3.1. Chemical Synthesis
3.1.1. General Methods
Melting points were determined in open capillary tubes with a MFB 595010 M Gallenkamp. 400 MHz 1H/100.6 MHz 13C-NMR spectra were recorded on a Varian Mercury 400 (Varian Inc., Palo Alto, CA, USA). The chemical shifts are reported in ppm (δ scale) relative to internal tetramethylsilane, and coupling constants are reported in Hertz (Hz). Assignments given for the NMR spectra of the new compounds were carried out on the basis of COSY 1H/13C (gHSQC sequences) experiments. IR spectra were run on Perkin-Elmer Spectrum RX I spectrophotometer (Waltham, MA, USA). Absorption values are expressed as wave-numbers (cm−1); only significant absorption bands are given. High-resolution mass spectrometry (HRMS) analyses were performed with an LC/MSD TOF Agilent Technologies spectrometer (Agilent Technologies Inc., Santa Clara, CA, USA). Column chromatography was performed either on silica gel 60 Å (35–70 mesh) (Merck, Darmstadt, Germany) or on aluminum oxide, neutral, 60 Å (50–200 µm, Brockmann I) (Merck, Darmstadt, Germany). Thin-layer chromatography was performed with aluminum-backed sheets with silica gel 60 F254 (Merck, Darmstadt, Germany, ref 1.05554), and spots were visualized with UV light and 1% aqueous solution of KMnO4. The analytical samples of all of the new compounds which were subjected to pharmacological evaluation possessed purity ≥95% as evidenced by their elemental analyses. The elemental analyses were carried out in a Flash 1112 series Thermo Finnigan (San Jose, CA, USA) elemental microanalyzator (A5) to determine C, H, and N.
3.1.2. (4-Azatetracyclo[5.3.2.02,6.08,10]dodec-11-en-4-yl)(phenyl)methanone 4
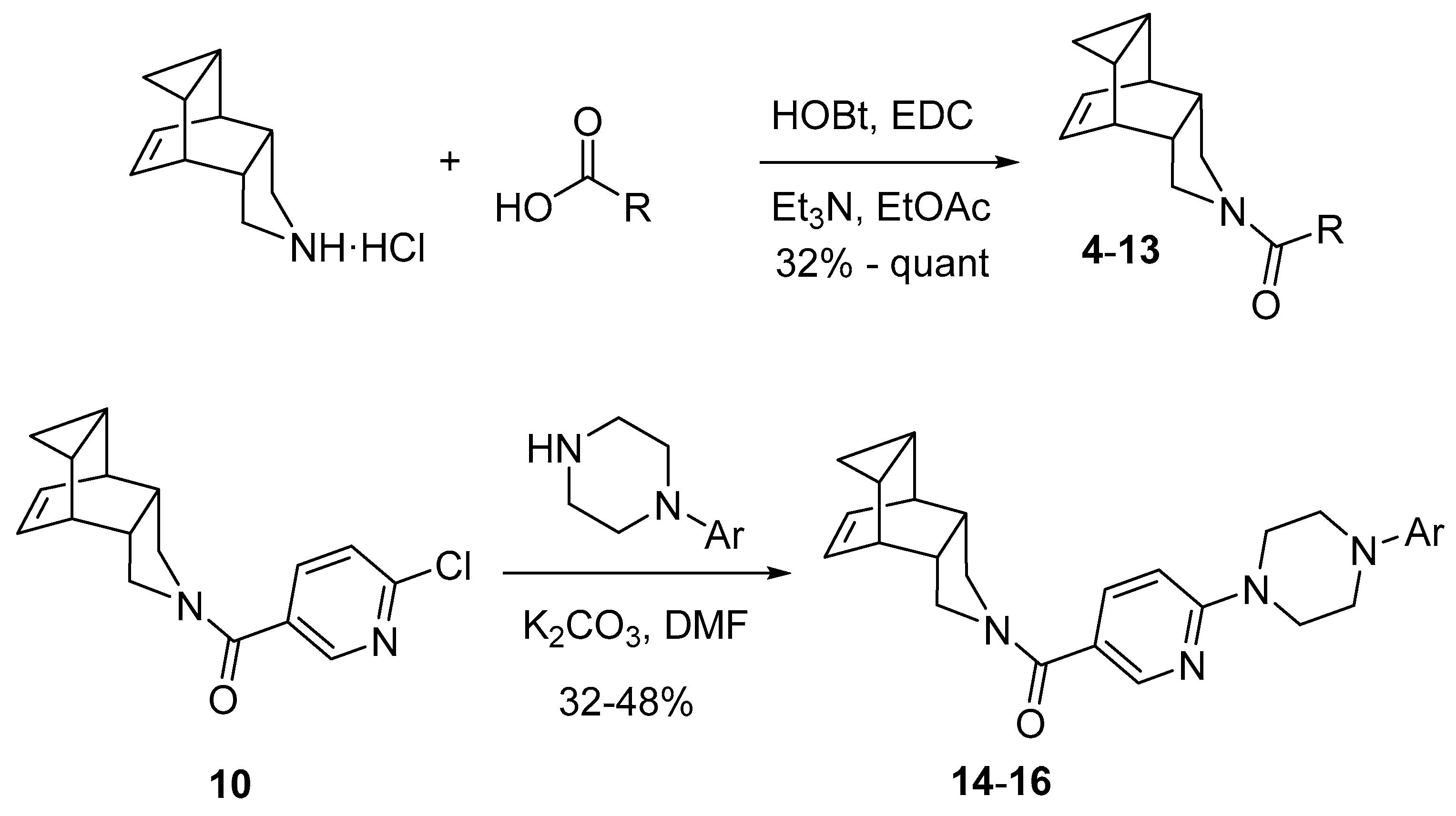
To a solution of 4-azapentacyclo[5.3.2.02,6.08,10]dodec-11-ene hydrochloride (400 mg, 2.07 mmol) in EtOAc (20 mL) were added benzoic acid (230 mg, 1.88 mmol), HOBt (381 mg, 2.82 mmol), EDC (437 g, 2.82 mmol), and triethylamine (1.2 mL, 8.27 mmol). The reaction mixture was stirred at room temperature overnight. To the resulting suspension was then added water (20 mL) and the phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (20 mL) and brine (20 mL), dried over anh. Na2SO4 and filtered. Evaporation in vacuo of the organics gave 4 as an orange oil (479 mg, 96% yield). Column chromatography (hexane/EtOAc mixture) gave 4 as a white solid (385 mg), m.p. 65–66 °C. IR (ATR) ν: 660, 700, 715, 763, 794, 814, 847, 986, 1029, 1135, 1170, 1231, 1378, 1423, 1572, 1618, 2845, 2865, 2921, 2946 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.10–0.18 (complex signal, 2H, 9′-H2), 0.82–0.98 (complex signal, 2H, 8′-H and 10′-H), 2.50–2.66 (complex signal, 2H, 2′-H and 6′-H), 2.71 (m, 1H, 1′-H or 7′-H), 2.91 (m, 1H, 7′-H or 1′-H), 3.09 (dd, J = 11.6 Hz, J’ = 4.4 Hz, 1 H, 3′-Ha or 5′-Ha), 3.42–3.56 (complex signal, 2H, 5′-Ha or 3′-Ha and 3′-Hb or 5′-Hb), 3.74 (dd, J = 13.0 Hz, J’ = 8.6 Hz, 1 H, 5′-Hb or 3′-Hb), 5.70 (m, 1H, 11′-H or 12′-H), 5.86 (m, 1H, 12′-H or 11′-H), 7.32–7.41 (complex signal, 5H, Ar-H). 13C-NMR (100.5 MHz, CDCl3) δ: 3.9 (CH2, C9′), 9.2 (CH, C8′ or C10′), 10.2 (CH, C10′ or C8′), 35.5 (CH, C1′ or C7′), 35.6 (CH, C7′ or C1′), 42.8 (CH, C2′ or C6′), 44.8 (CH, C6′ or C2′), 49.3 (CH2, C3′ or C5′), 53.3 (CH2, C5′ or C3′), 126.8 [CH, C2(6)], 128.1 [CH, C3(5)], 128.2 (CH, C11′ or C12′), 129.2 (CH, C12′ or C11′), 129.4 (CH, C4), 137.4 (C, C1), 168.9 (C, CO). Calcd. for C18H19NO: C, 81.47; H, 7.22; N, 5.28. Found: C, 81.52; H, 7.34; N 5.25.
3.1.3. (4-Azatetracyclo[5.3.2.02,6.08,10]dodec-11-en-4-yl)(pyridin-2-yl)methanone 5
To a solution of 4-azapentacyclo[5.3.2.02,6.08,10]dodec-11-ene hydrochloride (400 mg, 2.07 mmol) in EtOAc (20 mL) were added picolinic acid (231 mg, 1.88 mmol), HOBt (381 mg, 2.82 mmol), EDC (437 mg, 2.82 mmol), and triethylamine (1.2 mL, 8.27 mmol). The reaction mixture was stirred at room temperature overnight. To the resulting suspension was then added water (20 mL) and the phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (20 mL) and brine (20 mL), dried over anh. Na2SO4 and filtered. Evaporation in vacuo of the organics gave 5 as an orange oil (466 mg, 93% yield). Column chromatography (hexane/EtOAc mixture) gave 5 as a white solid (326 mg), m.p. 110–111 °C. IR (ATR) ν: 682, 720, 753, 796, 814, 844, 912, 988, 1041, 1082, 1142, 1165, 1201, 1226, 1269, 1294, 1302, 1340, 1378, 1400, 1441, 1474, 1562, 1585, 1618, 2850, 2870, 2926, 3007, 3048 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.10–0.19 (complex signal, 2H, 9′-H2), 0.84–0.97 (complex signal, 2H, 8′-H and 10′-H), 2.57–2.69 (complex signal, 2H, 2′-H and 6′-H), 2.75 (m, 1H, 1′-H or 7′-H), 2.90 (m, 1H, 7′-H or 1′-H), 3.31 (dd, J = 12.4 Hz, J’ = 4.8 Hz, 1H, 3′-Ha or 5′-Ha), 3.42 (dd, J = 13.0 Hz, J’ = 4.8 Hz, 1H, 5′-Ha or 3′-Ha), 3.82 (dd, J = 13.6 Hz, J’ = 8.8 Hz, 1H, 3′-Hb or 5′-Hb), 3.85 (dd, J = 12.8 Hz, J’ = 8.8 Hz, 1H, 5′-Hb or 3′-Hb), 5.71 (m, 1H, 11′-H or 12′-H), 5.83 (m, 1H, 12′-H or 11′-H), 7.30 (ddd, J = 12.4 Hz, J’ = 4.8 Hz, J″ = 1.6 Hz, 1H, 5-H), 7.67–7.80 (complex signal, 2H, 4-H and 3-H), 8.54 (ddd, J = 4.8 Hz, J’ = 1.6 Hz, J″ = 1.0 Hz, 1H, 6-H). 13C-NMR (100.5 MHz, CDCl3) δ: 4.1 (CH2, C9′), 10.0 (CH, C8′ or C10′), 10.2 (CH, C10′ or C8′), 35.4 (CH, C1′ and C7′), 42.5 (CH, C2′ or C6′), 45.2 (CH, C6′ or C2′), 50.2 (CH2, C3′ or C5′), 52.8 (CH2, C5′ or C3′), 123.6 (CH, C3), 124.3 (CH, C5), 128.6 (CH, C11′ or C12′), 129.1 (CH, C12′ or C11′), 136.7 (CH, C4), 147.9 (CH, C6), 154.8 (C, C2), 165.8 (C, CO). Calcd. for C17H18N2O: C, 76.66; H, 6.81; N, 10.52. Found: C, 76.47; H, 7.01; N, 10.21.
3.1.4. (4-Azatetracyclo[5.3.2. 02,6.08,10]dodec-11-en-4-yl)(tert-butyl)methanone 6
To a solution of 4-azapentacyclo[5.3.2.02,6.08,10]dodec-11-ene hydrochloride (200 mg, 1.03 mmol) in EtOAc (10 mL) were added pivalic acid (105 mg, 0.94 mmol), HOBt (190 mg, 1.41 mmol), EDC (218 mg, 1.41 mmol), and triethylamine (0.6 mL, 4.14 mmol). The reaction mixture was stirred at room temperature overnight. To the resulting suspension was then added water (10 mL) and the phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (10 mL) and brine (10 mL), dried over anh. Na2SO4 and filtered. Evaporation in vacuo of the organics gave 6 as a yellowish solid (216 mg, 94% yield). The analytical sample was obtained by crystallization from hot EtOAc (69 mg), m.p. 91–92 °C. IR (ATR) ν: 720, 756, 766, 809, 829, 849, 912, 943, 988, 1036, 1069, 1094, 1165, 1193, 1239, 1274, 1340, 1362, 1380, 1405, 1461, 1476, 1507, 1610, 2870, 2896, 2936, 2951, 2992 cm–1. 1H-NMR (400 MHz, CDCl3) δ: 0.10–0.18 (complex signal, 2H, 9′-H2), 0.91 [m, 2H, 8′(10′)-H], 1.18 [s, 9H, C(CH3)3], 2.56 [m, 2H, 2′(6′)-H], 2.84 [m, 2H, 1′(7′)-H], 3.27 [dd, J = 11.8 Hz, J’ = 4.2 Hz, 2H, 3′(5′)-Ha], 3.63 [m, 2H, 3′(5′)-Hb], 5.74 [t, J = 4.0 Hz, 2H, 11′(12′)-H]. 13C-NMR (100.5 MHz, CDCl3) δ: 3.9 (CH2, C9′), 10.1 [CH, C8′(10′)], 27.5 [CH3, C(CH3)3], 35.6 [CH, C1′(7′)], 38.6 [C, C(CH3)3], 51.7 [CH2, C3′(5′)], 128.6 [CH, C11′(12′)], 175.7 (C, CO). The signal of C2′(6′) was not observed. Anal. Calcd. for C16H23NO: C, 78.32; H, 9.45; N, 5.71. Found: C, 78.16; H, 9.52; N, 5.86.
3.1.5. (4-Azatetracyclo[5.3.2.02,6.08,10]dodec-11-en-4-yl)(thien-2-yl)methanone 7
To a solution of 4-azapentacyclo[5.3.2.02,6.08,10]dodec-11-ene hydrochloride (200 mg, 1.03 mmol) in EtOAc (10 mL) were added 2-thiophenecarboxylic acid (121 mg, 0.94 mmol), HOBt (190 mg, 1.41 mmol), EDC (218 mg, 1.41 mmol), and triethylamine (0.6 mL, 4.14 mmol). The reaction mixture was stirred at room temperature overnight. To the resulting suspension was then added water (10 mL) and the phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (10 mL) and brine (10 mL), dried over anh. Na2SO4 and filtered. Evaporation in vacuo of the organics gave 7 as a yellowish solid (219 mg, 86% yield). The analytical sample was obtained by crystallization from hot EtOAc (87 mg), m.p. 104–105 °C. IR (ATR) ν: 667, 703, 720, 738, 786, 814, 849, 890, 915, 950, 1008, 1031, 1057, 1087, 1132, 1239, 1254, 1279, 1312, 1352, 1380, 1403, 1431, 1519, 1580, 1598, 2921, 2936, 3002, 3037 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.12–0.20 (complex signal, 2H, 9′-H2), 0.86–1.02 (complex signal, 2H, 8′-H and 10′-H), 2.63 (m, 1H, 2′-H or 6′-H), 2.73 (m, 1H, 6′-H or 2′-H), 2.81–2.98 (complex signal, 2H, 1′-H and 7′-H), 3.36–3.48 (complex signal, 2H, 3′-Ha and 5′-Ha), 3.72–3.96 (complex signal, 2H, 3′-Hb and 5′-Hb), 5.75 (m, 1H, 11′-H or 12′-H), 5.83 (m, 1H, 12′-H or 11′-H), 7.03 (dd, J = 5.0 Hz, J’ = 3.4, 1 H, 4-H), 7.40 (dd, J = 3.4 Hz, J’ = 1.0 Hz, 1H, 3-H or 5-H), 7.43 (dd, J = 5.0 Hz, J’ = 1.0 Hz, 1H, 5-H or 3-H). 13C-NMR (100.5 MHz, CDCl3) δ: 4.1 (CH2, C9′), 10.0 (CH, C8′ or C10′), 10.1 (CH, C10′ or C8′), 35.6 (broad s, CH, C1′ and C7′), 42.1 (CH, C2′ or C6′), 45.4 (CH, C6′ or C2′), 50.9 (CH2, C3′ or C5′), 52.9 (CH2, C5′ or C3′), 126.8 (CH, C3), 128.3 (broad s, CH, C11′ or C12′), 129.1 (CH, C4), 129.3 (CH, C5), 129.4 (broad s, CH, C12′ or C11′), 139.4 (C, C2), 161.2 (C, CO). Anal. Calcd. for C16H17NOS: C, 70.81; H, 6.31; N, 5.16. Found: C, 70.70; H, 6.28; N, 5.12.
3.1.6. (4-Amino-3,5-dichlorophenyl)(4-azatetracyclo[5.3.2. 02,6.08,10]dodec-11-en-4-yl)methanone 8
To a solution of 4-azapentacyclo[5.3.2.02,6.08,10]dodec-11-ene hydrochloride (200 mg, 1.03 mmol) in EtOAc (10 mL) were added 3,5-dichloro-4-aminobenzoic acid (194 mg, 0.94 mmol), HOBt (190 mg, 1.41 mmol), EDC (218 mg, 1.41 mmol), and triethylamine (0.6 mL, 4.14 mmol). The reaction mixture was stirred at room temperature overnight. To the resulting suspension was then added water (10 mL) and the phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (10 mL) and brine (10 mL), dried over anh. Na2SO4 and filtered. Evaporation in vacuo of the organics gave 8 as a yellowish solid (322 mg, 89% yield). The analytical sample was obtained by crystallization from hot EtOAc, m.p. 186–187 °C. IR (ATR) ν: 680, 718, 743, 763, 783, 809, 844, 864, 892, 915, 955, 991, 1034, 1097, 1173, 1223, 1246, 1297, 1347, 1416, 1469, 1501, 1537, 1595, 2875, 2921, 3194, 3240, 3301, 3458 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.10–0.19 (complex signal, 2H, 9′-H2), 0.84–0.98 (complex signal, 2H, 8′-H and 10′-H), 2.52–2.64 (complex signal, 2H, 2′-H and 6′-H), 2.76 (m, 1H, 1′-H or 7′-H), 2.88 (m, 1H, 7′-H or 1′-H), 3.16 (m, 1H, 3′-Ha or 5′-Ha), 3.44 (m, 1H, 5′-Ha or 3′-Ha), 3.58 (m, 1H, 3′-Hb or 5′-Hb), 3.68 (m, 1H, 5′-Hb or 3′-Hb), 4.63 (s, 2H, NH2), 5.70 (m, 1H, 11′-H or 12′-H), 5.83 (m, 1H, 12′-H or 11′-H), 7.30 [s, 2H, 2(6)-H]. 13C-NMR (100.5 MHz, CDCl3) δ: 4.0 (CH2, C9′), 10.0 (CH, C8′ or C10′), 10.1 (CH, C10′ or C8′), 35.5 (CH, C1′ and 7′), 42.6 (CH, C2′ or C6′), 45.0 (CH, C6′ or C2′), 49.6 (CH2, C3′ or C5′), 53.6 (CH2, C5′ or C3′), 118.7 [C, C3(5)], 126.8 (C, C1), 127.2 [CH, C2(6)], 128.2 (CH, C11′ or C12′), 129.3 (CH, C12′ or C11′), 141.3 (C, C4), 166.4 (C, CO). Anal. Calcd. for C18H18Cl2N2O: C, 61.90; H, 5.20; N, 8.02. Found: C, 62.10; H, 5.20; N, 7.92.
3.1.7. (4-Azatetracyclo[5.3.2. 02,6.08,10]dodec-11-en-4-yl)(cyclohex-3-en-1-yl)methanone 9
To a solution of 4-azapentacyclo[5.3.2.02,6.08,10]dodec-11-ene hydrochloride (200 mg, 1.03 mmol) in EtOAc (10 mL) were added 3-cyclohexene carboxylic acid (119 mg, 0.94 mmol), HOBt (190 mg, 1.41 mmol), EDC (218 mg, 1.41 mmol), and triethylamine (0.6 mL, 4.14 mmol). The reaction mixture was stirred at room temperature overnight. To the resulting suspension was then added water (10 mL) and the phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (10 mL) and brine (10 mL), dried over anh. Na2SO4 and filtered. Evaporation in vacuo of the organics gave RL-135 as a yellowish solid (259 mg, quantitative yield). Column chromatography (hexane/EtOAc mixture) gave 9 as a white solid (199 mg), m.p. 78–79 °C. IR (ATR) ν: 682, 710, 763, 816, 839, 854, 887, 915, 940, 981, 1016, 1034, 1087, 1135, 1168, 1203, 1221, 1274, 1292, 1332, 1355, 1380, 1431, 1620, 1651, 2870, 2926, 3022 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.10–0.19 (complex signal, 2H, 9′-H2), 0.86–0.96 (complex signal, 2H, 8′-H and 10′-H), 1.56–2.38 (complex signal, 6H, 2-Hax, 5-Hax, 6-Hax, 2-Heq, 5-Heq, 6-Heq), 2.47 (m, 1H, 1-H), 2.56 (m, 1H, 2′-H or 6′-H), 2.67 (m, 1H, 6′-H or 2′-H), 2.81–2.89 (complex signal, 2H, 1′-H and 7′-H), 3.11–3.24 (complex signal, 2H, 3′-Ha and 5′-Ha), 3.52–3.64 (complex signal, 2H, 3′-Hb and 5′-Hb), 3.50–3.64 (complex signal, 2H, 3′-Hb and 5′-Hb), 5.61–5.84 (complex signal, 4H, 11′-H, 12′-H, 3-H and 4-H). 13C-NMR (100.5 MHz, CDCl3) δ: 4.03 and 4.06 (CH2, C9′), 9.9 (CH, C8′ or C10′), 10.1 (CH, C10′ or C8′), 24.96 and 24.99 (CH2, C5 or C6), 25.1 and 25.2 (CH2, C6 or C5), 27.40 and 27.43 (CH2, C2), 35.6 (CH, C1′ or C7′), 35.7 (CH, 7′ or C1′), 38.36 and 38.38 (CH, C1), 42.7 (CH, C2′ or C6′), 44.72 and 44.73 (CH, C6′ or C2′), 49.68 and 49.74 (CH2, C3′ or C5′), 50.6 (CH2, C5′ or C3′), 125.88 and 125.94 (CH, C3 or C4), 126.3 and 126.4 (CH, C4 or C3), 128.1 (CH, C11′ or C12′), 129.5 and 129.6 (CH, C12′ or C11′), 173.66 and 173.69 (C, CO). Anal. Calcd, for C18H23NO: C, 80.26; H, 8.61; N, 5.20. Found: C, 80.24; H, 8.73; N 5.19.
3.1.8. (4-Azatetracyclo[5.3.2. 02,6.08,10]dodec-11-en-4-yl) (6-chloropyridin-3-yl)methanone 10
To a solution of 4-azapentacyclo[5.3.2.02,6.08,10]dodec-11-ene hydrochloride (500 mg, 2.58 mmol) in EtOAc (25 mL) were added 6-choloronicotinic acid (370 mg, 2.35 mmol), HOBt (477 mg, 3.53 mmol), EDC (547 mg, 3.53 mmol), and triethylamine (1.4 mL, 10.34 mmol). The reaction mixture was stirred at room temperature overnight. To the resulting suspension was then added water (25 mL) and the phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (25 mL) and brine (25 mL), dried over anh. Na2SO4 and filtered. Evaporation in vacuo of the organics gave 10 as a yellowish solid (664 mg, 86% yield). Column chromatography (hexane/EtOAc mixture) gave 10 as a white solid (457 mg), m.p. 101–102 °C. IR (ATR) ν: 712, 736, 759, 793, 814, 835, 924, 940, 985, 1030, 1097, 1129, 1156, 1174, 1215, 1239, 1251, 1271, 1283, 1350, 1372, 1430, 1455, 1563, 1583, 1612, 2914, 2948, 3002 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.12–0.20 (complex signal, 2H, 9′-H2), 0.85–0.98 (complex signal, 2H, 8′-H and 10′-H), 2.54–2.68 (complex signal, 2H, 2′-H and 6′-H), 2.75 (m, 1 H, 1′-H or 7′-H), 2.92 (m, 1 H, 7′-H or 1′-H), 3.10 (dd, J = 10.8 Hz, J’ = 3.2 Hz, 1 H, 5′-Ha or 3′-Ha), 3.42–3.58 (complex signal, 2H, 3′-Ha or 5′-Ha and 5′-Hb or 3′-Hb), 3.71 (m, 1H, 3′-Hb or 5′-Hb), 5.69 (t, J = 7.2 Hz, 1H, 11′-H or 12′-H), 5.86 (t, J = 7.2 Hz, 1H, 12′-H or 11′-H), 7.35 (dd, J = 8.4 Hz, J’ = 0.8 Hz, 1H, 5-H), 7.71 (dd, J = 8.4 Hz, J’ = 2.6 Hz, 1H, 4-H), 8.43 (dd, J = 2.6 Hz, J’ = 0.8 Hz, 1H, 2-H). 13C-NMR (100.5 MHz, CDCl3) δ: 3.9 (CH2, C9′), 9.8 (CH, C8′ or C10′), 10.1 (CH, C10′ or C8′), 35.5 (CH, C1′ or C7′), 35.6 (CH, C7′ or C1′), 42.7 (CH, C2′ or C6′), 44.8 (CH, C6′ or C2′), 49.7 (CH2, C3′ or C5′), 53.4 (CH2, C5′ or C3′), 124.1 (CH, C5), 128.1 (CH, C11′ or C12′), 129.4 (CH, C12′ or C11′), 131.8 (C, C3), 137.7 (CH, C4), 148.1 (CH, C2), 152.3 (C, C6), 165.2 (C, CO). Anal. Calcd. for C17H17ClN2O: C, 67.88; H, 5.70; N, 9.31. Found: C, 68.14; H, 5.84; N, 9.00.
3.1.9. (4-Azatetracyclo[5.3.2. 02,6.08,10]dodec-11-en-4-yl)(2-chloropyridin-4-yl)methanone, 11
To a solution of 4-azapentacyclo[5.3.2.02,6.08,10]dodec-11-ene hydrochloride (500 mg, 2.58 mmol) in EtOAc (25 mL) were added 6-choloronicotinic acid (370 mg, 2.35 mmol), HOBt (477 mg, 3.53 mmol), EDC (547 mg, 3.53 mmol) and triethylamine (1.4 mL, 10.34 mmol). The reaction mixture was stirred at room temperature overnight. To the resulting suspension was then added water (25 mL) and the phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (25 mL) and brine (25 mL), dried over anh. Na2SO4 and filtered. Evaporation in vacuo of the organics gave 11 as a yellowish solid (614 mg, 79% yield). Column chromatography (hexane/EtOAc mixture) gave 11 as a white solid (445 mg), m.p. 135–136 °C. IR (ATR) ν: 669, 708, 720, 741, 753, 771, 817, 844, 915, 942, 987, 1041, 1091, 1118, 1163, 1176, 1203, 1232, 1245, 1269, 1287, 1342, 1373, 1437, 1464, 1476, 1530, 1593, 1632, 2868, 2932, 3003, 3057 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.12–0.22 (complex signal, 2H, 9′-H2), 0.85–1.00 (complex signal, 2H, 8′-H and 10′-H), 2.56–2.70 (complex signal, 2H, 2′-H and 6′-H), 2.76 (m, 1H, 1′-H or 7′-H), 2.92 (m, 1H, 7′-H or 1′-H), 3.01 (dd, J = 11.0 Hz, J’ = 3.8 Hz, 1H, 5′-Ha or 3′-Ha), 3.40–3.52 (complex signal, 2H, 3′-Ha or 5′-Ha and 5′-Hb or 3′-Hb), 3.68 (m, 1H, 3′-Hb or 5′-Hb), 5.71 (m, 1H, 11′-H or 12′-H), 5.87 (m, 1H, 12′-H or 11′-H), 7.18 (dd, J = 5.0 Hz, J’ = 1.4 Hz, 1H, 5-H), 7.30 (dd, J = 1.4 Hz, J’ = 0.8 Hz, 1H, 3-H), 8.42 (dd, J = 5.0 Hz, J’ = 0.8 Hz, 1H, 6-H). 13C-NMR (100.5 MHz, CDCl3) δ: 4.0 (CH2, C9′), 9.8 (CH, C8′ or C10′), 10.1 (CH, C10′ or C8′), 35.5 (CH, C1′ or C7′), 35.6 (CH, C7′ or C1′), 42.7 (CH, C2′ or C6′), 44.6 (CH, C6′ or C2′), 49.6 (CH2, C3′ or C5′), 53.1 (CH2, C5′ or C3′), 119.8 (CH, C5), 121.9 (CH, C3), 128.2 (CH, C11′ or C12′), 129.4 (CH, C12′ or C11′), 147.6 (C, C4), 150.1 (CH, C6), 151.9 (C, C2), 164.8 (C, CO). Anal. Calcd. for C17H17ClN2O: C, 67.88; H, 5.70; N, 9.31. Found: C, 67.98; H, 5.77; N, 9.09.
3.1.10. (4-Azatetracyclo[5.3.2. 02,6.08,10]dodec-11-en-4-yl)(1-methylpiperidin-4-yl)methanone 12
To a solution of 4-azapentacyclo[5.3.2.02,6.08,10]dodec-11-ene hydrochloride (200 mg, 1.03 mmol) in EtOAc (10 mL) were added 1-methylpiperidine-4-carboxylic acid (135 mg, 0.94 mmol), HOBt (190 mg, 1.41 mmol), EDC (218 g, 1.41 mmol), and triethylamine (0.6 mL, 4.14 mmol). The reaction mixture was stirred at room temperature overnight. To the resulting suspension was then added water (10 mL) and the phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (10 mL) and brine (10 mL), dried over anh. Na2SO4 and filtered. Evaporation in vacuo of the organics gave a yellowish solid (129 mg). Column chromatography (hexane/EtOAc/methanol mixture) gave 12 as a yellowish solid (86 mg, 32% yield). The analytical sample was obtained by crystallization from hot EtOAc (50 mg), m.p. 100–101 °C. IR (ATR) ν: 718, 767, 819, 835, 850, 876, 915, 987, 1013, 1041, 1067, 1090, 1129, 1150, 1191, 1214, 1250, 1276, 1305, 1359, 1374, 1431, 1447, 1625, 2780, 2857, 2914, 2940, 3328 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.10–0.18 (complex signal, 2H, 9′-H2), 0.86–0.96 (complex signal, 2H, 8′-H and 10′-H), 1.58–1.68 (complex signal, 2H, 3-Hax and 5-Hax), 1.71–1.87 (complex signal, 2H, 3-Heq and 5-Heq), 1.88–1.98 (complex signal, 2H, 2-Hax and 6-Hax), 2.18 (tt, J = 11.2 Hz, J’ = 3.6 Hz, 1 H, 1-H), 2.24 (s, 3 H, N-CH3), 2.55 (m, 1H, 2′-H or 6′-H), 2.66 (m, 1H, 6′-H or 2′-H), 2.81–2.96 (complex signal, 4H, 1′-H, 7′-H, 2-Heq and 6-Heq), 3.11 (dd, J = 11.0 Hz, J’ = 5.0 Hz, 1H, 3′-Ha or 5′-Ha), 3.16 (dd, J = 13.0 Hz, J’ = 5.0 Hz, 1H, 5′-Ha or 3′-Ha), 3.50–3.64 (complex signal, 2 H, 3′-Hb and 5′-Hb), 5.74 (complex signal, 2 H, 11′-H and 12′-H). 13C-NMR (100.5 MHz, CDCl3) δ: 4.1 (CH2, C9′), 9.4 (CH, C8′ or C10′), 10.2 (CH, C10′ or C8′), 27.9 (CH2, C3 or C5), 28.0 (CH2, C5 or C3), 35.6 (CH, C1′ or C7′), 35.7 (CH, 7′ or C1′), 39.9 (CH, C4), 42.7 (CH, C2′ or C6′), 44.8 (CH, C6′ or C2′), 46.4 (CH3, N–CH3), 49.8 (CH2, C3′ or C5′), 50.6 (CH2, C5′ or C3′), 55.2 (CH2, C2 or C6), 55.3 (CH2, C6 or C2), 128.1 (CH, C11′ or C12′), 129.6 (CH, C12′ or C11′), 172.9 (C, CO). HRMS-ESI+ m/z [M + H]+: Calcd. for [C18H26N2O+H]+: 287.2118, found: 287.2113.
3.1.11. 1-[[4-(4-Azatetracyclo[5.3.2. 02,6.08,10]dodec-11-en-4-yl)carbonyl]piperidin-1-yl]ethan-1-one 13
To a solution of 4-azapentacyclo[5.3.2.02,6.08,10]dodec-11-ene hydrochloride (200 mg, 1.03 mmol) in EtOAc (10 mL) were added 1-acetyl-4-piperidinecarboxylic acid (161 mg, 0.94 mmol), HOBt (190 mg, 1.41 mmol), EDC (218 g, 1.41 mmol), and triethylamine (0.6 mL, 4.14 mmol). The reaction mixture was stirred at room temperature overnight. To the resulting suspension was then added water (10 mL) and the phases were separated. The organic phase was washed with saturated aqueous NaHCO3 solution (10 mL) and brine (10 mL), dried over anh. Na2SO4 and filtered. Evaporation in vacuo of the organics gave a yellowish solid (182 mg). Column chromatography (hexane/EtOAc mixture) gave 13 as a white solid (134 mg, 45% yield), m.p. 134–135 °C. IR (ATR) ν: 605, 703, 762, 814, 829, 920, 956, 977, 997, 1041, 1098, 1116, 1168, 1222, 1271, 1307, 1356, 1426, 1620, 1640, 2852, 2925, 2992 cm–1. 1H-NMR (400 MHz, CDCl3) δ: 0.11–0.19 (complex signal, 2H, 9′-H2), 0.88–0.98 (complex signal, 2H, 8′-H and 10′-H), 1.50–1.84 (complex signal, 4H, 3-H2, 5-H2), 2.06 (s, 3H, COCH3), 2.46 (tt, J = 10.6 Hz, J’ = 4.0 Hz, 1 H, 4-H), 2.52–2.74 (complex signal, 3 H, 2′-H, 6′-H and 2-Hax or 6-Hax), 2.81–2.89 (complex signal, 2H, 1′-H and 7′-H), 3.05 (m, 1H, 6-Hax or 2-Hax), 3.11–3.22 (complex signal, 2H, 3′-Ha and 5′-Ha), 3.50–3.64 (complex signal, 2H, 3′-Hb or 5′-Hb), 3.84 (dm, J = 13.6 Hz, 1H, 2-Heq or 6-Heq), 4.54 (dm, J = 13.6 Hz, 1H, 6-Heq or 2-Heq), 5.75 (complex signal, 2H, 11′-H and 12′-H). 13C-NMR (100.5 MHz, CDCl3) δ: 4.1 (CH2, C9′), 9.9 (CH, C8′ or C10′), 10.1 (CH, C10′ or C8′), 21.4 (CH3, COCH3), 27.6 and 27.7 (CH2, C3 or C5), 28.1 and 28.2 (CH2, C5 or C3), 35.6 (CH, C1′ or C7′), 35.7 (CH, 7′ or C1′), 40.1 (CH, C4), 40.9 and 41.0 (CH2, C2 or C6), 42.62 and 42.64 (CH, C2′ or C6′), 44.69 and 44.70 (CH, C6′ or C2′), 45.7 and 45.8 (CH2, C6 or C2), 49.8 (CH2, C3′ or C5′), 50.63 and 50.64 (CH2, C5′ or C3′), 128.0 (CH, C11′ or C12′), 129.65 and 129.68 (CH, C12′ or C11′), 168.8 (C, COCH3), 171.82 and 171.85 (C, CO). Anal. Calcd. for C19H26N2O2: C 72.58; H, 8.34; N, 8.91. Found: C, 72.65; H 8.60; N 8.48.
3.1.12. (4-Azatetracyclo[5.3.2.02,6.08,10]dodec-11-en-4-yl)[6-(4-phenylpiperazin-1-yl)pyridin-3-yl]methanone 14
To a solution of 10 (100 mg, 0.33 mmol) and 1-phenylpiperazine (60 mg, 0.37 mmol) in DMF (0.5 mL) was added solid K2CO3 (82 mg, 0.59 mmol). The resulting suspension was stirred at 90 °C for 48 h. Water (5 mL) and dichloromethane (DCM) (5 mL) were added and the phases were separated. The aqueous phase was then extracted with further DCM (2 × 5 mL). The organics were dried over anh. Na2SO4, filtered and evaporated in vacuo to give a yellowish solid (137 mg). Column chromatography (hexane/EtOAc mixture) gave 14 as a white solid (56 mg, 39% yield). The analytical sample was obtained by washing this solid with cold pentane (45 mg), m.p. 90–91 °C. IR (ATR) ν: 661, 695, 739, 754, 814, 822, 845, 948, 987, 1013, 1028, 1041, 1095, 1152, 1227, 1310, 1349, 1349, 1395, 1413, 1491, 1594, 1617, 2847, 2919, 2997 cm–1. 1H-NMR (400 MHz, CDCl3) δ: 0.12–0.20 (complex signal, 2H, 9-H2), 0.86–1.00 (complex signal, 2H, 8-H and 10-H), 2.54–2.66 (complex signal, 2H, 2-H and 6-H), 2.76 (m, 1H, 1-H or 7-H), 2.90 (m, 1H, 7-H or 1-H), 3.18–3.36 [complex signal, 6H, 3-Ha, 5-Ha, 2″(6″)-H2], 3.38–3.84 [complex signal, 6H, 3-Hb, 5-Hb, 3″(5″)-H2], 5.70 (m, 1H, 11-H or 12-H), 5.84 (m, 1H, 12-H or 11-H), 6.66 (d, J = 8.8 Hz, 1H, 5′-H), 6.90 (t, J = 7.2 Hz, 1 H, 4‴-H), 6.97 [d, J = 8.6 Hz, 2 H, 2‴(6‴)-H], 7.28 [dd, J = 8.6 Hz, J’ = 7.2 Hz, 2 H, 3‴(5‴)-H], 7.66 (dd, J = 8.8 Hz, J’ = 2.4 Hz, 1 H, 4′-H), 8.31 (d, J = 2.4 Hz, 1 H, 2′-H). 13C-NMR (100.5 MHz, CDCl3) δ: 3.9 (CH2, C9), 10.2 (broad s, CH, C8 and C10), 35.5 (CH, C1 and C7), 42.6 (CH, C2 or C6), 44.9 [CH2, C3″(5″)], 45.0 (CH, C6 or C2), 49.1 [CH2, C2″(6″)], 49.6 (CH2, C3 or C5), 53.6 (CH2, C5 or C3), 105.8 (CH, C5′), 116.4 [CH, C2‴(6‴)], 120.2 (CH, C4‴), 121.9 (C, C3′), 128.2 (CH, C11 or C12), 129.19 [CH, C3‴(5‴)], 129.24 (CH, C12 or C11), 137.5 (CH, C4′), 147.5 (CH, C2′), 151.1 (C, C1‴), 159.3 (C, C6′), 167.1 (C, CO). HRMS-ESI+ m/z [M + H]+ calcd. for [C27H30N4O+H]+: 427.2494, found: 427.2492.
3.1.13. (4-Azatetracyclo[5.3.2.02,6.08,10]dodec-11-en-4-yl)[6-[4-(4-trifluoromethyl)phenylpiperazin-1-yl]pyridin-3-yl]methanone 15
To a solution of 10 (100 mg, 0.33 mmol) and 1-(4-trifluoromethylphenyl)piperazine (85 mg, 0.37 mmol) in DMF (0.5 mL) was added solid K2CO3 (82 mg, 0.59 mmol). The resulting suspension was stirred at 90 °C for 48 h. Water (5 mL) and DCM (5 mL) were added and the phases were separated. The aqueous phase was then extracted with further DCM (2 × 5 mL). The organics were dried over anh. Na2SO4, filtered and evaporated in vacuo to give a yellowish solid (161 mg). Column chromatography (hexane/EtOAc mixture) gave 15 as a white solid (52 mg, 32% yield). The analytical sample was obtained by washing with cooled pentane (38 mg), m.p. 157–158 °C. IR (ATR) ν: 667, 711, 721, 744, 770, 806, 824, 909, 951, 971, 984, 1039, 1070, 1106, 1157, 1199, 1230, 1330, 1354, 1390, 1429, 1493, 1522, 1594, 1615, 2847, 2919 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.10–0.18 (complex signal, 2H, 9-H2), 0.84–0.98 (complex signal, 2H, 8-H and 10-H), 2.54–2.66 (complex signal, 2H, 2-H and 6-H), 2.75 (m, 1H, 1-H or 7-H), 2.90 (m, 1H, 7-H or 1-H), 3.26 (m, 1H, 3-Ha or 5-Ha), 3.41 [t, J = 5.4 Hz, 4H, 2″(6″)-H2], 3.48 (m, 1H, 5-Ha or 3-Ha), 3.56–3.82 [complex signal, 6H, 3-Hb, 5-Hb, 3″(5″)-H2], 5.69 (m, 1H, 11-H or 12-H), 5.85 (m, 1H, 12-H or 11-H), 6.65 (d, J = 8.8 Hz, 1H, 5′-H), 6.95 [d, J = 8.6 Hz, 2H, 2‴(6‴)-H], 7.50 [d, J = 8.6 Hz, 2H, 3‴(5‴)-H], 7.66 (dd, J = 8.8 Hz, J’ = 2.2 Hz, 1H, 4′-H), 8.31 (d, J = 2.2 Hz, 1H, 2′-H). 13C-NMR (100.5 MHz, CDCl3) δ: 3.9 (CH2, C9), 10.2 (broad s, CH, C8 and C10), 35.6 (CH, C1 and C7), 42.7 (CH, C2 or C6), 44.5 [CH2, C3″(5″)], 45.1 (CH, C6 or C2), 47.7 [CH2, C2″(6″)], 49.6 (CH2, C3 or C5), 53.6 (CH2, C5 or C3), 105.8 (CH, C5′), 114.6 [CH, C2‴(6‴)], 120.8 (q, J = 32 Hz, C, C4‴), 122.1 (C, C3′), 124.6 (q, J = 269 Hz, C, CF3), 126.4 [q, J = 4 Hz, CH, C3‴(5‴)], 128.1 (CH, C11 or C12), 129.3 (CH, C12 or C11), 137.5 (CH, C4′), 147.5 (CH, C2′), 153.0 (C, C1‴), 159.1 (C, C6′), 167.0 (C, CO). HRMS-ESI + m/z [M + H]+ calcd. for [C28H29F3N4O + H]+: 495.2396, found: 495.2369.
3.1.14. 4-[[4-[5-(4-Azatetracyclo[5.3.2. 02,6.08,10]dodec-11-en-4-yl)carbonyl]pyridin-2-yl]piperazin-1-yl]benzonitrile, 16
To a solution of 10 (100 mg, 0.33 mmol) and 4-piperazinobenzonitrile (69 mg, 0.37 mmol) in DMF (0.5 mL) was added solid K2CO3 (82 mg, 0.59 mmol). The resulting suspension was stirred at 90°C for 48 h. Water (5 mL) and DCM (5 mL) were added and the phases were separated. The aqueous phase was then extracted with further DCM (2 x 5 mL). The organics were dried over anh. Na2SO4, filtered and evaporated in vacuo to give a yellowish solid (181 mg). Column chromatography (hexane/EtOAc mixture) gave 16 as a white solid (72 mg, 48% yield), m.p. 160–161 °C. IR (ATR) ν: 656, 692, 713, 742, 773, 811, 912, 951, 1008, 1039, 1176, 1235, 1312, 1392, 1426, 1511, 1537, 1555, 1599, 1648, 1666, 2847, 2925 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.10–0.22 (complex signal, 2H, 9‴-H2), 0.84–1.00 (complex signal, 2H, 8‴-H and 10‴-H), 2.54–2.66 (complex signal, 2H, 2‴-H and 6‴-H), 2.75 (m, 1H, 1‴-H or 7‴-H), 2.90 (m, 1H, 7‴-H or 1‴-H), 3.15–3.56 (complex signal, 6H, 3‴-Ha, 5‴-Ha, 3′(5′)-H2), 3.58–3.84 (complex signal, 6H, 3‴-Hb, 5‴-Hb, 2′(6′)-H2), 5.69 (m, 1H, 11‴-H or 12‴-H), 5.84 (m, 1H, 12‴-H or 11‴-H), 6.63 (d, J = 8.8 Hz, 1 H, 3″-H), 6.87 [dm, J = 8.8 Hz, 2 H, 3(5)-H], 7.51 [dm, J = 8.8 Hz, 2 H, 2(6)-H], 7.67 (dd, J = 8.8 Hz, J’ = 2.2 Hz, 1 H, 4″-H), 8.31 (d, J = 2.2 Hz, 1 H, 6″-H). 13C-NMR (100.5 MHz, CDCl3) δ: 3.9 (CH2, C9‴), 10.1 (broad singlet, CH, C8‴ and C10‴), 35.5 (CH, C1‴ and C7‴), 42.7 (CH, C2‴ or C6‴), 44.2 [CH2, C2′(6′)], 45.0 (CH, C6‴ or C2‴), 46.6 [CH2, C3′(5′)], 49.6 (CH2, C3‴ or C5‴), 53.5 (CH2, C5‴ or C3‴), 100.5 (C, C1), 105.7 (CH, C3″), 114.0 [CH, C3(5)], 119.9.2 (C, CN), 122.2 (C, C5″), 128.1 (CH, C11‴ or C12‴), 129.3 (CH, C12‴ or C11‴), 133.5 [CH, C2(6)], 137.6 (CH, C4″), 147.4 (CH, C6″), 152.9 (C, C4), 158.8 (C, C2″), 166.9 (C, CO). HRMS-ESI + m/z [M + H]+ calcd. for [C28H29N5O+H]+: 452.2445, found: 452.2444.





{kind=link}















