Semi-Preparative Separation of 10 Caffeoylquinic Acid Derivatives Using High Speed Counter-Current Chromatogaphy Combined with Semi-Preparative HPLC from the Roots of Burdock (Arctium lappa L.)

Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of HSCCC Conditions
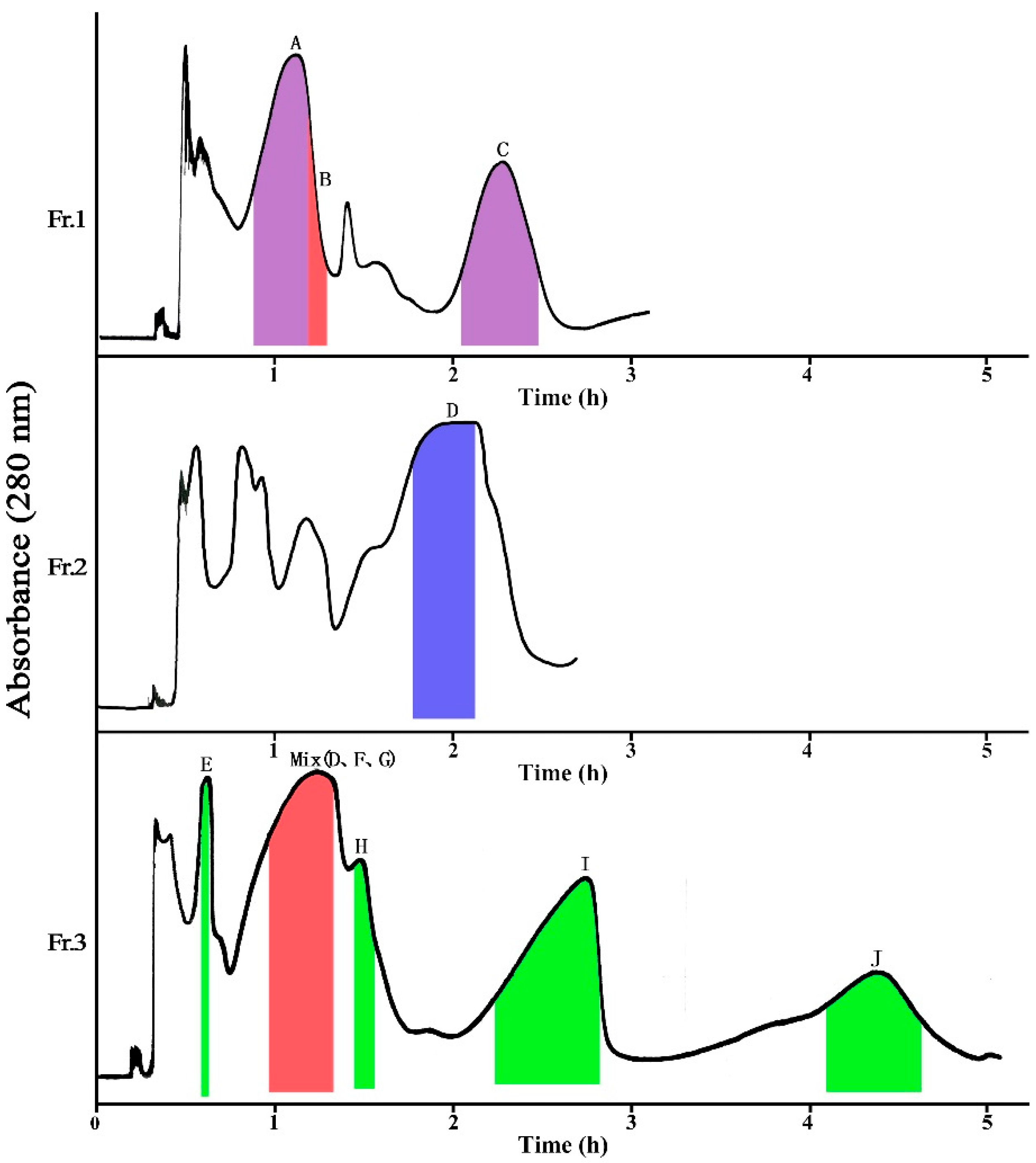
2.1.1. Separation of Fr. 1 by HSCCC
2.1.2. Separation of Fr. 2 by HSCCC
2.1.3. Separation of Fr. 3 by HSCCC
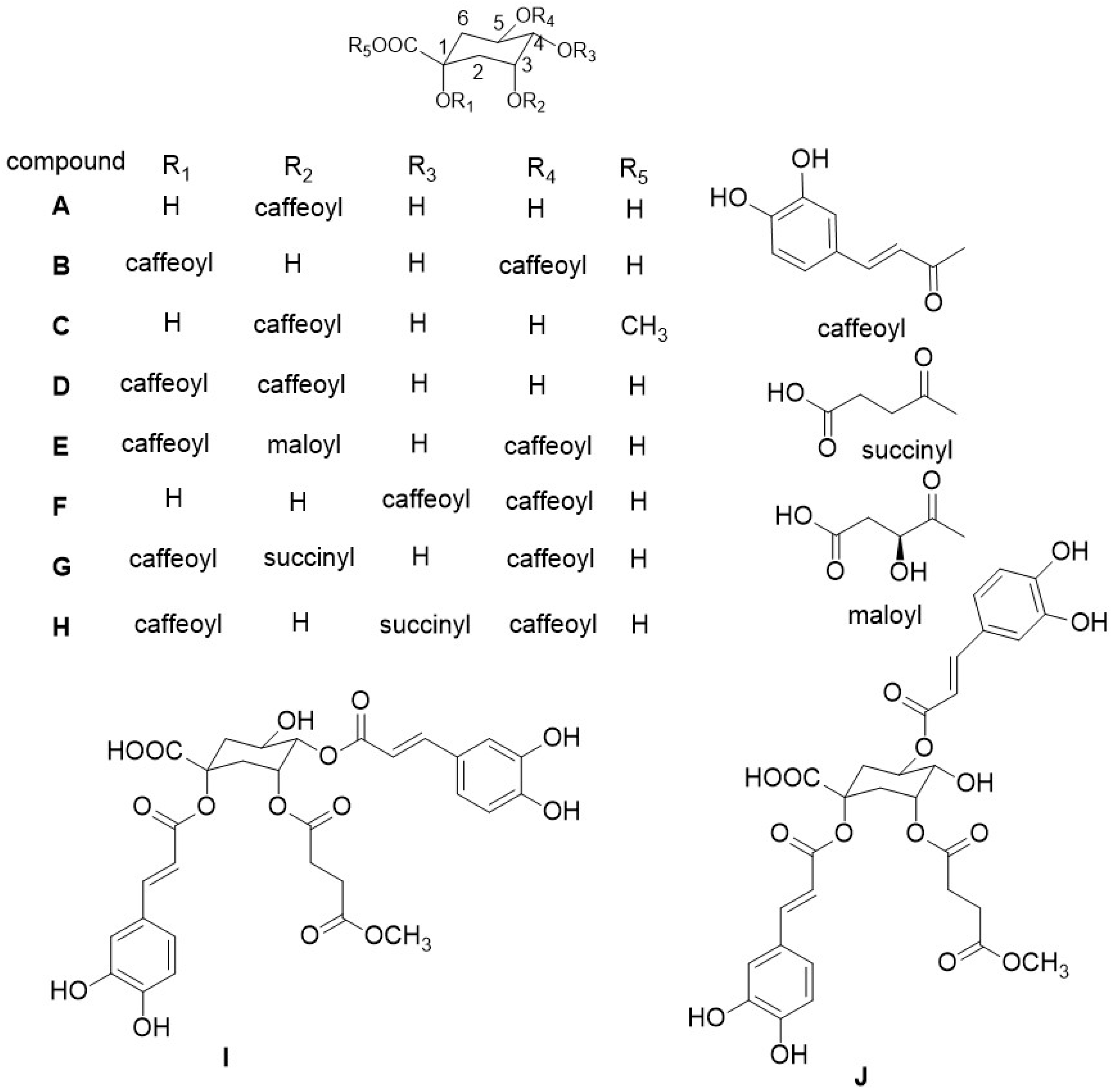
2.2. Identification of Compounds
2.2.1. Identification of Known Compounds
2.2.2. Identification of New Compounds
3. Experimental
3.1. Material and Reagents
3.2. Apparatus
3.3. Pre-Processing of Crude Sample
3.4. Optimization of HPLC Conditions
3.5. Selection and Preparation of the Two-Phase Solvent System
3.6. Separation Procedure
3.7. Semi-Preparative HPLC Separations
3.8. Identification of Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tian, X.; Sui, S.; Huang, J.; Bai, J.P.; Ren, T.S.; Zhao, Q.C. Neuroprotective effects of Arctium lappa L. roots against glutamate-induced oxidative stress by inhibiting phosphorylation of p38, JNK and ERK 1/2 MAPKs in PC12 cells. Environ. Toxicol. Pharmacol. 2014, 38, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Maruta, Y.; Kawabata, J.; Niki, R. Antioxidative caffeoylquinic acid derivatives in the roots of Burdock (Arctium lappa L.). J. Agric. Food Chem. 1995, 43, 2592–2595. [Google Scholar] [CrossRef]
- Yang, W.S.; Lee, S.R.; Yong, J.J.; Park, D.W.; Cho, Y.M.; Joo, H.M.; Kim, I.; Seu, Y.B.; Sohn, E.H.; Kang, S.C. Antiallergic Activity of Ethanol Extracts of Arctium lappa L. Undried Roots and Its Active Compound, Oleamide, in Regulating FcεRI-Mediated and MAPK Signaling in RBL-2H3 Cells. J. Agric. Food Chem. 2016, 64, 3564–3573. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Lu, J.M.; Yang, J.J.; Chuang, S.C.; Ujiie, T. Anti-inflammatory and radical scavenge effects of Arctium lappa. Am. J. Chin. Med. 1996, 24, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lin, C.H.; Lin, C.C.; Lin, Y.H.; Chen, C.F.; Chen, I.C.; Wang, L.Y. Hepatoprotective effects of Arctium lappa Linne on liver injuries induced by chronic ethanol consumption and potentiated by carbon tetrachloride. J. Biomed. Sci. 2002, 9, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Kuhnert, N. Identification and characterization of five new classes of chlorogenic acids in burdock (Arctium lappa L.) roots by liquid chromatography/tandem mass spectrometry. Food Funct. 2011, 2, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, J.; LI, X.P.; Zhou, C.X.; Sun, H.D.; Hao, X.J.; Xiao, P.P. Advances in caffeoylquinic acid research. China J. Chin. Mater. Med. 2006, 31, 869–874. [Google Scholar]
- Jiang, X.W.; Bai, J.P.; Zhang, Q.; Hu, X.L.; Tian, X.; Zhu, J.; Liu, J.; Meng, W.H.; Zhao, Q.C. Caffeoylquinic acid derivatives from the roots of Arctium lappa, L. (burdock) and their structure–activity relationships (SARs) of free radical scavenging activities. Phytochem. Lett. 2016, 15, 159–163. [Google Scholar] [CrossRef]
- Eunju, L.; Jusun, K.; Hyunpyo, K.; Jehyun, L.; Kang, S.S. Phenolic constituents from the flower buds of Lonicera japonica and their 5-lipoxygenase inhibitory activities. Food Chem. 2010, 120, 134–139. [Google Scholar]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Skalickawoźniak, K.; Garrard, I. A comprehensive classification of solvent systems used for natural product purifications in countercurrent and centrifugal partition chromatography. Nat. Prod. Rep. 2015, 32, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.D.N.; Jerz, G.; Figueiredo, F.D.S.; Winterhalter, P.; Leitao, G.G. Solvent system selectivities in counter current chromatography using Salicornia gaudichaudiana metabolites as practical example withoff-line electrospray mass-spectrometry injection profiling. J. Chromatogr. A 2015, 1385, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Li, A.F.; Zhang, Y.Q.; Sun, A.L.; Liu, R.M. Preparative isolation and purification of phenolic acids from the dried buds of lonicera japonica thumb by high-speed counter-current chromatography in gradient elution mode. J. Liq. Chromatogr. Relat. Technol. 2012, 35, 1933–1944. [Google Scholar]
- Wang, Y.F.; Liu, B. Preparative isolation and purification of dicaffeoylquinic acids from the Ainsliaea fragrans champ by high-speed counter-current chromatography. Phytochem. Anal. 2007, 18, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Paek, J.H.; Lim, S.S. Preparative isolation of aldose reductase inhibitory compounds from Nardostachys chinensis by elution-extrusion counter-current chromatography. Arch. Pharm. Res. 2014, 37, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.L.; Liu, Q.; Yu, J.G.; Jiang, X.Y.; Wu, Z.L.; Wang, M.L.; Chen, M.; Chen, X.Q. One-step separation of nine structural analogues from Poria cocos (Schw.) Wolf. via tandem high-speed counter-current chromatography. J. Chromatogr. B 2015, 1004, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.L.; Wang, H.M.; Ni, F.Y.; Wang, X.J.; Zhao, Y.W.; Huang, W.Z.; Wang, Z.Z.; Xiao, W. Study on anti-inflammatory activities of phenolic acids from Lonicerae Japonicae Flos. Chin. Tradit. Herb. Drugs 2015, 46, 490–495. [Google Scholar]
- Carnat, A.; Heitz, A.; Fraisse, D.; Carnat, A.P.; Lamaison, J.L. Major dicaffeoylquinic acids from Artemisia vulgaris. Fitoterapia 2000, 71, 587–589. [Google Scholar] [CrossRef]
- Wang, J.; Xu, Q.L.; Zhou, Z.Y.; Tan, J.W. Caffeoylquinic acid derivatives from stems of Akebia trifoliate. J. Chin. Med. Mater. 2014, 37, 1190–1193. [Google Scholar]
- Sun, Y.; Ma, X.B.; Liu, J.X. Compounds from fraction with cardiovascular activity of chrysanthemum indicum. China J. Chin. Mater. Med. 2012, 37, 61–65. [Google Scholar]
- Zou, X.W.; Liu, D.; Liu, Y.P.; Fu, Y.H.; Zhang, X.Z.; Xiu, Z.L.; Xiao, H.B. Isolation and characterization of two new phenolic acids from cultured cells of Saussurea involucrate. Phytochem. Lett. 2014, 7, 133–136. [Google Scholar] [CrossRef]
- Zhu, X.F.; Zhang, H.X.; Lo, R. Three di-O-caffeoylquinic acid derivatives from the heads of Cynara scolymus L. Nat. Prod. Res. 2009, 23, 527–532. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |


{kind=link}
{kind=link}
{kind=link}
| Sample | Solvent System (Pet-EtOAc-MeOH-H2O, v/v) | KD | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | H | I/J | ||
| Fr. 1 | 0:5:0:5 | 0.55 | 0.58 | 4.82 | ||||||
| 0:5:0.5:5 | 0.47 | 0.52 | 2.21 | |||||||
| Fr. 2 | 0:5:1:5 | 26.88 | ||||||||
| 1:5:1:5 | 4.00 | |||||||||
| 1:4:1:4 | 2.03 | |||||||||
| Fr. 3 | 1:4:1:4 | 0.50 | 2.16 | 2.29 | 2.25 | 7.88 | ||||
| 1:4:2:3 | 0.19 | 0.29 | 0.39 | 0.41 | 0.76 | |||||
| Position | Compound I | Compound J | ||
|---|---|---|---|---|
| δH (ppm, Mult, J in Hz) | δC (ppm) | δH (ppm, Mult, J in Hz) | δC (ppm) | |
| 1 | 79.7 | 78.8 | ||
| 2 | 2.31–2.47 (m, overlap) | 32.3 | 2.34–2.58 (m, overlap) | 31.4 |
| 3 | 5.38 (d, 3.6) | 69.1 | 5.26 (m) | 69.5 |
| 4 | 4.88 (dd, 3.2, 8.8) | 74.7 | 3.85 (dd, 3.2, 8.8) | 71.3 |
| 5 | 4.14 (m) | 63.9 | 5.23 (m ) | 68.7 |
| 6 | 1.92 (m), 2.31–2.47 (m, overlap) | 39.4 (overlap) | 1.93 (m), 2.34–2.58 (m, overlap) | 35.9 |
| 7 | 172.6 | 172.0 | ||
| 1′ | 171.4 | 171.7 | ||
| 2′ | 2.31–2.47 (m, overlap) | 28.8 | 2.34–2.58 (m, overlap) | 28.3 |
| 3′ | 2.31–2.47 (m, overlap) | 29.3 | 2.34–2.58 (m, overlap) | 28.8 |
| 4′ | 172.4 | 171.2 | ||
| -OCH3 | 3.48 (s) | 51.8 | 3.51 (s) | 51.3 |
| 1″ | 126.0 | 125.3 | ||
| 2″ | 7.07 (1H, br s) | 115.4 | 7. 07 (br s) | 114.7 |
| 3″ | 145.6 (overlap) | 145.8 | ||
| 4″ | 148.9 | 148.2 | ||
| 5″ | 6.78 (d, 8.4) | 116.3 | 6.79 (d, 8.0) | 115.7 |
| 6″ | 7.03 (d, 8.4) | 121.9 | 7. 03 (dd, 8.0) | 121.3 |
| 7″ | 7.50 (d, 15.6) | 146.1 | 7.51 (d, 16.0) | 145.2 |
| 8″ | 6.31 (d, 15.6) | 114.5 | 6.25 (d, 16.0) | 113.7 |
| 9″ | 166.5 | 165.1 | ||
| 1″′ | 125.9 | 125. 4 | ||
| 2′′′ | 7.06 (br s) | 115.3 | 7. 07(br s) | 114. 8 |
| 3′′′ | 145.6 (overlap) | 145.8 | ||
| 4′′′ | 149.0 | 148.3 | ||
| 5′′′ | 6.77 (d, 8.4) | 116.2 | 6.80 (d, 8.0) | 115.7 |
| 6′′′ | 7.00 (d, 8.4) | 121.8 | 7.05 (dd, 8.0) | 121.3 |
| 7′′′ | 7.47 (d, 15.6) | 146.1 | 7. 52 (d, 16.0) | 145.3 |
| 8′′′ | 6.23 (d, 15.6) | 114.5 | 6. 29 (d, 16.0) | 114.0 |
| 9′′′ | 165.8 | 165.9 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Z.; Wang, X.; Liu, P.; Li, M.; Dong, H.; Qiao, X. Semi-Preparative Separation of 10 Caffeoylquinic Acid Derivatives Using High Speed Counter-Current Chromatogaphy Combined with Semi-Preparative HPLC from the Roots of Burdock (Arctium lappa L.). Molecules 2018, 23, 429. https://doi.org/10.3390/molecules23020429
Zheng Z, Wang X, Liu P, Li M, Dong H, Qiao X. Semi-Preparative Separation of 10 Caffeoylquinic Acid Derivatives Using High Speed Counter-Current Chromatogaphy Combined with Semi-Preparative HPLC from the Roots of Burdock (Arctium lappa L.). Molecules. 2018; 23(2):429. https://doi.org/10.3390/molecules23020429
Chicago/Turabian StyleZheng, Zhenjia, Xiao Wang, Pengli Liu, Meng Li, Hongjing Dong, and Xuguang Qiao. 2018. "Semi-Preparative Separation of 10 Caffeoylquinic Acid Derivatives Using High Speed Counter-Current Chromatogaphy Combined with Semi-Preparative HPLC from the Roots of Burdock (Arctium lappa L.)" Molecules 23, no. 2: 429. https://doi.org/10.3390/molecules23020429