [CrossRef]
- As suggested by preliminary DFT calculations (B97D/6-31G(d) method), the reason behind the observed poor reactivity of these substrates could be a strong - interaction of polyaromatic molecules with graphite-like structure of activated carbon, where the formation of 1:1 or 1:2 sandwich complexes might interfere with the reduction of the nitro group. The stability of such 1:1 and 1:2 complexes increases with the number of condensed aromatic rings in nitroarene substrates. For instance, the relative energies of 1:1 complexes of nitrobenzene, 1-amino and 2-aminonaphthalene, 1-amino and 2-aminoanthracene and 1-aminopyrene, with coronene as a graphite-like model structure are −13.6, −18.3, −16.1, −21.0, −21.7 and −24.1 kcal mol–1, respectively. By employing the M062X/6-31G(d) method, these values were found to follow the same trend, but with less pronounced relative energy difference: −12.1, −15.7, −16.5, −17.9, −18.9 and −19.3 kcal mol–1.
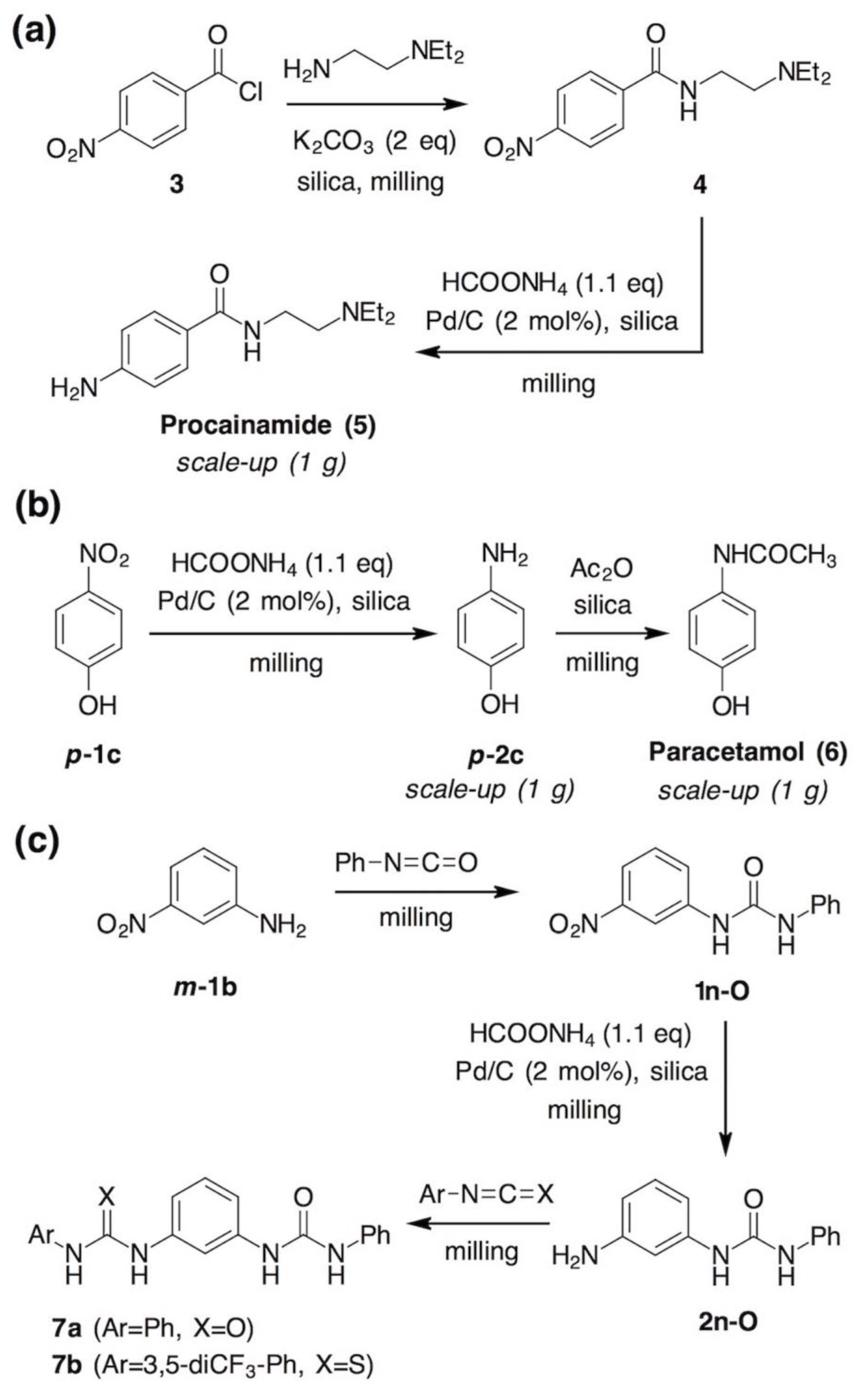
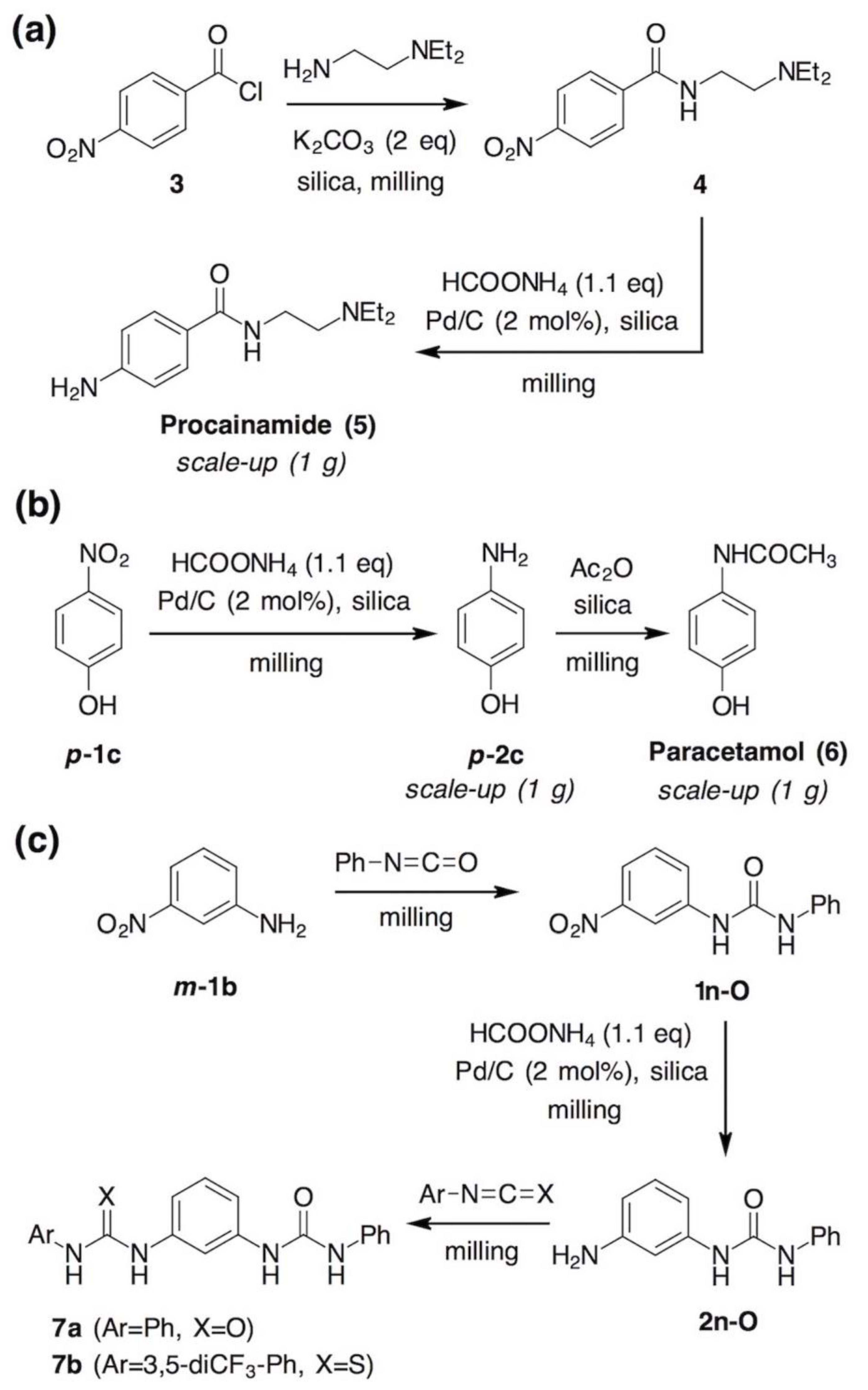
- Shannon, S.K.; Peacock, M.J.; Kates, S.A.; Barany, G. Solid-Phase Synthesis of Lidocaine and Procainamide Analogues Using Backbone Amide Linker (BAL) Anchoring. J. Comb. Chem. 2003, 5, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Loots, L.; Friščić, T. Towards medicinal mechanochemistry: Evolution of milling from pharmaceutical solid form screening to the synthesis of active pharmaceutical ingredients (APIs). Chem. Commun. 2016, 52, 7760–7781. [Google Scholar] [CrossRef]
- Métro, T.-X.; Bonnamour, J.; Reidon, T.; Sarpoulet, J.; Martinez, J.; Lamaty, F. Mechanosynthesis of amides in the total absence of organic solvent from reaction to product recovery. Chem. Commun. 2012, 48, 11781–11783. [Google Scholar] [CrossRef]
- Bonnamour, J.; Métro, T.-X.; Martinez, J.; Lamaty, F. Environmentally benign peptide synthesis using liquid-assisted ball-milling: Application to the synthesis of Leu-enkephalin. Green Chem. 2013, 15, 1116–1120. [Google Scholar] [CrossRef]
- Estévez, V.; Villacampa, M.; Menéndez, J.C. Concise synthesis of atorvastatin lactone under high-speed vibration milling conditions. Org. Chem. Front. 2014, 1, 458–463. [Google Scholar] [CrossRef]
- Tan, D.; Štrukil, V.; Mottillo, C.; Friščić, T. Mechanosynthesis of pharmaceutically relevant sulfonyl-(thio)ureas. Chem. Commun. 2014, 50, 5248–5250. [Google Scholar] [CrossRef]
- Rajput, L.; Banerjee, R. Mechanochemical Synthesis of Amide Functionalized Porous Organic Polymers. Cryst. Growth Des. 2014, 14, 2729–2732. [Google Scholar] [CrossRef]
- Yang, Y.; Bu, F.; Liu, J.; Shakir, I.; Xu, Y. Mechanochemical synthesis of two-dimensional aromatic polyamides. Chem. Commun. 2017, 53, 7481–7484. [Google Scholar] [CrossRef]
- Perlovich, G.L.; Volkova, T.V.; Bauer-Brandl, A. Polymorphism of paracetamol. J. Therm. Anal. Calorim. 2007, 89, 767–774. [Google Scholar] [CrossRef]
- Di Martino, P.; Conflant, P.; Drache, M.; Huvenne, J.-P.; Guyot-Hermann, A.-M. Preparation and physical characterization of forms II and III of paracetamol. J. Therm. Anal. 1997, 48, 447–458. [Google Scholar] [CrossRef]
- Telford, R.; Seaton, C.C.; Clout, A.; Buanz, A.; Gaisford, S.; Williams, G.R.; Prior, T.J.; Okoye, C.H.; Munshi, T.; Scowen, I.J. Stabilisation of metastable polymorphs: The case of paracetamol form III. Chem. Commun. 2016, 52, 12028–12031. [Google Scholar] [CrossRef] [PubMed]
- Štrukil, V.; Igrc, M.D.; Eckert-Maksić, M.; Friščić, T. Click Mechanochemistry: Quantitative Synthesis of “Ready to Use” Chiral Organocatalysts by Efficient Two-Fold Thiourea Coupling to Vicinal Diamines. Chem. Eur. J. 2012, 18, 8464–8473. [Google Scholar] [CrossRef] [PubMed]
- Štrukil, V.; Igrc, M.D.; Fábián, L.; Eckert-Maksić, M.; Childs, S.L.; Reid, D.G.; Duer, M.J.; Halasz, I.; Mottillo, C.; Friščić, T. A model for a solvent-free synthetic organic research laboratory: Click-mechanosynthesis and structural characterization of thioureas without bulk solvents. Green Chem. 2012, 14, 2462–2473. [Google Scholar] [CrossRef]
- Štrukil, V.; Margetić, D.; Igrc, M.D.; Eckert-Maksić, M.; Friščić, T. Desymmetrisation of aromatic diamines and synthesis of non-symmetrical thiourea derivatives by click-mechanochemistry. Chem. Commun. 2012, 48, 9705–9707. [Google Scholar] [CrossRef] [PubMed]
- Neat grinding of equimolar amounts of m-phenylenediamine (m-2b) and phenyl isocyanate for 30 minutes using one 12 mm stainless steel ball led to a mixture of unreacted diamine (21%), isocyanate (4%), amino-urea 2n-O (21%) and bis(urea) 7a (54%), based on 1H NMR analysis. For comparison, sterically more hindered o-phenylenediamine (o-2b) afforded the respective amino-urea in 78% under the same conditions.
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Product | Yield 2 | Product | Yield |
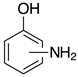
 | o-2b, 99% (99%) m-2b, 98% (96%) p-2b, 95% (96%) |  | 2n-S, n.r. (X=S) 2n-O, 96% (X=O) |
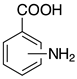
 | o-2c, 99% m-2c, 94% p-2c, 99% | 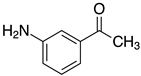 | 2o, 95% 3 |
 | o-2d, 98% m-2d, 99% p-2d, 97% | 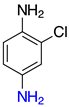 | p-2p, 17% |
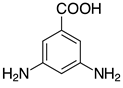 | 2e, 99% | 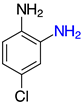 | o-2p, 42% |
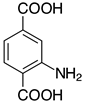 | 2f, 99% | 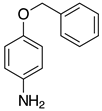 | 2q, 37% |
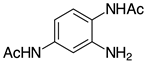 | 2g, 94% | 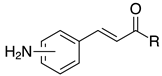 | m-2r, 99% (R=Ph) p-2r, 99% (R=Ph) |
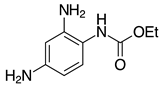 | 2h, 93% | 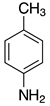 | 2s, 95% |
 | 2i, 99% | 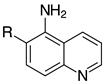 | 2t, 95% (R=H) 2u, 84% (R=Me) |
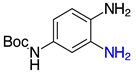 | 2j, 99% | 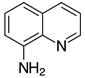 | 2v, 90% |
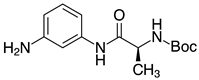 | 2k, 99% | 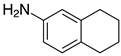 | 2w, 40% (97%) 3 |
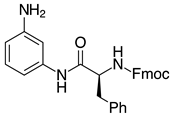 | 2l-Phe, 93% 3 |  | 2x, 18% (37%) |
 | 2l-Val, 90% 3 | 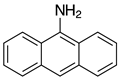 | 2y, traces |
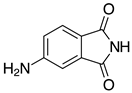 | 2m, 97% | 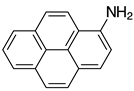 | 2z, n.r. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portada, T.; Margetić, D.; Štrukil, V. Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives. Molecules 2018, 23, 3163. https://doi.org/10.3390/molecules23123163
Portada T, Margetić D, Štrukil V. Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives. Molecules. 2018; 23(12):3163. https://doi.org/10.3390/molecules23123163
Chicago/Turabian StylePortada, Tomislav, Davor Margetić, and Vjekoslav Štrukil. 2018. "Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives" Molecules 23, no. 12: 3163. https://doi.org/10.3390/molecules23123163
APA StylePortada, T., Margetić, D., & Štrukil, V. (2018). Mechanochemical Catalytic Transfer Hydrogenation of Aromatic Nitro Derivatives. Molecules, 23(12), 3163. https://doi.org/10.3390/molecules23123163