Qualitative and Quantitative Analysis of C-glycosyl-flavones of Iris lactea Leaves by Liquid Chromatography/Tandem Mass Spectrometry

Abstract
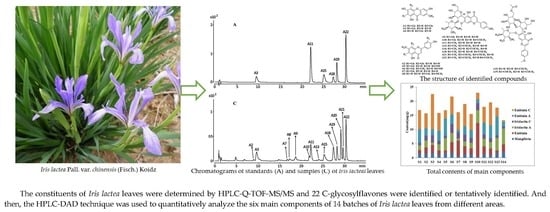
:
1. Introduction
2. Results and Discussion
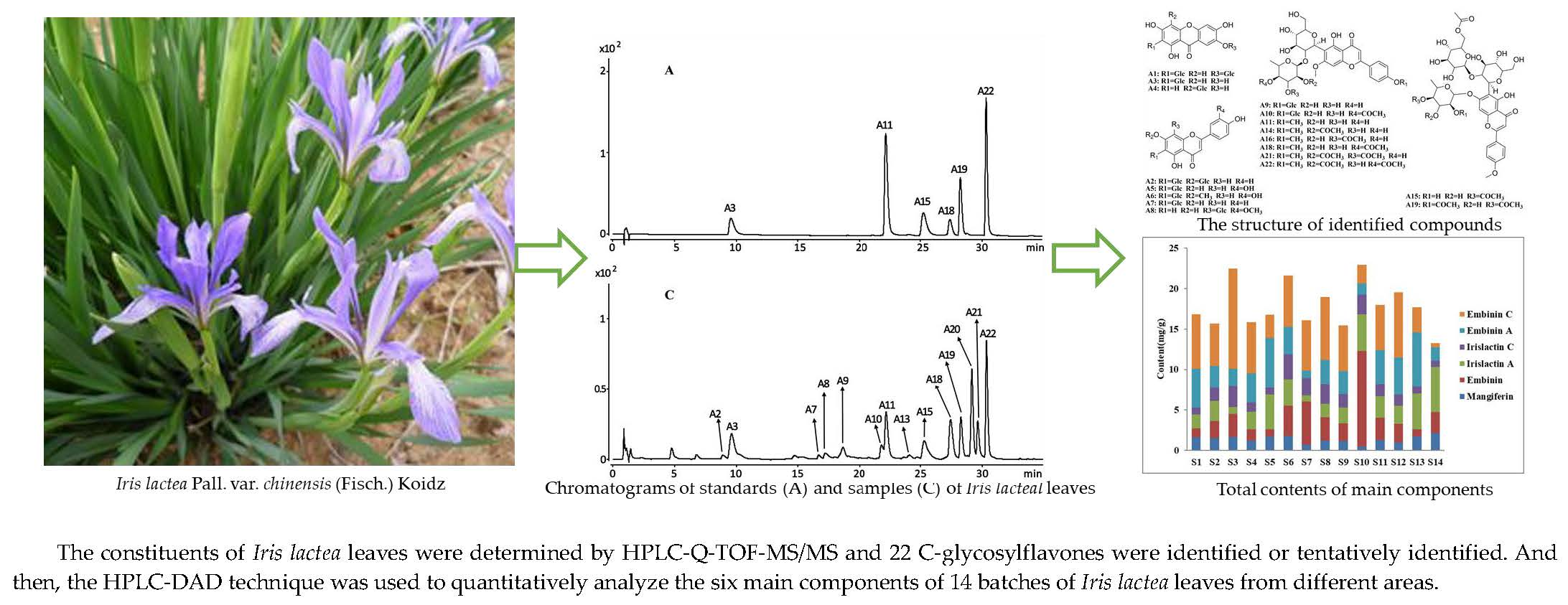
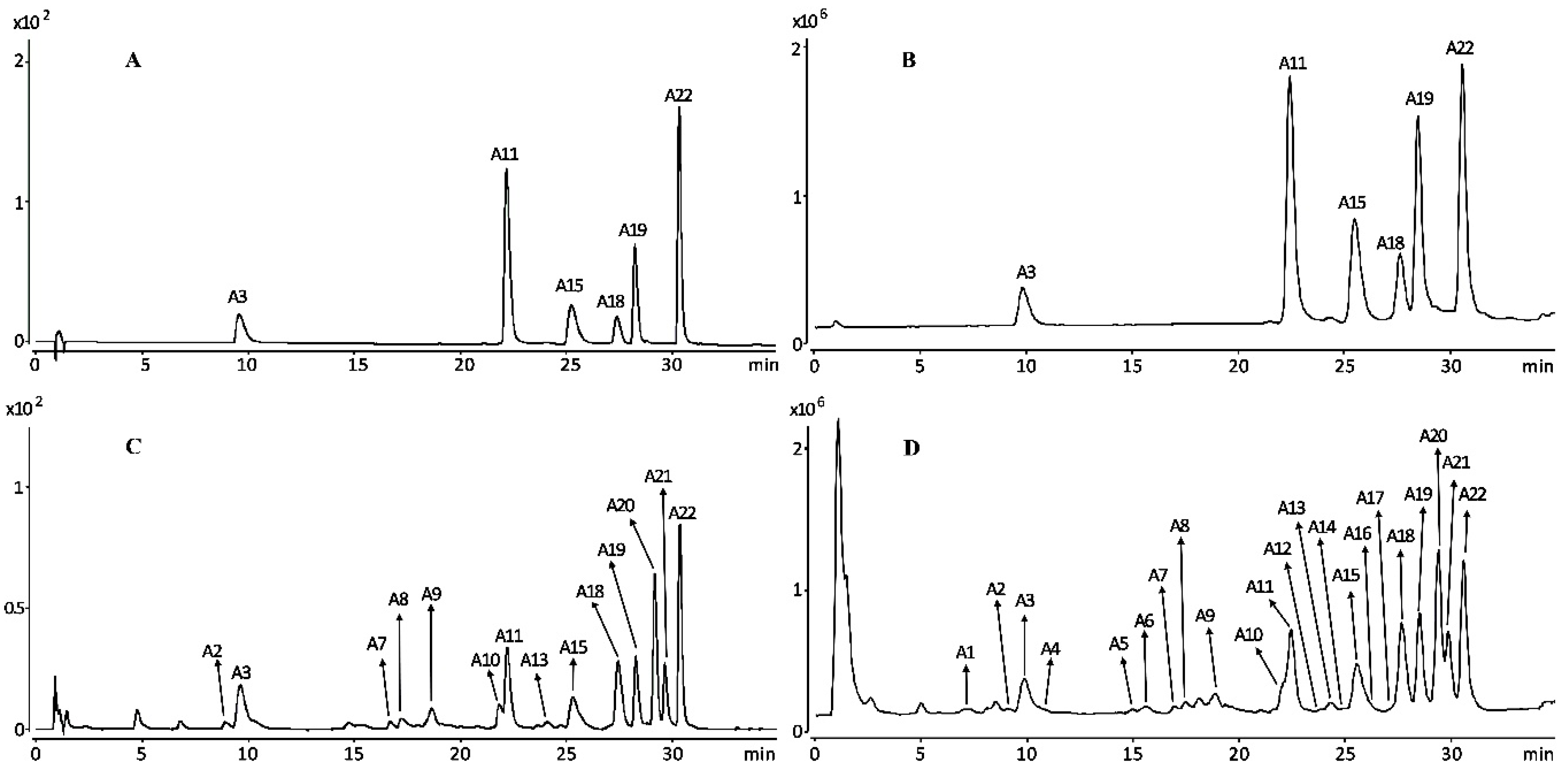
2.1. Compound Identifications
2.2. Optimization of the Extraction Process
10−3 BC − 9.9 × 10−4 A2 − 4.44 × 10−3 B2 − 4.82 × 10−4 C2
2.3. Optimization of Chromatographic Conditions
2.4. Method Validation
2.4.1. Linearity and Limits of Detection (LOD) and Quantitation (LOQ)
2.4.2. Precision, Repeatability, Stability and Recovery
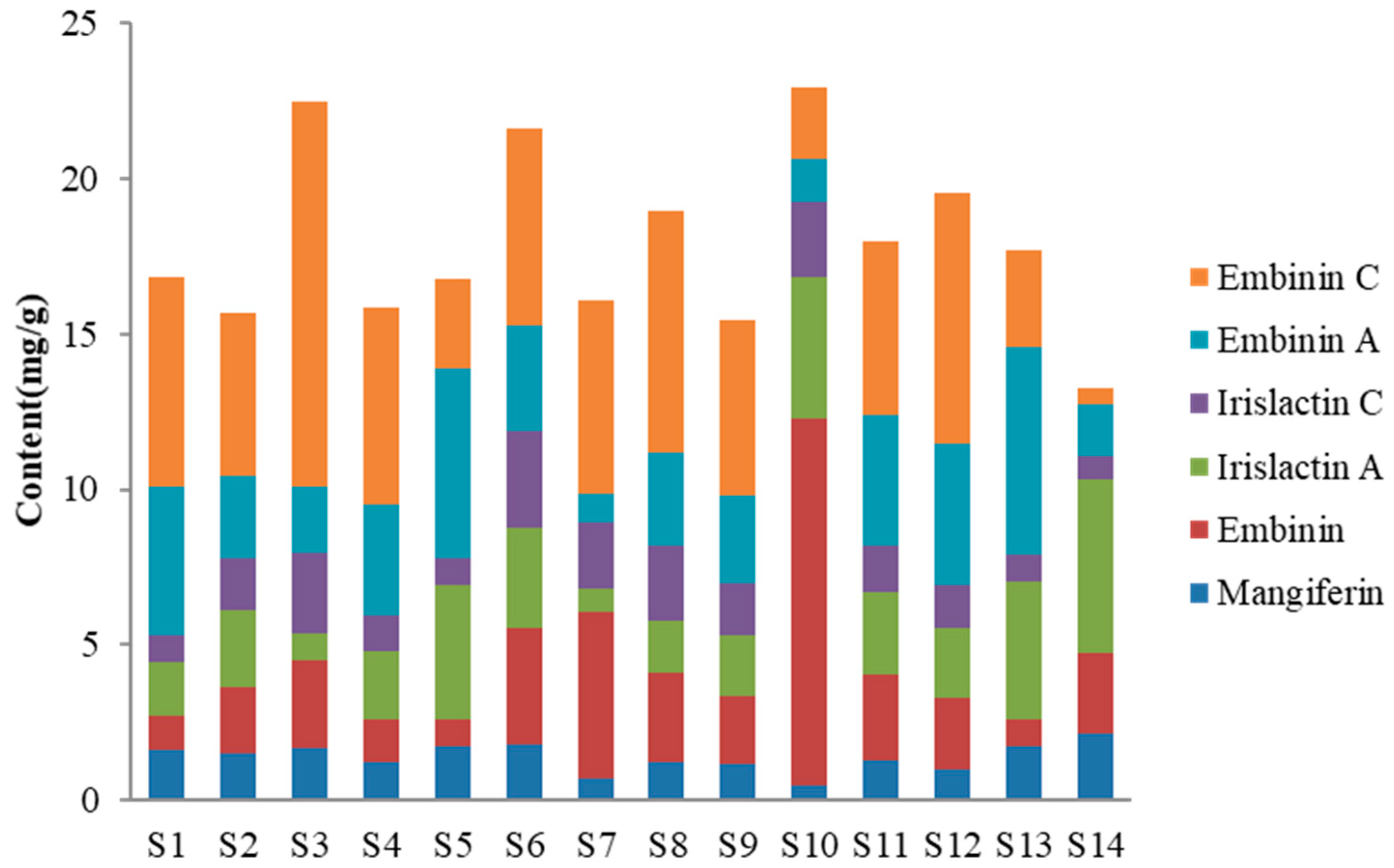
2.5. Quantitative Analysis of HPLC-DAD for Flavonoids of I. lactea Leaves
3. Materials and Methods
3.1. Chemicals and Plant Material
3.2. Preparation of Samples and Standard Solutions
3.3. Qualitative Analysis of HPLC-Q-TOF-MS/MS for Chemical Constituents of I. lactea Leaves
3.4. Optimization of the Extraction Process
3.4.1. Single Factor Experiments
3.4.2. Box-Behnken Response-Surface Design Experiment
3.5. Method Validation
3.5.1. Preparation of Sample Solution
3.5.2. Linearity, LOD and LOQ
3.5.3. Precision, Repeatability, Stability and Recovery
3.6. Quantitative Analysis of HPLC-DAD for Flavonoids of I. lactea Leaves
3.7. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- The Editorial Committee of Chinese Materia Medica, State Administration of Traditional Chinese Medicine. Chinese Materia Medica; Shanghai Scientific and Technical Publishers: Shanghai, China, 1999; Volume 22, pp. 271–275. [Google Scholar]
- Lv, H.; Wang, H.; He, Y.; Ding, C.; Wang, X.; Suo, Y. Separation and purification of four oligostilbenes from iris lactea, pall. var. chinensis, (fisch.) koidz by high-speed counter-current chromatography. J. Chromatogr. B 2015, 988, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.G.; Hou, D.Y.; Weng, X.; Wang, C.Y. Process Optimization for Ultrasonic Extraction of Lavonoids and Determination of Antioxidation Effect in Iris lactea Pall.Var. chinensis Koidz. Mod. Agric. Sci. Technol. 2014, 2, 301–303. [Google Scholar] [CrossRef]
- Liu, C.X.; Li, Q.S.; Gao, L.Y. Studies on the Determination of Irisquinone A and B in Biological Samples. Chin. Tradit. Herb. Drugs 1998, 29, 533–535. [Google Scholar]
- Colin, D.; Lancon, A.; Delmas, D.; Lizard, G.; Abrossinow, J.; Kahn, E.; Jannin, B.; Latruffe, N. Antiproliferative activities of resveratrol and related compounds in human hepatocyte derived HepG2 cells are associated with biochemical cell disturbance revealed by fluorescence analyses. Biochime 2008, 90, 1674–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Sun, W.; Yongchun, Y.U. The impact of radiosensitizer irisquinone on lung metastasis in H22-bearing mice. Jiangsu Med. J. 2008, 34, 176–178. [Google Scholar] [CrossRef]
- Zhang, F.G.; Li, D.H.; Qi, J.; Liu, C.X. In Vitro Anticancer Effects of Pallasone A and Its Induced Apoptosis on Leukemic K562 Cells. Chin. Pharm. J. 2010, 22, 1716–1719. [Google Scholar]
- Fu, L.W.; Li, X.B.; Liang, Y.J.; Feng, H.L.; Zhang, Y.M.; Pang, Q.C. Effect of irisquinone on cytotoxicity to the cancer cells with multidrug resistance and its mechanism. Chin. Pharmacol. Bull. 2001, 17, 234–236. [Google Scholar] [CrossRef]
- Zhou, Y.Q.; Bian, X.H. Clinical Study on NPC Radiosensitization of Irrisquinones. Acta Univ. Med. Nanjing 2001, 21, 328–330. [Google Scholar] [CrossRef]
- Wang, X.W. Irisquinone: Antineoplastic, radiosensitizer. Drugs Future 1999, 24, 613–617. [Google Scholar] [CrossRef]
- Meng, Y.; Qin, M.J.; Qi, B.Q.; Xie, G.Y. Four new C-glycosylflavones from the leaves of Iris lactea Pall. var. chinensis (Fisch.) Koidz. Phytochem. Lett. 2017, 22, 33–38. [Google Scholar] [CrossRef]
- Zhou, T.R.; Ge, G.T.; Jia, Y.S.; Hou, M.L.; Wang, W.; Nuo, M.; Ba, D.L.H. The effect advantage natural grassland on mixed grass group of silage quality. Grassl. Prataculture 2015, 27, 19–26. [Google Scholar] [CrossRef]
- Wu, X.A.; Zhao, Y.M.; Yu, N.J. Flavone C-glycosides from Trollius ledebouri reichb. J. Asian Nat. Prod. Res. 2006, 8, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.X.; Lin, S.; Xu, W.; Huang, M.Q.; Chu, J.F.; Xiao, F.; Lin, J.M.; Peng, J. Qualitative and Quantitative Analysis of the Major Constituents in Shexiang Tongxin Dropping Pill by HPLC-Q-TOF-MS/MS and UPLC-QqQ-MS/MS. Molecules 2015, 20, 18597–18619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liu, X.; Yang, J.; Han, Q.B.; Song, J.Z.; Li, S.L.; Qiao, C.F.; Ding, L.S.; Xu, H.X. Analysis of caged xanthones from the resin of Garcinia hanburyi using ultra-performance liquid chromatography/electrospray ionization quadrupole time-of-flight tandem mass spectrometry. Anal. Chim. Acta 2008, 629, 104–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, Y.; Kiyota, T.; Draghici, C.; Gao, J.M.; Yeboah, F.; Acoca, S.; Jarussophon, S.; Purisima, E. Molecular Formula Analysis by an MS/MS/MS Technique To Expedite Dereplication of Natural Products. Anal. Chem. 2007, 79, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; Li, Z.K.; Wang, W.; Sooranna, S.R.; Shi, Y.T.; Chen, Y.; Wu, C.Q.; Zeng, J.G.; Tang, Q.; Xie, H.Q. Chemical Profiles and Simultaneous Quantification of Aurantii fructus by Use of HPLC-Q-TOF-MS Combined with GC-MS and HPLC Methods. Molecules 2018, 23, 2189. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.Y.; Zhu, Y.; Shu, P.; Qin, X.Y.; Wu, G.; Wang, Q.; Qin, M.J. Phenolic metabolite profiles and antioxidants assay of three Iridaceae medicinal plants for traditional Chinese medicine “She-gan” by on-line HPLC–DAD coupled with chemiluminescence (CL) and ESI-Q-TOF-MS/MS. J. Pharm. Biomed. 2014, 98, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Iswaldi, I.; Arráez-Román, D.; Rodríguez-Medina, I.; Beltran-Debon, R.; Joven, J.; Segura-Carretero, A.; Fernandez-Gutierrez, A. Identification of phenolic compounds in aqueous and ethanolic rooibos extracts (Aspalathus linearis) by HPLC-ESI-MS (TOF/IT). Anal. Bioanal. Chem. 2011, 400, 3643–3654. [Google Scholar] [CrossRef] [PubMed]
- Sethi, M.L.; Taneja, S.C.; Dhar, K.L.; Atal, C.K. Three isoflavone-glycosides from juniperus macropoda. Phytochemistry 1983, 22, 289–292. [Google Scholar] [CrossRef]
- Liu, S.; Yan, J.; Xing, J.; Song, F.; Liu, Z.; Liu, S. Characterization of compounds and potential neuraminidase inhibitors from the n-butanol extract of Compound Indigowoad Root Granule using ultrafiltration and liquid chromatography–tandem mass spectrometry. J. Pharm. Biomed. 2012, 59, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Yabuya, T.; Kitajima, J.; Iwashina, T. Identification of novel C-glycosylflavones and their contribution to flower colour of the Dutch iris cultivars. Plant Physiol. Biochem. 2013, 72, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Kawase, A.; Yagishita, K. On the Structure of a New C-Glycosyl Flavone, Embinin, Isolated from the Petals of Iris germanica Linnaeous. Agric. Biol. Chem. 1968, 32, 537–538. [Google Scholar] [CrossRef]
- Pryakhina, N.I.; Sheichenko, V.I.; Blinova, K.F. Acylated C-glycosides of Iris lactea. Chem. Nat. Compd. 1984, 20, 554–559. [Google Scholar] [CrossRef]
- Shen, W.J.; Qin, M.J.; Shu, P.; Zhang, C.F. Two new C-glycosylflavones from the leaves of Iris lactea var. chinensis. Chin. Chem. Lett. 2008, 19, 821–824. [Google Scholar] [CrossRef]
- Bandyukova, V.A.; Yugin, V.A. Natural flavonoid C-glycosides. Chem. Nat. Compd. 1981, 17, 1–21. [Google Scholar] [CrossRef]
- Wen, L.R.; Zhao, Y.P.; Jiang, Y.M.; Yu, L.M.; Zeng, X.F.; Yang, J.L.; Tian, M.M.; Liu, H.L.; Yang, B. Identification of a flavonoid C-glycoside as potent antioxidant. Free Radic. Biol. Med. 2017, 110, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Thao, N.P.; Luyen, B.T.T.; Widowati, W.; Fauziah, N.; Maesaroh, M.; Herlina, T.; Manzoor, Z.; Ali, I.; Koh, Y.S.; Kim, Y.H. Anti-inflammatory Flavonoid C-Glycosides from Piper aduncum Leaves. Planta Med. 2016, 82, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Li, P.; Li, P.; Yan, R.; Zhang, X.Q.; Wang, Y.; Zhang, X.T.; Ye, W.C.; Zhang, Q.W. α-Glucosidase Inhibitory Effect and Simultaneous Quantification of Three Major Flavonoid Glycosides in Microctis folium. Molecules 2013, 18, 4221–4232. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.R.; Correia-Da-Silva, M.; Silva, P.M.A.; Ribeiro, D.; Emília, S.; Bousbaa, H.; Pinto, M. Synthesis of new glycosylated flavonoids with inhibitory activity on cell growth. Molecules 2018, 23, 1093. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Zhang, J.; Zhang, X.L.; Huang, X.J.; Wu, X.; Zhang, Q.W.; Li, Y.L.; Ye, W.C. Flavone C-glycosides from the leaves of Lophatherum gracile and their in vitro antiviral activity. Planta Med. 2011, 78, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.J.; Xu, W.; Zhang, W.D.; Zhang, C.; Liu, R.H.; Shen, Y.H.; Li, H.L.; Wang, X.L.; Wang, X.W.; Pan, Q.Q.; et al. Quantitative LC/MS/MS method and in vivo pharmacokinetic studies of vitexin rhamnoside, a bioactive constituent on cardiovascular system from hawthorn. Biomed. Chromatogr. 2010, 21, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Hawas, U.W.; Soliman, G.M.; Abou ElKassem, L.T.; Farrag, A.R.; Mahmoud, K.; León, F. A new flavonoid C-glycoside from Solanum elaeagnifolium with hepatoprotective and curative activities against paracetamol-induced liver injury in mice. Z. Naturforsch. C J. Biosci. 2013, 68, 19–28. [Google Scholar] [CrossRef]
- Jung, I.H.; Lee, H.E.; Park, S.J.; Ahn, Y.J.; Kwon, G.Y.; Woo, H.; Lee, S.Y.; Kim, J.S.; Jo, Y.W.; Jang, D.S.; et al. Ameliorating effect of spinosin, a C-glycoside flavonoid, on scopolamine-induced memory impairment in mice. Pharmacol. Biochem. Behav. 2014, 120, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Sekar, M. Molecules of Interest–Mangiferin—A Review. Annu. Res. Rev. Biol. 2015, 5, 307–320. [Google Scholar] [CrossRef]
- Benard, O.; Chi, Y. Medicinal Properties of Mangiferin, Structural Features, Derivative Synthesis, Pharmacokinetics and Biological Activities. Mini-Rev. Med. Chem. 2015, 15, 582–594. [Google Scholar] [CrossRef] [PubMed]
- D’Archivio, A.A.; Maggi, M.A.; Ruggieri, F.; Carlucci, M.; Ferrone, V.; Carlucci, G. Optimisation by response surface methodology of microextraction by packed sorbent of non steroidal anti-inflammatory drugs and ultra-high performance liquid chromatography analysis of dialyzed samples. J. Pharmaceut. Biomed. 2016, 125, 114–121. [Google Scholar] [CrossRef]
- Berger-Brito, I.; Machour, N.; Morin, C.; Portet-Koltalo, F. Experimental Designs for Optimizing Multi-residual Microwave-assisted Extraction and Chromatographic Analysis of Oxygenated (Hydroxylated, Quinones) Metabolites of PAHs in Sediments. Chromatographia 2018, 81, 1401–1412. [Google Scholar] [CrossRef]
- D’Archivio, A.A.; Maggi, M.A. Investigation by response surface methodology of the combined effect of pH and composition of water-methanol mixtures on the stability of curcuminoids. Food Chem. 2017, 219, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.L.C.; Bruns, R.E.; Ferreira, H.S.; Matos, G.D.; David, J.M.; Brandão, G.C.; Silva, E.G.P.D.; Portugal, L.A.; Reis, P.S.D.; Souza, A.S.; et al. Box-behnken design: An alternative for the optimization of analytical methods. Anal. Chim. Acta 2007, 597, 179–186. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds mangiferin, embinin, irislactin C, embinin A, irislactin A and embinin C are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
| Compound | Tr (time) | UV (nm) | Quasi-Molecular (Error, ppm) | Molecular Formula | m/z Calculated | MS/MS Fragments | Proposed Compound | References |
|---|---|---|---|---|---|---|---|---|
| A1 | 7.089 | 239, 257, 320, 360 | 583.1261 (−2.55) [M − H]− | C25H28O16 | 583.1246 | 565, 493, 463, 331, 301, 259 | neomangiferin | [18] |
| A2 | 9.188 | 268, 320 | 593.1514(−0.3) [M − H]− | C27H30O15 | 593.1512 | 575, 503, 473, 341, 311, 282, 119 | apigenin 7-O-glucoside-6C-glucoside | [19,20] |
| A3 | 9.863 | 239, 260, 320, 360 | 421.0785 (−2.04) [M − H]− | C19H18O11 | 421.0762 | 403, 301, 331, 285, 271, 259, 243, 215 | mangiferin | [18] |
| A4 | 10.586 | 239, 260, 320, 360 | 421.0779 (−0.53) [M − H]− | C19H18O11 | 421.0776 | 331, 301, 285, 271, 258, 243, 215 | isomangiferin | [18] |
| A5 | 15.002 | 268, 352 | 447.0934 (−2.49) [M − H]− | C21H20O11 | 447.0929 | 429, 357, 327, 331, 299, 133 | luteolin 6-C-β-d-glucoside | [21] |
| A6 | 15.561 | 252 (sh*), 272, 318 | 461.1073 (3.49) [M − H]− | C22H22O11 | 461.1091 | 446, 313, 298, 285, 133 | swertiajaponin | [19] |
| A7 | 16.899 | 267, 336 | 431.0988 (−0.05) [M − H]− | C21H20O10 | 431.0986 | 341, 323, 311, 283, 117 | Saponaretin | [21] |
| A8 | 17.613 | 256, 332 | 461.1079 (2.33) [M − H]− | C22H22O11 | 461.1089 | 371, 341, 298 | scoparin | [19] |
| A9 | 18.899 | 270, 324 | 799.2299(0.49) [M + HCOO]− | C34H42O19 | 754.2320 | 753, 659, 633, 591, 427, 307 | Swertisin 2″-O-rhamnoside-4′-O-glucoside | [22] |
| A10 | 22.065 | 270, 327 | 841.2448(−4.79) [M + HCOO]− | C36H44O20 | 796.2426 | 795, 659, 633, 591, 427, 307 | Swertisin 2″-O-(4′′′-acetylrhamnoside)-4′-O-glucoside | [22] |
| A11 | 22.581 | 270, 338 | 605.1905 (−4.81) [M − H]− | C29H34O14 | 605.1876 | 485, 459, 441, 423, 381, 363, 351, 339, 321, 307, 163, 103 | embinin | [23] |
| A12 | 23.888 | 270, 332 | 883.2489(−2.03) [M + HCOO]− | C38H46O21 | 838.2532 | 837, 675, 633, 555, 513, 427, 307 | The isomer of irislactin C | [11] |
| A13 | 24.163 | 270, 332 | 883.2529(1.08) [M + HCOO]− | C38H46O21 | 838.2532 | 837, 675, 633, 555, 513, 427, 307 | The isomer of irislactin C | [11] |
| A14 | 24.920 | 268, 332 | 647.1994(−1.94) [M − H]− | C31H36O15 | 647.1979 | 605, 587, 459, 441, 381, 339, 145, 101 | 2′′′-acetyl-embinin | [11,24] |
| A15 | 25.505 | 268, 330 | 883.2489(2.11) [M + HCOO]− | C38H46O21 | 838.2532 | 837, 633, 513, 427, 307 | irislactin C | [11] |
| A16 | 26.331 | 270, 330 | 647.1979(−0.42) [M − H]− | C31H36O15 | 647.1978 | 605, 527, 459, 381, 351,339 127, 101 | 3′′′-acetyl-embinin | [11,24] |
| A17 | 27.260 | 268, 328 | 883.2526(−2.51) [M + HCOO]− | C38H46O21 | 838.2532 | 837, 675, 633, 555, 513, 427, 307 | The isomer of irislactin C | [11] |
| A18 | 27.776 | 268, 330 | 647.1960(3.34) [M − H]− | C31H36O15 | 647.1977 | 605, 587, 459, 441, 381, 339, 145, 101 | embinin A | [11,24] |
| A19 | 28.636 | 270, 328 | 925.2577(3.87) [M + HCOO]− | C40H48O22 | 880.2637 | 879, 675, 633, 555, 427, 307 | irislactin A | [25] |
| A20 | 29.290 | 268, 330 | 925.2628(−2.77) [M + HCOO]− | C40H48O22 | 880.2637 | 879, 675, 633, 427, 307 | The isomer of irislactin A | [25] |
| A21 | 29.857 | 268, 328 | 689.2145 (0.17) [M − H]− | C33H38O16 | 689.2146 | 647, 605, 587, 527, 459, 441, 351, 127, 113 | irislactin B | [25] |
| A22 | 30.700 | 246, 326 | 689.2079(−1.02) [M − H]− | C33H38O16 | 689.2074 | 647, 605, 587, 527, 459, 441, 351, 145, 109 | embinin C | [11] |
| Analyte | Calibration Curves | R2 | Liner Range (µg·mL−1) | LOD (ng·mL−1) | LOQ (ng·mL−1) |
|---|---|---|---|---|---|
| Mangiferin | y = 37119x − 13.767 | 0.9998 | 3.74–22.44 | 26.7 | 93.5 |
| Embinin | y = 25969x − 13.174 | 0.9999 | 4.40–198.00 | 11.5 | 16.5 |
| Irislactin C | y = 19575x − 1.0423 | 0.9999 | 2.21–100.00 | 3.9 | 8.3 |
| Irislactin A | y = 21296x + 0.0506 | 0.9998 | 2.52–113.40 | 16.7 | 31.5 |
| Embinin A | y = 33469x − 3.9068 | 0.9999 | 3.36–37.10 | 23.6 | 84.0 |
| Embinin C | y = 25250x − 2.1531 | 0.9999 | 4.00–180.00 | 8.9 | 15.0 |
| No. | Mangiferin | Embinin | Irislactin C | Irislactin A | Embinin A | Embinin C |
|---|---|---|---|---|---|---|
| S1 | 1.60 ± 0.01 | 1.14 ± 0.01 | 1.68 ± 0.01 | 0.88 ± 0.01 | 4.79 ± 0.03 | 6.74 ± 0.08 |
| S2 | 1.52 ± 0.01 | 2.14 ± 0.01 | 2.46 ± 0.01 | 1.64 ± 0.00 | 2.66 ± 0.02 | 5.26 ± 0.02 |
| S3 | 1.71 ± 0.00 | 2.78 ± 0.01 | 0.90 ± 0.01 | 2.57 ± 0.01 | 2.15 ± 0.01 | 12.38 ± 0.02 |
| S4 | 1.24 ± 0.02 | 1.39 ± 0.01 | 2.15 ± 0.03 | 1.16 ± 0.01 | 3.60 ± 0.05 | 6.29 ± 0.06 |
| S5 | 1.72 ± 0.01 | 0.91 ± 0.00 | 4.31 ± 0.04 | 0.85 ± 0.01 | 6.09 ± 0.03 | 2.90 ± 0.00 |
| S6 | 1.80 ± 0.01 | 3.75 ± 0.01 | 3.20 ± 0.02 | 3.11 ± 0.01 | 3.41 ± 0.03 | 6.32 ± 0.02 |
| S7 | 0.69 ± 0.01 | 5.39 ± 0.01 | 0.75 ± 0.00 | 2.12 ± 0.01 | 0.92 ± 0.01 | 6.21 ± 0.07 |
| S8 | 1.23 ± 0.02 | 2.85 ± 0.04 | 1.67 ± 0.02 | 2.43 ± 0.02 | 2.99 ± 0.02 | 7.80 ± 0.10 |
| S9 | 1.16 ± 0.01 | 2.19 ± 0.03 | 1.95 ± 0.02 | 1.68 ± 0.01 | 2.80 ± 0.03 | 5.66 ± 0.01 |
| S10 | 0.48 ± 0.01 | 11.78 ± 0.03 | 4.57 ± 0.03 | 2.42 ± 0.07 | 1.36 ± 0.03 | 2.30 ± 0.01 |
| S11 | 1.31 ± 0.05 | 2.75 ± 0.01 | 2.66 ± 0.00 | 1.44 ± 0.02 | 4.23 ± 0.05 | 5.60 ± 0.02 |
| S12 | 1.01 ± 0.01 | 2.29 ± 0.02 | 2.22 ± 0.01 | 1.39 ± 0.00 | 4.54 ± 0.02 | 8.08 ± 0.03 |
| S13 | 1.72 ± 0.03 | 0.88 ± 0.01 | 4.43 ± 0.06 | 0.86 ± 0.02 | 6.67 ± 0.05 | 3.14 ± 0.01 |
| S14 | 2.16 ± 0.01 | 2.59 ± 0.03 | 5.56 ± 0.02 | 0.77 ± 0.01 | 1.68 ± 0.03 | 0.49 ± 0.02 |
| No. | Habitat | Collection | Collection Time | No. | Habitat | Collection | Collection Time |
|---|---|---|---|---|---|---|---|
| S1 | Jiangsu | Nanjing | 2015.04 | S8 | Shaanxi | Xi’an | 2015.05 |
| S2 | Jiangsu | Nanjing | 2015.04 | S9 | Beijing | Dongcheng | 2015.05 |
| S3 | Jiangsu | Nanjing | 2015.05 | S10 | Beijing | Haidian | 2015.05 |
| S4 | Henan | Zhengzhou | 2015.04 | S11 | Shandong | Zaozhuang | 2015.05 |
| S5 | Tianjin | Jixian | 2015.04 | S12 | Shandong | Zaozhuang | 2015.05 |
| S6 | Tianjin | Tianjin | 2015.04 | S13 | Liaoning | Huludao | 2015.05 |
| S7 | Shanghai | Shanghai | 2015.04 | S14 | Liaoning | Chaoyang | 2014.09 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.; Meng, Y.; Zhu, Y.; Wu, G.; Yuan, J.; Qin, M.; Xie, G. Qualitative and Quantitative Analysis of C-glycosyl-flavones of Iris lactea Leaves by Liquid Chromatography/Tandem Mass Spectrometry. Molecules 2018, 23, 3359. https://doi.org/10.3390/molecules23123359
Chen D, Meng Y, Zhu Y, Wu G, Yuan J, Qin M, Xie G. Qualitative and Quantitative Analysis of C-glycosyl-flavones of Iris lactea Leaves by Liquid Chromatography/Tandem Mass Spectrometry. Molecules. 2018; 23(12):3359. https://doi.org/10.3390/molecules23123359
Chicago/Turabian StyleChen, Dan, Yu Meng, Yan Zhu, Gang Wu, Jun Yuan, Minjian Qin, and Guoyong Xie. 2018. "Qualitative and Quantitative Analysis of C-glycosyl-flavones of Iris lactea Leaves by Liquid Chromatography/Tandem Mass Spectrometry" Molecules 23, no. 12: 3359. https://doi.org/10.3390/molecules23123359
APA StyleChen, D., Meng, Y., Zhu, Y., Wu, G., Yuan, J., Qin, M., & Xie, G. (2018). Qualitative and Quantitative Analysis of C-glycosyl-flavones of Iris lactea Leaves by Liquid Chromatography/Tandem Mass Spectrometry. Molecules, 23(12), 3359. https://doi.org/10.3390/molecules23123359