
Activity Landscape and Molecular Modeling to Explore the SAR of Dual Epigenetic Inhibitors: A Focus on G9a and DNMT1




Abstract
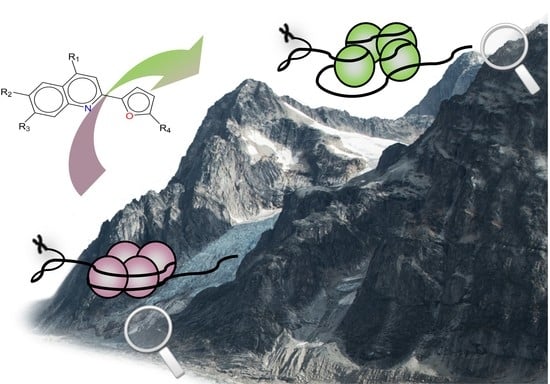
:
1. Introduction
2. Results and Discussion
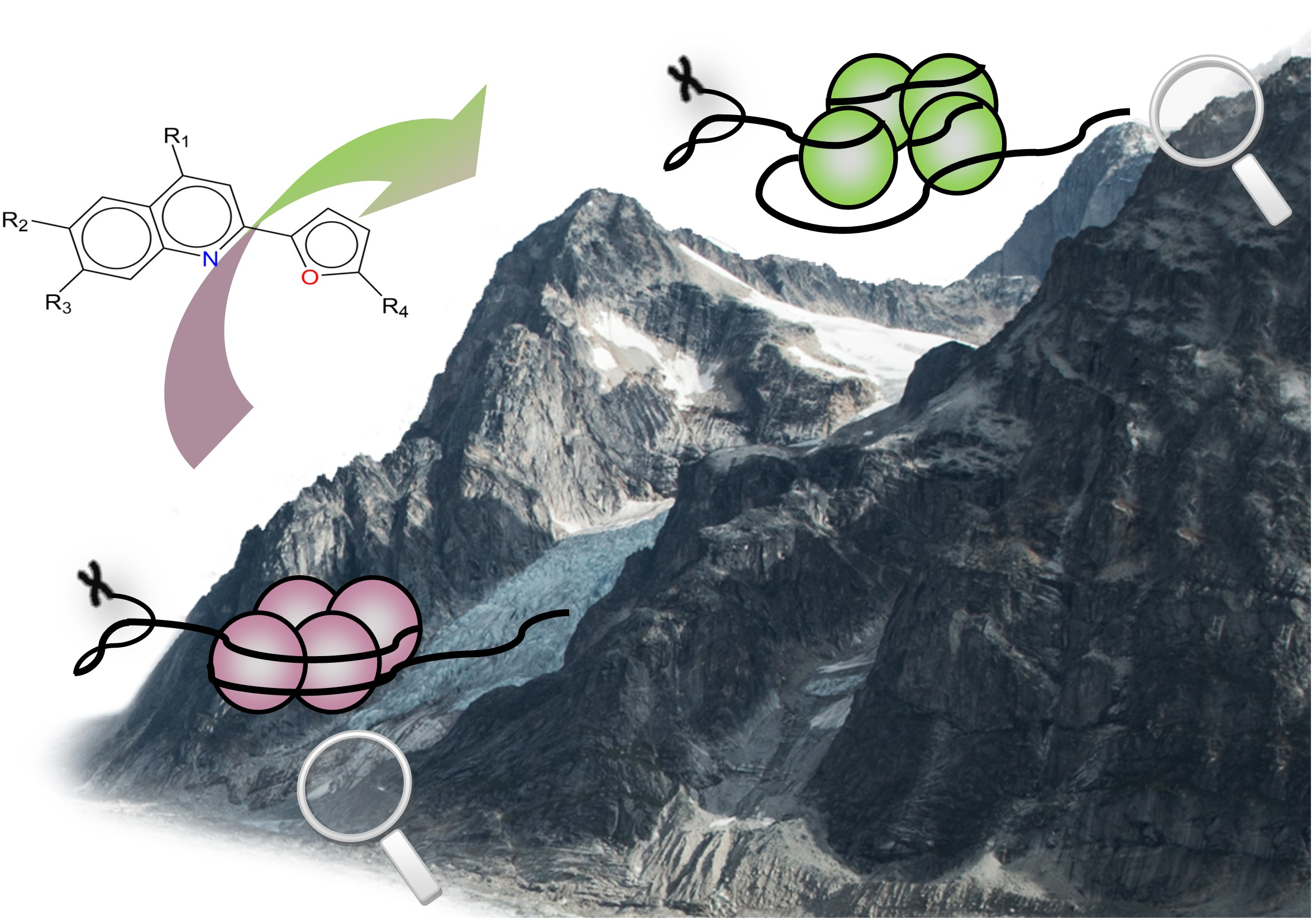
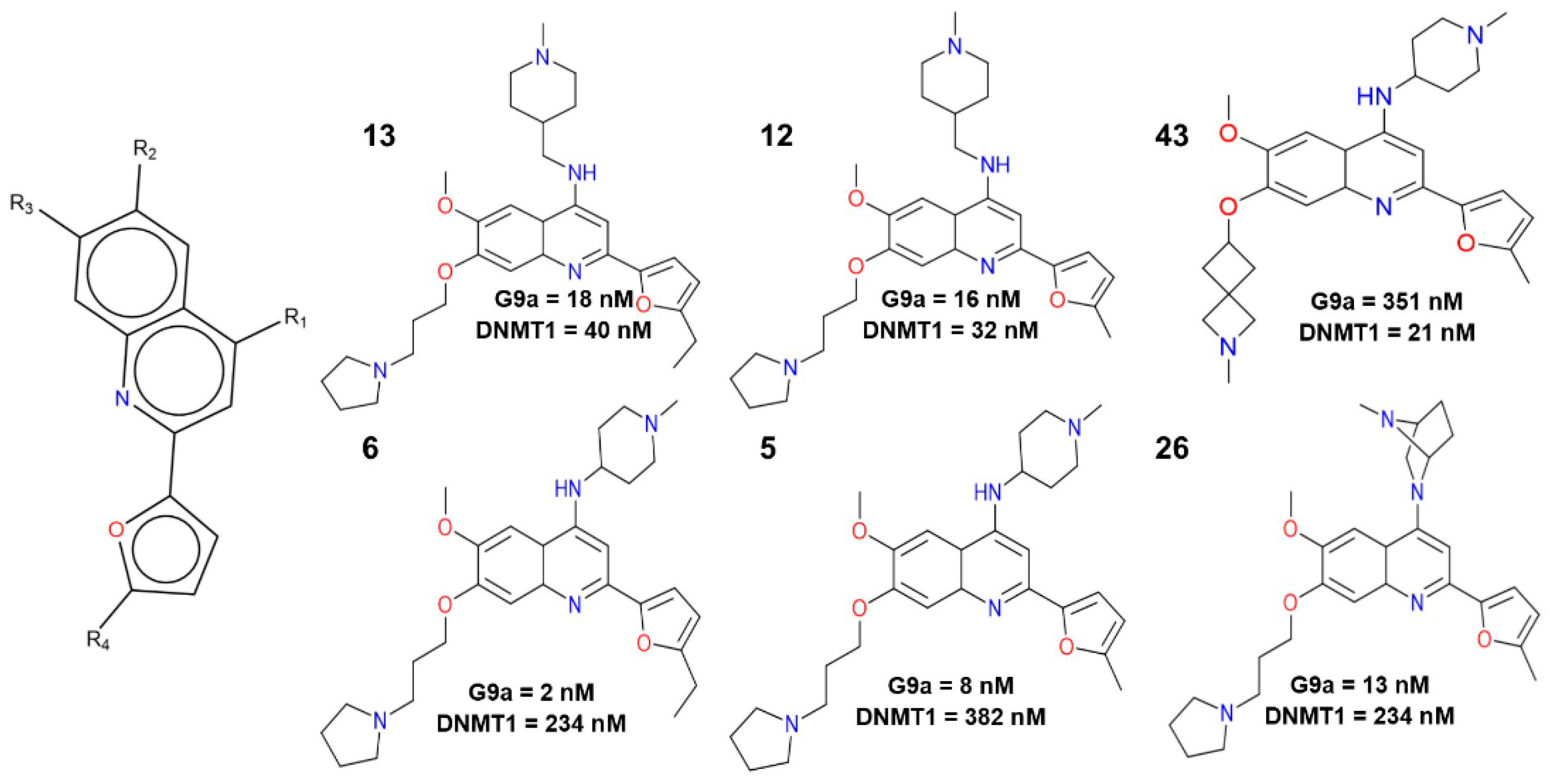
2.1. Qualitative Analysis of the SAR with SAReport
2.2. Activity Landscape
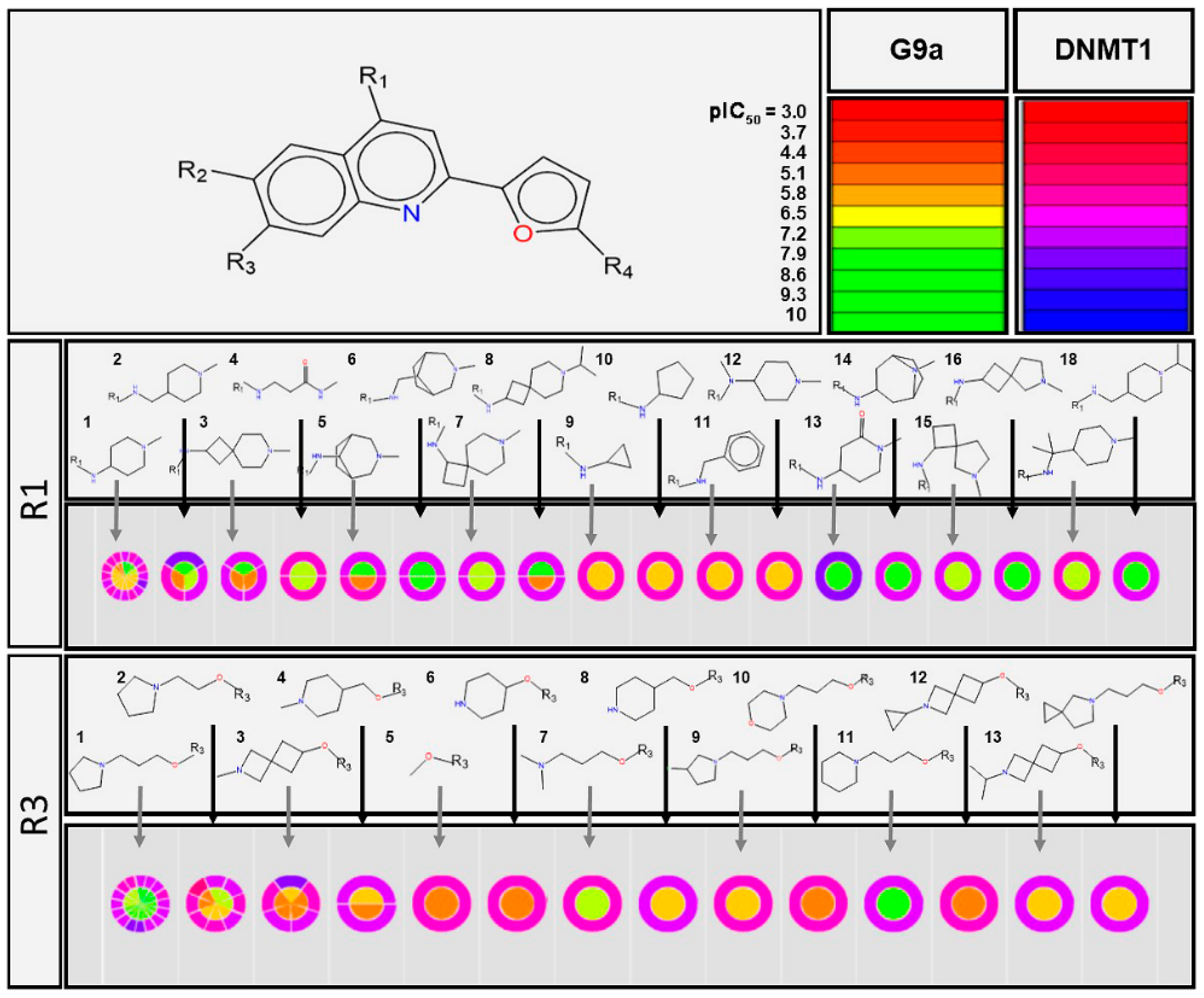
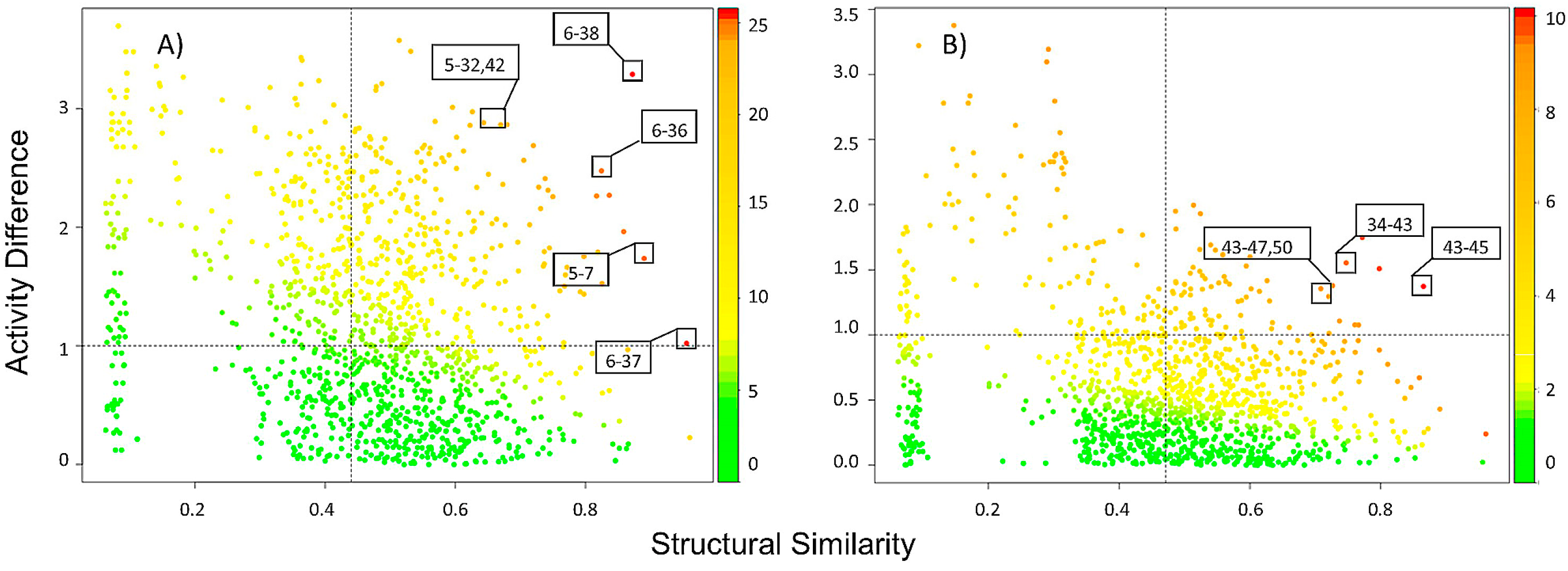
2.2.1. SAS Maps
2.2.2. Quantitative Analysis of SAS Maps
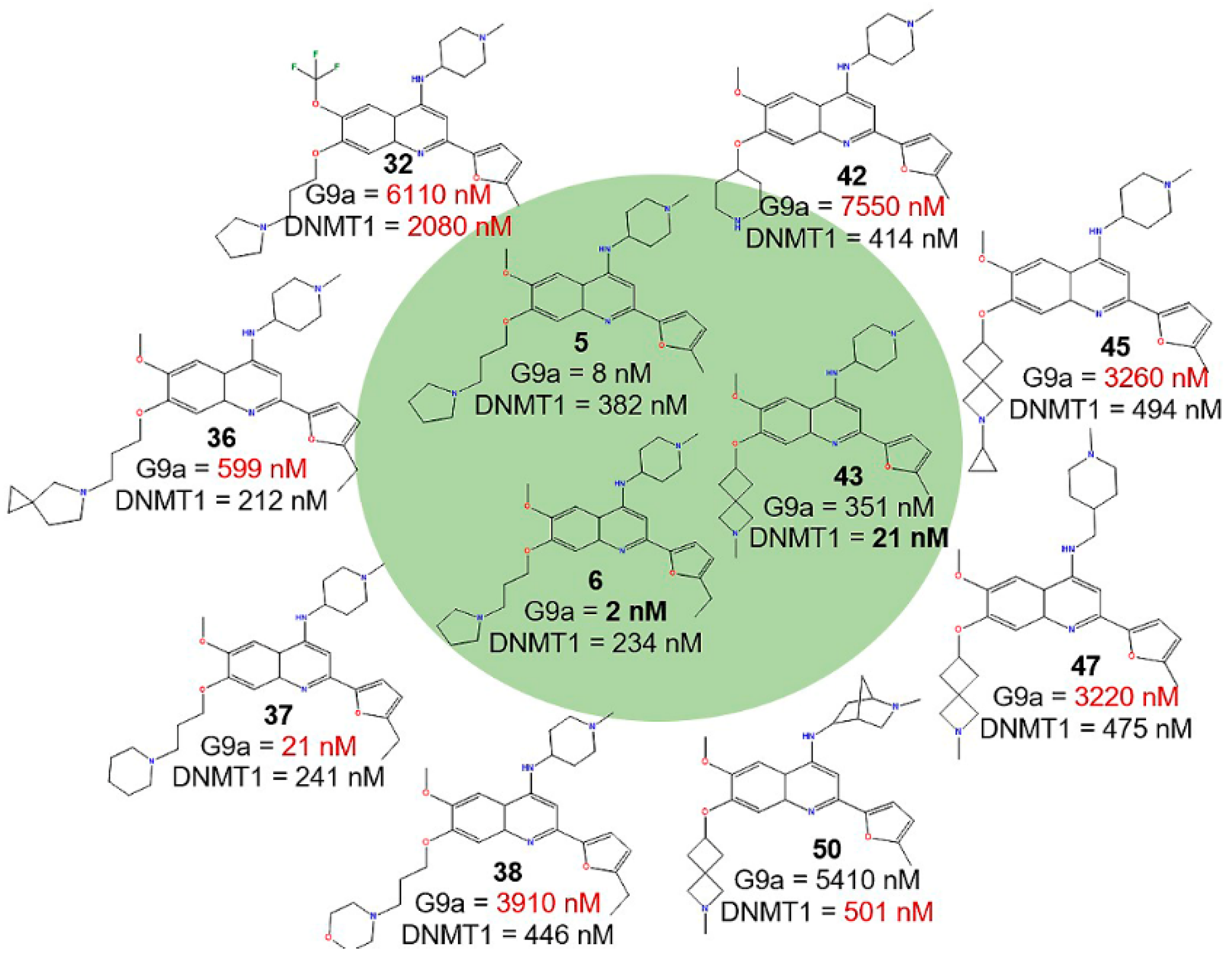
2.2.3. Activity Cliff Generators
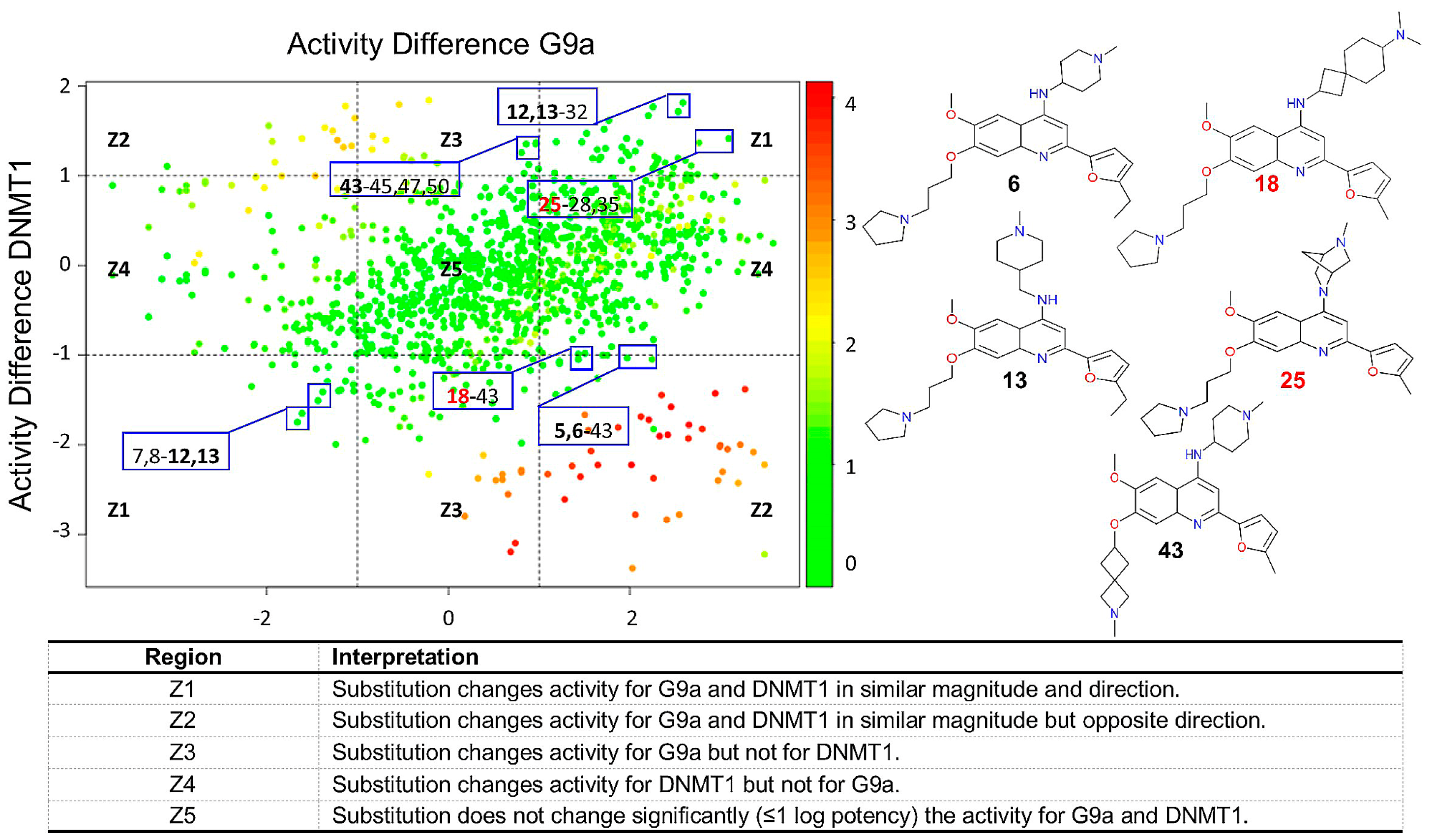
2.2.4. DAD Maps
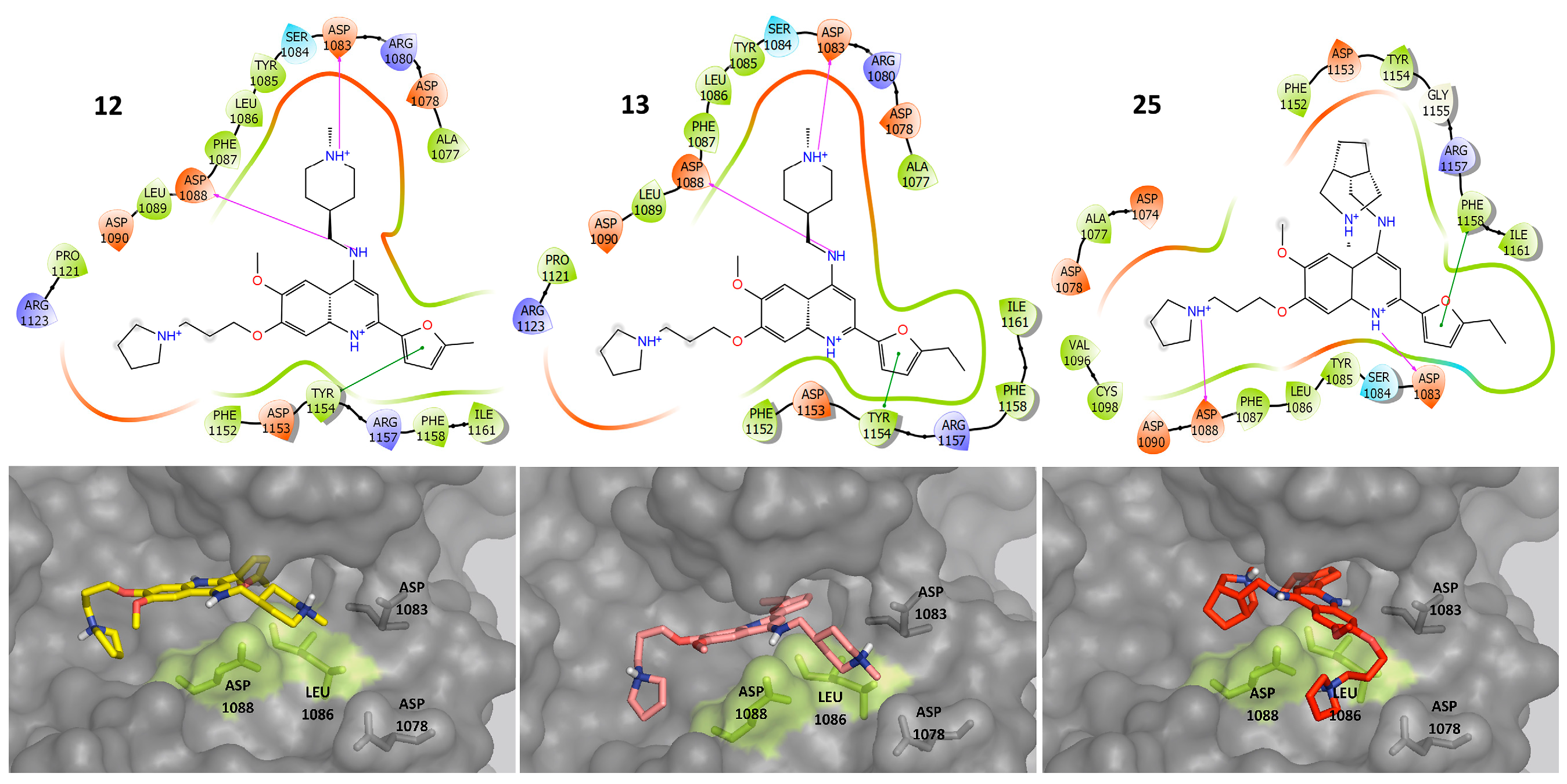
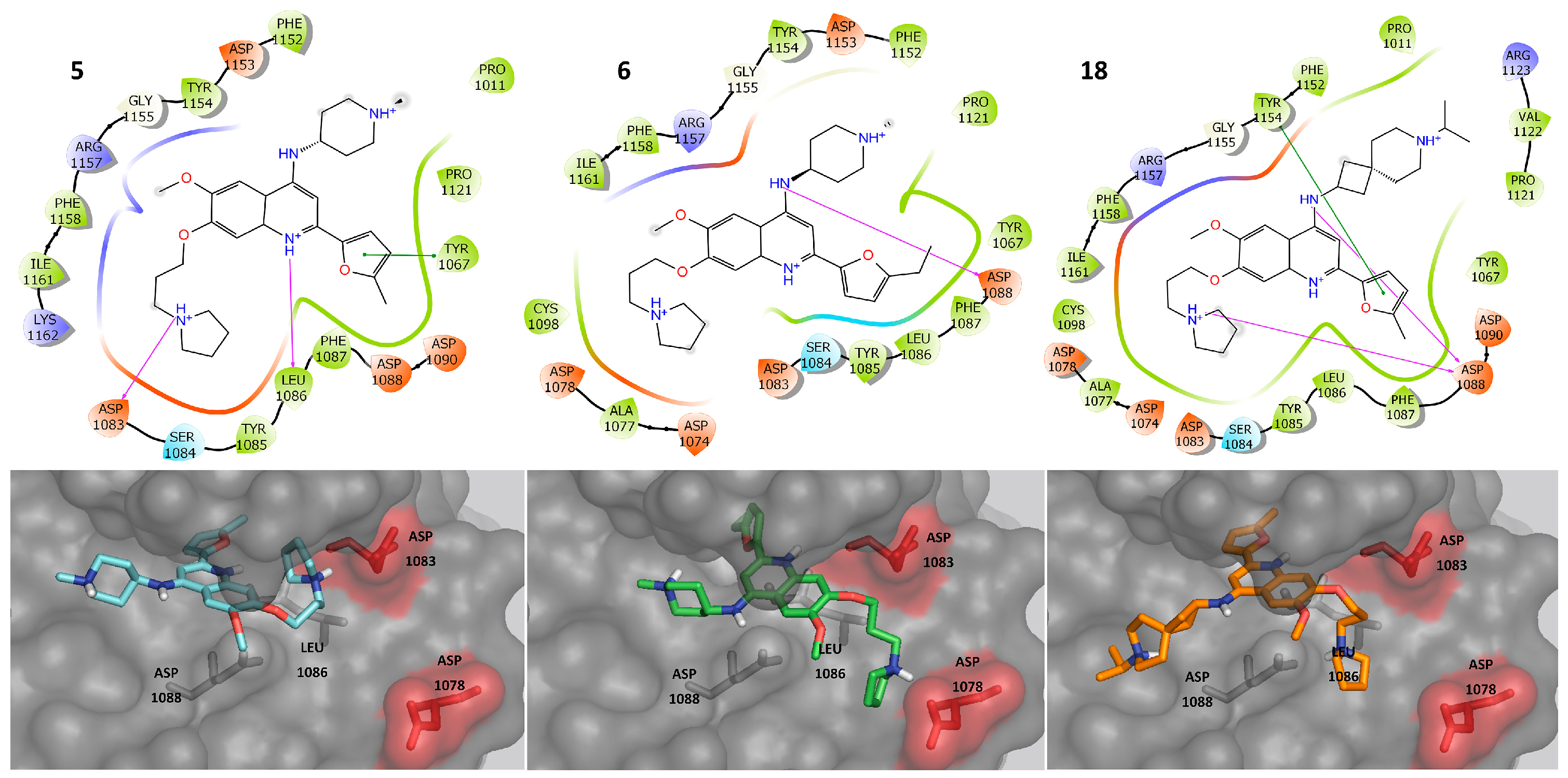
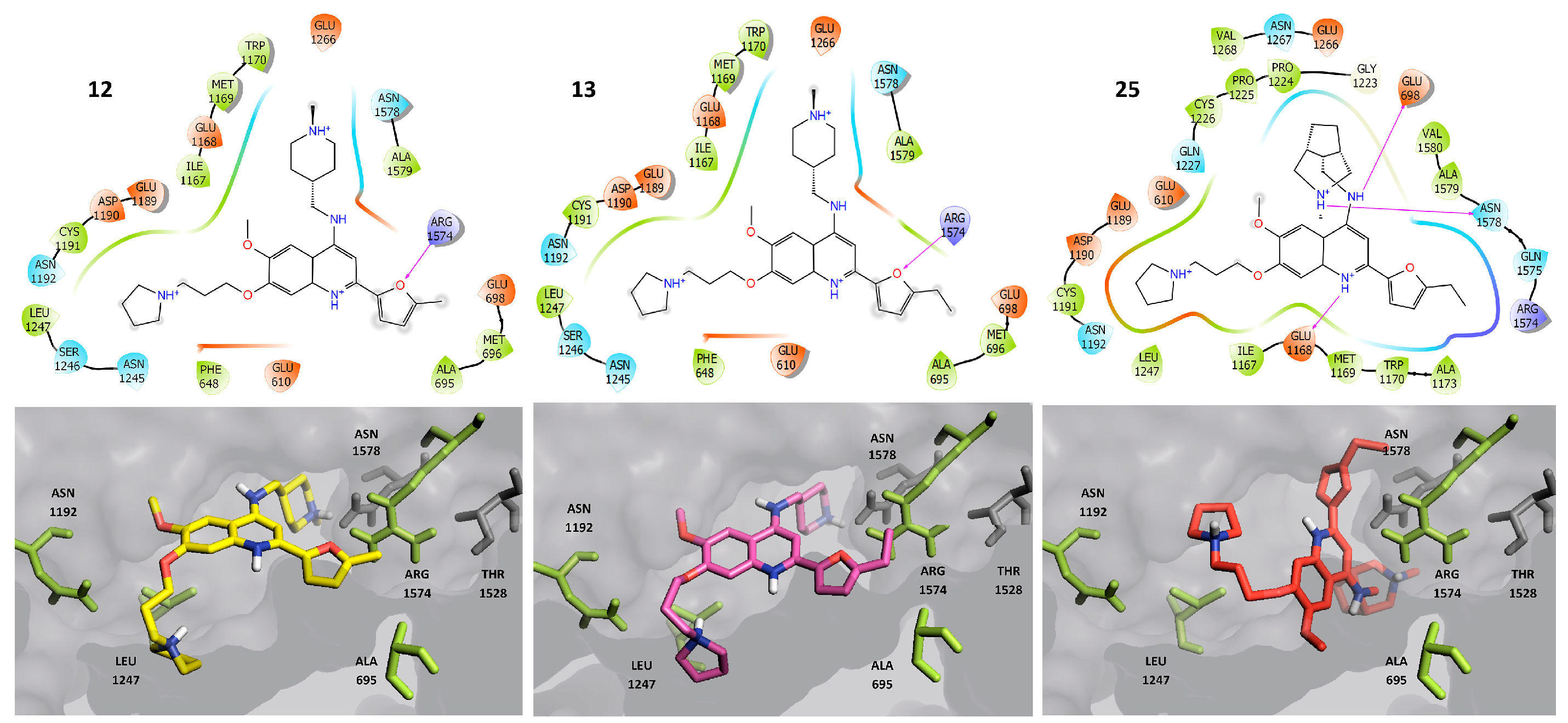
2.3. Molecular Docking
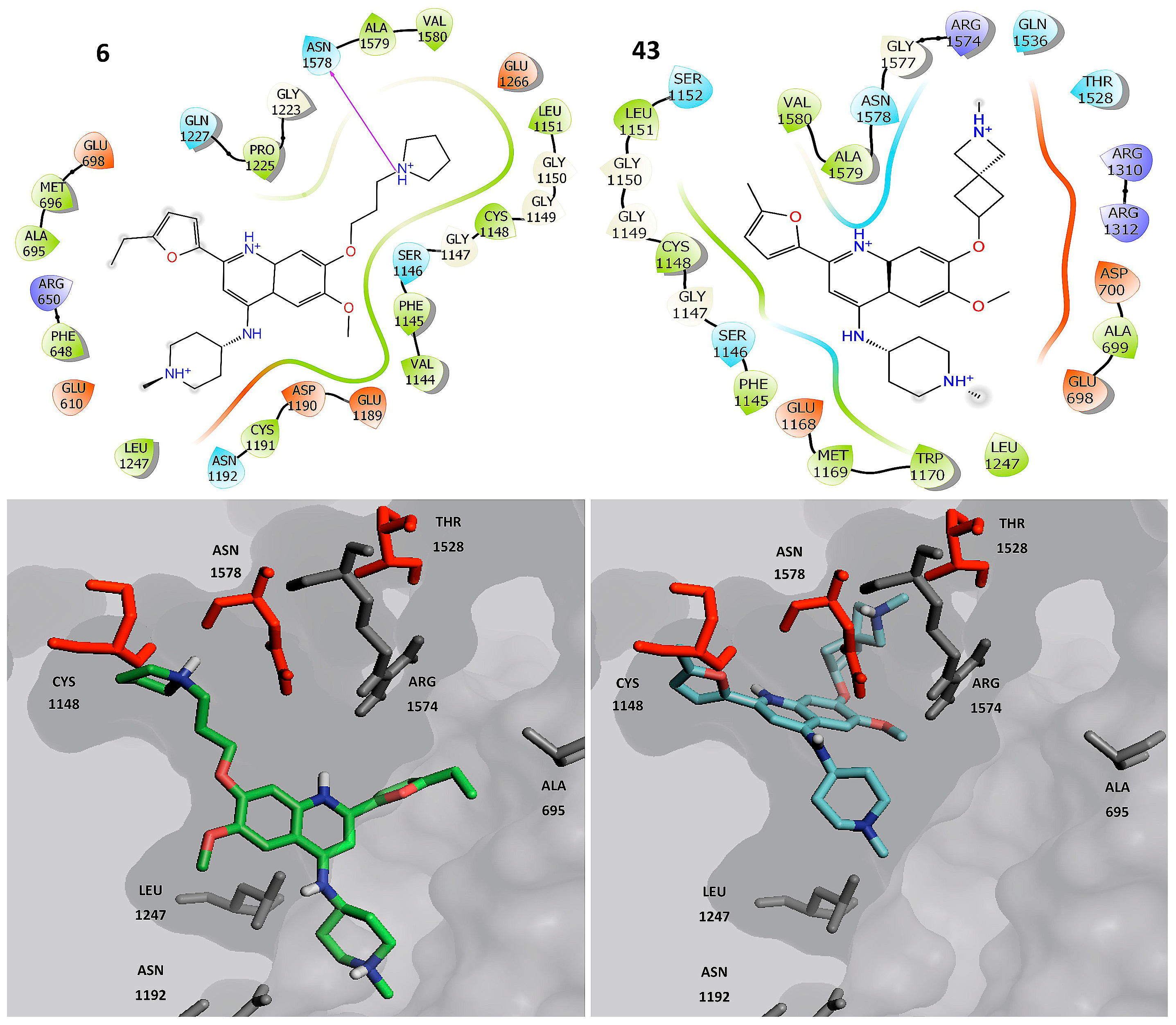
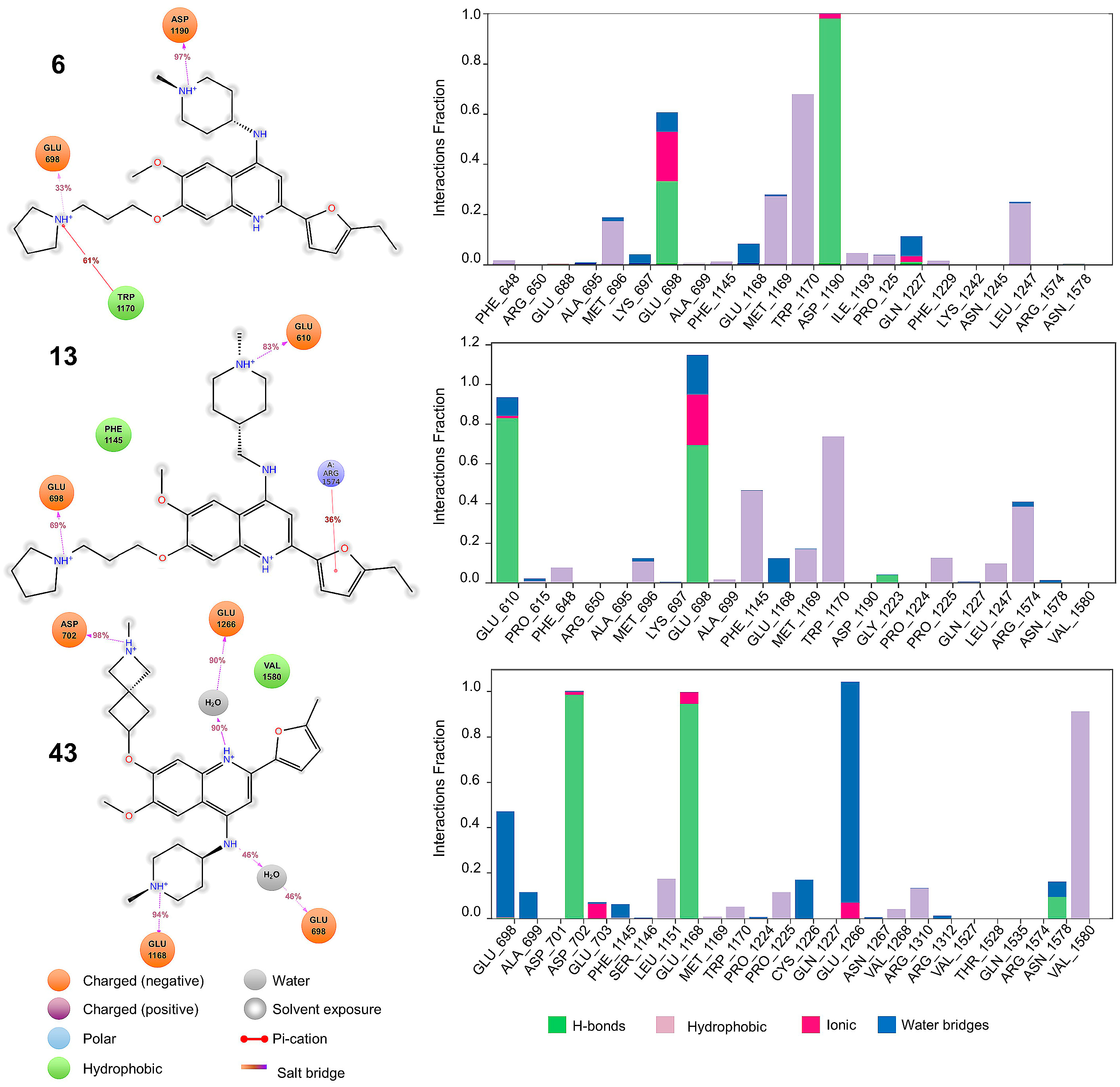
2.4. Molecular Dynamics
3. Materials and Methods
3.1. Data Set
3.2. Software and Online Resources
3.3. Activity Landscape
3.4. Molecular Docking
3.4.1. Protein Preparation
3.4.2. Ligand Preparation
3.4.3. Molecular Docking
3.4.4. Search for Ideal Conditions
3.5. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rabal, O.; Sánchez-Arias, J.A.; José-Enériz, E.S.; Agirre, X.; De Miguel, I.; Garate, L.; Miranda, E.; Sáez, E.; Roa, S.; Martínez-Climent, J.A.; et al. Detailed exploration around 4-aminoquinolines chemical space to navigate the lysine methyltransferase G9a and DNA methyltransferase biological spaces. J. Med. Chem. 2018, 61, 6546–6573. [Google Scholar] [CrossRef]
- Cruz-Monteagudo, M.; Medina-Franco, J.L.; Pérez-Castillo, Y.; Nicolotti, O.; Cordeiro, M.N.D.S.; Borges, F. Activity cliffs in drug discovery: Dr Jekyll or Mr Hyde? Drug Discov. Today 2014, 19, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Villanueva, J.; Méndez-Lucio, O.; Soria-Arteche, O.; Medina-Franco, J.L. Activity cliffs and activity cliff generators based on chemotype-related activity landscapes. Mol. Divers. 2015, 19, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
- Ramya Chandar Charles, M.; Hsieh, H.-P.; Selvaraj Coumar, M. Delineating the active site architecture of G9a lysine methyltransferase through substrate and inhibitor binding mode analysis: A molecular dynamics study. J. Biomol. Struct. Dyn. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Yongye, A.B.; Byler, K.; Santos, R.; Martínez-Mayorga, K.; Maggiora, G.M.; Medina-Franco, J.L. Consensus models of activity landscapes with multiple chemical, conformer, and property representations. J. Chem. Inf. Model. 2011, 51, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2018; Available online: http://www.chemcomp.com (accessed on 31 August 2018).
- González-Medina, M.; Méndez-Lucio, O.; Medina-Franco, J.L. Activity landscape plotter: A web-based application for the analysis of structure-activity relationships. J. Chem. Inf. Model. 2017, 57, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Huey, R. AutoDock4 and AutoDockTools4: Automated docking with selective receptor felexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Maggiora, G.M. On outliers and activity cliffs—Why QSAR often disappoints. J. Chem. Inf. Model. 2006, 46, 1535. [Google Scholar] [CrossRef]
- Medina-Franco, J.L. Scanning structure–Activity relationships with structure–Activity similarity and related maps: From consensus activity cliffs to selectivity switches. J. Chem. Inf. Model. 2012, 52, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Villanueva, J.; Santos, R.; Hernández-Campos, A.; Giulianotti, M.A.; Castillo, R.; Medina-Franco, J.L. Structure-activity relationships of benzimidazole derivatives as antiparasitic agents: Dual activity-difference (DAD) maps. Medchemcomm 2011, 2, 44–49. [Google Scholar] [CrossRef]
- Guha, R.; Van Drie, J.H. Structure—Activity landscape index: Identifying and quantifying activity cliffs. J. Chem. Inf. Model. 2008, 48, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Barsyte-Lovejoy, D.; Liu, F.; Rival-Gervier, S.; Allali-Hassani, A.; Labrie, V.; Wigle, T.J.; Dimaggio, P.A.; Wasney, G.A.; Siarheyeva, A.; et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 2011, 7, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, K.; Suetake, I.; Yamashita, E.; Suga, M.; Narita, H.; Nakagawa, A.; Tajima, S. Structural insight into maintenance methylation by mouse DNA methyltransferase 1 (Dnmt1). Proc. Natl. Acad. Sci. 2011, 108, 9055–9059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Joo, K.; Lee, J.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins Struct. Funct. Bioinforma. 2009. [CrossRef] [PubMed]
- Cho, A.E.; Guallar, V.; Berne, B.J.; Friesner, R. Importance of accurate charges in molecular docking: Quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 2005, 26, 865–967. [Google Scholar] [CrossRef]
- Liu, F.; Chen, X.; Allali-Hassani, A.; Quinn, A.M.; Wigle, T.J.; Wasney, G.A.; Dong, A.; Senisterra, G.; Chau, I.; Siarheyeva, A.; et al. Protein lysine methyltransferase G9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J. Med. Chem. 2010, 53, 5844–5857. [Google Scholar] [CrossRef]
- Song, J.; Teplova, M.; Ishibe-Murakami, S.; Patel, D.J. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 2012, 335, 709–712. [Google Scholar] [CrossRef]
- Ciceri, P.; Müller, S.; O’Mahony, A.; Fedorov, O.; Filippakopoulos, P.; Hunt, J.P.; Lasater, E.A.; Pallares, G.; Picaud, S.; Wells, C.; et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 2014, 10, 305–312. [Google Scholar] [CrossRef]
- Zang, J.; Liang, X.; Huang, Y.; Jia, Y.; Li, X.; Xu, W.; Chou, C.J.; Zhang, Y. Discovery of novel pazopanib-based HDAC and VEGFR dual inhibitors targeting cancer epigenetics and angiogenesis simultaneously. J. Med. Chem. 2018, 61, 5304–5322. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Chen, W.; Wang, X.; Yang, X.; Xu, T.; Wang, P.; Zhang, W.; Rao, Y.; Miao, C.; Sheng, C. Small molecule inhibitors simultaneously targeting cancer metabolism and epigenetics: Discovery of novel nicotinamide phosphoribosyltransferase (NAMPT) and histone deacetylase (HDAC) dual inhibitors. J. Med. Chem. 2017, 60, 7965–7983. [Google Scholar] [CrossRef] [PubMed]
- San José-Enériz, E.; Agirre, X.; Rabal, O.; Vilas-Zornoza, A.; Sanchez-Arias, J.A.; Miranda, E.; Ugarte, A.; Roa, S.; Paiva, B.; Estella-Hermoso de Mendoza, A.; et al. Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies. Nat. Commun. 2017, 8, 15424. [Google Scholar] [CrossRef] [PubMed]
- Naveja, J.J.; Medina-Franco, J.L. Insights from pharmacological similarity of epigenetic targets in epipolypharmacology. Drug Discov. Today 2018, 23, 141–150. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Target | Fingerprint | Activity Cliff | Smooth SAR | Similarity Cliff | Not Descriptive |
|---|---|---|---|---|---|
| G9a | MACCS | 268 (21.8%) | 320 (26.2%) | 290 (23.7%) | 347 (28.3%) |
| PubChem | 432 (35.3%) | 468 (38.2%) | 142 (11.6%) | 183 (14.9%) | |
| ECFP4 | 324 (26.5%) | 439 (35.8%) | 171 (13.9%) | 291 (23.8%) | |
| Consensus * | 341 (27.8%) | 409 (33.4%) | 201 (16.4%) | 274 (22.4%) | |
| DNMT1 | MACCS | 55 (4.5%) | 532 (43.5%) | 488 (39.9%) | 147 (12.1%) |
| PubChem | 92 (7.6%) | 755 (61.9%) | 262 (21.5%) | 110 (9.0%) | |
| ECFP4 | 177 (14.5%) | 949 (77.6%) | 71 (5.8%) | 25 (2.1%) | |
| Consensus * | 108 (8.86%) | 745 (61.1%) | 274 (22.4%) | 94 (7.73%) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-López, E.; Prieto-Martínez, F.D.; Medina-Franco, J.L. Activity Landscape and Molecular Modeling to Explore the SAR of Dual Epigenetic Inhibitors: A Focus on G9a and DNMT1. Molecules 2018, 23, 3282. https://doi.org/10.3390/molecules23123282
López-López E, Prieto-Martínez FD, Medina-Franco JL. Activity Landscape and Molecular Modeling to Explore the SAR of Dual Epigenetic Inhibitors: A Focus on G9a and DNMT1. Molecules. 2018; 23(12):3282. https://doi.org/10.3390/molecules23123282
Chicago/Turabian StyleLópez-López, Edgar, Fernando D. Prieto-Martínez, and José L. Medina-Franco. 2018. "Activity Landscape and Molecular Modeling to Explore the SAR of Dual Epigenetic Inhibitors: A Focus on G9a and DNMT1" Molecules 23, no. 12: 3282. https://doi.org/10.3390/molecules23123282
APA StyleLópez-López, E., Prieto-Martínez, F. D., & Medina-Franco, J. L. (2018). Activity Landscape and Molecular Modeling to Explore the SAR of Dual Epigenetic Inhibitors: A Focus on G9a and DNMT1. Molecules, 23(12), 3282. https://doi.org/10.3390/molecules23123282