Formation of Nudicaulins In Vivo and In Vitro and the Biomimetic Synthesis and Bioactivity of O-Methylated Nudicaulin Derivatives

 ,
,  and
and
Abstract
:
1. Introduction
2. Results
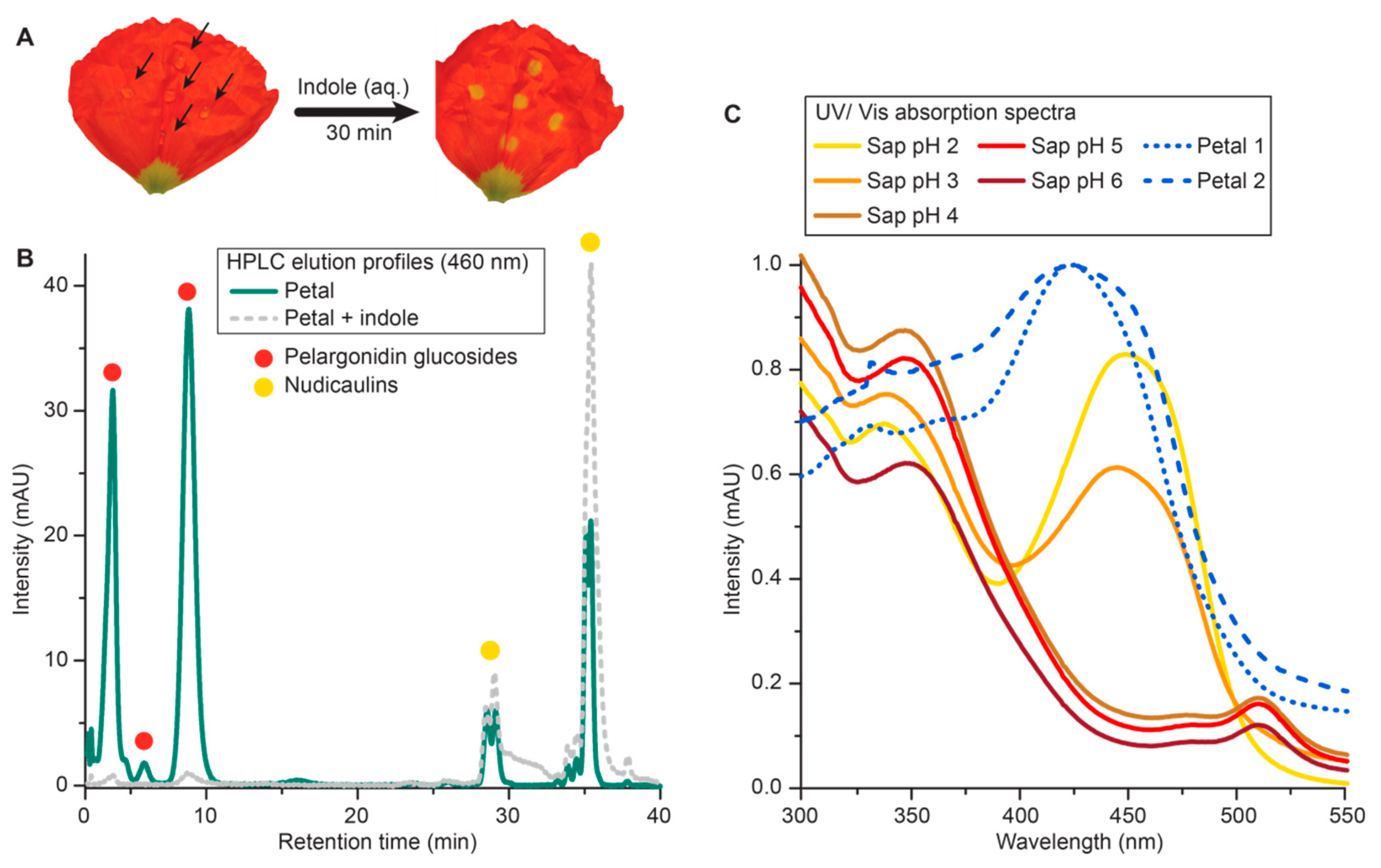
2.1. The Final Step of the Nudicaulin Biosynthesis
2.2. Synthesis of O-Methylated Nudicaulin Derivatives
2.3. Bioactivity
3. Discussion
3.1. The Final Step of the Nudicaulin Biosynthesis
3.2. Synthesis of O-Methylated Nudicaulin Derivatives
3.3. Bioactivity
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Plant Material
4.3. Final Step of Nudicaulin Biosynthesis
4.4. Synthetic Procedures and Analytical Data
4.4.1. 3,5,7,3′4′-Penta-O-Methylcyanidin (5)
4.4.2. General Procedure for the Preparation of Nudicaulin Derivatives (Racemic Mixtures)
4.4.3. 5,7,11,3′,4′-Penta-O-Methylnudicaulin (6)
4.4.4. 16-Methyl-5,7,11,3′,4′-Penta-O-Methylnudicaulin (7)
4.4.5. 17-Methyl-5,7,11,3′,4′-Penta-O-Methylnudicaulin (8)
4.4.6. 18-Methyl-5,7,11,3′,4′-Penta-O-Methylnudicaulin (9)
4.4.7. 17-Fluoro-5,7,11,3′,4′-Penta-O-Methylnudicaulin (10)
4.4.8. 16-Hydroxy-5,7,11,3′,4′-Penta-O-Methylnudicaulin (11)
4.5. Cell Toxicity
4.6. Antimicrobial Activity
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Price, J.R.; Robinson, R.; Scott-Moncrieff, R. The yellow pigment of Papaver nudicaule. Part I. J. Chem. Soc. 1939, 0, 1465–1468. [Google Scholar] [CrossRef]
- Schliemann, W.; Schneider, B.; Wray, V.; Schmidt, J.; Nimtz, M.; Porzel, A.; Böhm, H. Flavonols and an indole alkaloid skeleton bearing identical acylated glycosidic groups from yellow petals of Papaver nudicaule. Phytochemistry 2006, 67, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Tatsis, E.C.; Schaumlöffel, A.; Warskulat, A.-C.; Massiot, G.; Schneider, B.; Bringmann, G. Nudicaulins, yellow flower pigments of Papaver nudicaule: Revised constitution and assignment of absolute configuration. Org. Lett. 2013, 15, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Tatsis, E.C.; Eylert, E.; Maddula, R.K.; Ostrozhenkova, E.; Svatoš, A.; Eisenreich, W.; Schneider, B. Biosynthesis of nudicaulins: A 13CO2-pulse/chase labeling study with Papaver nudicaule. ChemBioChem 2014, 15, 1645–1650. [Google Scholar] [CrossRef] [PubMed]
- Warskulat, A.-C.; Tatsis, E.C.; Dudek, B.; Kai, M.; Lorenz, S.; Schneider, B. Unprecedented utilization of pelargonidin and indole for the biosynthesis of pant indole alkaloids. ChemBioChem 2016, 17, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Kanchanapoom, T.; Kasai, R.; Chumsri, P.; Kraisintu, K.; Yamasaki, K. Lotthanongine, an unprecedented flavonoidal indole alkaloid from the roots of Thai medicinal plant Trigonostemon reidioides. Tetrahedron Lett. 2002, 43, 2941–2943. [Google Scholar] [CrossRef]
- Vepsäläinen, J.J.; Auriola, S.; Tukiainen, M.; Ropponen, N.; Callaway, J.C. Isolation and characterization of yuremamine, a new phytoindole. Planta Med. 2005, 71, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Calvert, M.B.; Sperry, J. Bioinspired total synthesis and structural revision of yuremamine, an alkaloid from the entheogenic plant Mimosa tenuiflora. J. Chem. Soc. Chem. Commun. 2015, 51, 6202–6205. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anticancer Agents 2002, 2, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Coderch, C.; Morreale, A.; Gago, F. Tubulin-based structure-affinity relationships for antimitotic vinca alkaloids. ACAMC 2012, 12, 219–225. [Google Scholar] [CrossRef]
- Martinez-Harms, J.; Warskulat, A.-C.; Dudek, B.; Kunert, G.; Lorenz, S.; Hansson, B.S.; Schneider, B. Biosynthetic and functional color-scent associations in flowers of Papaver nudicaule and its impact on pollinators. ChemBioChem 2018, 19, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Kurkdjian, A.; Guern, J. Intracellular pH: Measurement and importance in cell activity. Annu. Rev. Plant. Physiol. Plant. Mol. Biol. 1989, 40, 271–303. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Molsoft LLC. MolCart. 2018. Available online: http://www.molsoft.com (accessed on 20 July 2018).
- Kimura, Y.; Kato, R.; Oyama, K.-I.; Kondo, T.; Yoshida, K. Efficient preparation of various O-methylquercetins by selective demethylation. Nat. Prod. Commun. 2016, 11, 957–961. [Google Scholar] [PubMed]
- Kimura, Y.; Oyama, K.-i.; Kondo, T.; Yoshida, K. Synthesis of 8-aryl-3,5,7,3′,4′-penta-O-methylcyanidins from the corresponding quercetin derivatives by reduction with LiAlH4. Tetrahedron Lett. 2017, 58, 919–922. [Google Scholar] [CrossRef]
- Tatsis, E.C.; Böhm, H.; Schneider, B. Occurrence of nudicaulin structural variants in flowers of papaveraceous species. Phytochemistry 2013, 92, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Butelli, E.; Martin, C. Engineering anthocyanin biosynthesis in plants. Curr. Opin. Plant Biol. 2014, 19, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Kondo, T. Structure and molecular stacking of anthocyanins-flower color variation. Angew. Chem. Int. Ed. Engl. 1991, 30, 17–33. [Google Scholar] [CrossRef]
- Yoshida, K.; Mori, M.; Kondo, T. Blue flower color development by anthocyanins: From chemical structure to cell physiology. Nat. Prod. Rep. 2009, 26, 884–915. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Kitahara, S.; Ito, D.; Kondo, T. Ferric ions involved in the flower color development of the Himalayan blue poppy, Meconopsis grandis. Phytochemistry 2006, 67, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Oyama, K.-I.; Nakamura, S.; Yamakawa, D.; Tokuno, K.; Yoshida, K. Novel and efficient synthesis of cyanidin 3-O-β-d-glucoside from (+)-catechin via a flav-3-en-3-ol as a key intermediate. Org. Lett. 2006, 8, 3609–3612. [Google Scholar] [CrossRef] [PubMed]
- Krauth, F.; Dahse, H.-M.; Rüttinger, H.-H.; Frohberg, P. Synthesis and characterization of novel 1,2,4-triazine derivatives with antiproliferative activity. Bioorg. Med. Chem. 2010, 18, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Krieg, R.; Jortzik, E.; Goetz, A.-A.; Blandin, S.; Wittlin, S.; Elhabiri, M.; Rahbari, M.; Nuryyeva, S.; Voigt, K.; Dahse, H.-M.; et al. Arylmethylamino steroids as antiparasitic agents. Nat. Commun. 2017, 8, 14478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds 6–11 are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|
| δ, mult., J [Hz] | δ, mult., J [Hz] | δ, mult., J [Hz] | δ, mult., J [Hz] | δ, mult., J [Hz] | δ, mult., J [Hz] | |
| 3 | 5.39, s | 5.36, s | 5.36, s | 5.33, s | 5.36, s | 5.31, s |
| 6 | 6.30, d, 1.9 | 6.30, d, 1.9 | 6.30, d, 1.8 | 6.29, d, 1.9 | 6.30, d, 1.8 | 6.29, d, 1.9 |
| 8 | 6.25, d, 1.9 | 6.25, d, 1.9 | 6.25, d, 1.8 | 6.25, d, 1.9 | 6.24, d, 1.8 | 6.25, d, 1.9 |
| 15 | 8.34, d, 7.8 | 8.15, brs | 8.20, d, 8.2 | 8.15, dd, 2.1, 6.7 | 8.29, dd, 8.7, 4JH-15/F-17 = 4.9 | 7.71, d, 2.3 |
| 16 | 7.60, dd, 7.6, 7.8 | 7.42, brd, 8.2 | 7.47, overlap | 7.32, ddd, 8.7, 2.3, 3JH-16/F-17 = 8.7 | ||
| 17 | 7.68, dd, 7.6, 8.0 | 7.50, brd, 8.3 | 7.48, overlap | 7.07, dd, 2.3, 8.8 | ||
| 18 | 7.75, d, 8.0 | 7.62, d, 8.3 | 7.55, brs | 7.49, dd, 2.3, 3JH-18/F-17 = 8.4 | 7.53, d, 8.8 | |
| 2′ | 7.97, d, 2.0 | 7.96, d, 2.1 | 7.95, d, 2.0 | 7.94, d, 2.0 | 7.92, d, 2.0 | 7.90, d, 2.1 |
| 5′ | 7.37, d, 8.7 | 7.38, d, 8.7 | 7.35, d, 8.7 | 7.34, d, 8.6 | 7.34, d, 8.7 | 7.34, d, 8.7 |
| 6′ | 8.26, dd, 2.0, 8.7 | 8.25, dd, 2.1, 8.7 | 8.23, dd, 2.0, 8.7 | 8.23, dd, 2.0, 8.6 | 8.20, dd, 2.0, 8.7 | 8.18, dd, 2.1, 8.7 |
| 5-OMe | 4.01, s | 4.00, s | 4.00, s | 3.99, s | 3.99, s | 3.99, s |
| 7-OMe | 3.76, s | 3.76, s | 3.76, s | 3.75, s | 3.75, s | 3.76, s |
| 11-OMe | 3.66, s | 3.65, s | 3.65, s | 3.68, s | 3.65, s | 3.64, s |
| 3′-OMe | 3.99, s | 3.99, s | 3.99, s | 3.98, s | 3.98, s | 3.99, s |
| 4′-OMe | 4.08, s | 4.08, s | 4.07, s | 4.07, s | 4.06, s | 4.06, s |
| 16-Me | 2.52, s | |||||
| 17-Me | 2.54, s | |||||
| 18-Me | 2.64, s |
| Position | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|
| δ | δ | δ | δ | δ, J13C-19F [Hz] | δ | |
| 2 | 178.2 | 177.7 | 176.9 | 178.4 | 179.8 | 176.2 |
| 3 | 46.0 | 45.9 | 46.0 | 46.0 | 46.2 | 45.8 |
| 4 | 100.6 | 100.7 | 100.3 | 100.9 | 100.7 | 100.8 |
| 5 | 159.0 | 159.0 | 158.8 | 158.8 | 158.7 | 158.7 |
| 6 | 94.3 | 94.2 | 94.2 | 94.2 | 94.2 | 94.2 |
| 7 | 166.0 | 166.0 | 166.0 | 165.9 | 166.0 | 166.0 |
| 8 | 90.1 | 90.1 | 90.1 | 90.1 | 90.1 | 90.1 |
| 9 | 161.7 | 161.7 | 161.7 | 161.6 | 161.7 | 161.6 |
| 11 | 127.8 | 127.7 | 127.8 | 127.8 | 127.6 | 127.8 |
| 12 | 169.7 | 169.2 | 168.6 | 169.1 | 168.8 | 168.6 |
| 13 | 132.5 | 132.5 | 132.5 | 132.9 | 132.0 | 132.9 |
| 14 | 123.1 | 123.2 | 120.4 | 123.1 | 119.7, d, 2.5 | 124.5 |
| 15 | 125.8 | 126.2 | 125.6 | 123.3 | 127.2, d, 10.4 | 112.3 |
| 16 | 129.0 | 139.7 | 129.9 | 128.9 | 115.9, d, 24.0 | 159.5 |
| 17 | 132.1 | 133.0 | 143.8 | 133.7 | 165.2, d, 251.0 | 119.2 |
| 18 | 117.7 | 117.3 | 118.0 | 128.4 | 105.7, d, 27.4 | 118.4 |
| 19 | 148.9 | 146.9 | 149.3 | 147.8 | 151.2 | 141.5 |
| 1′ | 124.5 | 124.4 | 124.5 | 124.4 | 124.5 | 124.4 |
| 2′ | 116.4 | 116.3 | 116.3 | 116.2 | 116.3 | 116.0 |
| 3′ | 151.3 | 151.2 | 151.2 | 151.2 | 151.3 | 151.2 |
| 4′ | 158.9 | 158.0 | 158.6 | 158.7 | 158.9 | 158.3 |
| 5′ | 113.3 | 113.5 | 113.4 | 113.4 | 113.5 | 113.3 |
| 6′ | 132.0 | 131.8 | 131.7 | 131.7 | 131.9 | 131.1 |
| 5-OMe | 56.6 | 56.9 | 56.9 | 56.9 | 56.6 | 56.9 |
| 7-OMe | 56.5 | 56.5 | 56.5 | 56.4 | 56.4 | 56.4 |
| 11-OMe | 53.7 | 53.7 | 53.6 | 53.7 | 53.7 | 53.3 |
| 3′-OMe | 56.9 | 56.6 | 56.6 | 56.5 | 56.9 | 56.6 |
| 4′-OMe | 57.3 | 57.3 | 57.3 | 57.3 | 57.3 | 57.2 |
| 16-Me | 21.7 | |||||
| 17-Me | 22.0 | |||||
| 18-Me | 17.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dudek, B.; Schnurrer, F.; Dahse, H.-M.; Paetz, C.; Warskulat, A.-C.; Weigel, C.; Voigt, K.; Schneider, B. Formation of Nudicaulins In Vivo and In Vitro and the Biomimetic Synthesis and Bioactivity of O-Methylated Nudicaulin Derivatives. Molecules 2018, 23, 3357. https://doi.org/10.3390/molecules23123357
Dudek B, Schnurrer F, Dahse H-M, Paetz C, Warskulat A-C, Weigel C, Voigt K, Schneider B. Formation of Nudicaulins In Vivo and In Vitro and the Biomimetic Synthesis and Bioactivity of O-Methylated Nudicaulin Derivatives. Molecules. 2018; 23(12):3357. https://doi.org/10.3390/molecules23123357
Chicago/Turabian StyleDudek, Bettina, Florian Schnurrer, Hans-Martin Dahse, Christian Paetz, Anne-Christin Warskulat, Christiane Weigel, Kerstin Voigt, and Bernd Schneider. 2018. "Formation of Nudicaulins In Vivo and In Vitro and the Biomimetic Synthesis and Bioactivity of O-Methylated Nudicaulin Derivatives" Molecules 23, no. 12: 3357. https://doi.org/10.3390/molecules23123357
APA StyleDudek, B., Schnurrer, F., Dahse, H.-M., Paetz, C., Warskulat, A.-C., Weigel, C., Voigt, K., & Schneider, B. (2018). Formation of Nudicaulins In Vivo and In Vitro and the Biomimetic Synthesis and Bioactivity of O-Methylated Nudicaulin Derivatives. Molecules, 23(12), 3357. https://doi.org/10.3390/molecules23123357