1. Introduction
Creeping bentgrass (
Agrostis soionifera) is a perennial herb of the family Gramineae and belongs to the cool-season turfgrasses. Because of its strong adaptability, drought resistance, soft and delicate blades, rapid growth and strong ornamental characteristics, it was widely used on golf course fairways, grass tennis courts, courtyards and parks [
1,
2,
3]. However, creeping bentgrass has poor disease resistance and is easily infected by various diseases, including dollar spot, downy mildew, snow rot and smut, which causes serious decreases in turf quality and function, resulting in huge losses to turf production and management. For a long time, the resistant varieties and chemical fungicides have been used as prevention methods against lawn diseases [
4]. However, the traditional breeding cycle is too long. Most of the control effects of chemical fungicides are not ideal, and chemical pesticide accumulation in the soil will seriously pollute the environments. Thus, a new effective and environmentally friendly way to control the diseases is to use an inducer to induce disease resistance in turfgrass. Butanediol (BDO) is a new type of disease resistance-inducing factor, which has no toxicity, produces no pollution and provides durable disease resistance. Cortes-Barco and Hsiang reported the induction of disease-resistance in creeping bentgrass for the first time, and ISR produced by BDO effectively inhibited grass leaf diseases [
5]. Subsequently, BDO can induce disease-resistance in creeping bentgrass, and resistance effect is greater than that of the systemic acquired resistance (SAR) pathway by benzothiadiazole (BTH) induction [
6].
Plants induce an innate immunity that is highly flexible and involves a complex response mechanism that recognizes and counteracts different invaders. At first, plants can use their own physical and chemical barriers, and then activate some inducible defensive mechanisms. At present, several systemic defensive response mechanisms are known, such as SAR, which is activated by pathogens producing limited infections, for example, hypersensitive necrosis response [
7]; another is ISR, which is elicited upon the infection of roots by selecting a series of non-pathogenic rhizobacteria [
8,
9]. The ISR responses were found in several plants, such as
Arabidopsis thaliana, carnation, legumes, cucumber and potato [
10]. It has a broad-spectrum resistance to fungi, bacteria and viruses. The resistance to
Rhizoctonia solani in rice was activated by FP7 and PF1
Pseudomonas fluorescens strain [
11]. Additionally, in creeping bentgrass, the
P. fluorescens HP72 strain induced resistance to
R. solani [
12].
Ethylene plays roles in regulating several aspects of plant development, including fruit ripening, seed germination and leaf senescence. In addition, ethylene also has relation to responses to abiotic and biotic stresses. Ethylene plays a key role and responds to diverse types of induced resistances against pathogens through complex networks. Furthermore, recently, the important components in the ethylene-signaling pathway related to disease resistance were revealed by genetic analysis. As a signal molecule to transcription factors, ethylene activates downstream gene expression [
13,
14,
15,
16]. Thus, ethylene is also an important signal molecule in ISR. The ISR induced by Rhizobacteria is practically always dependent on jasmonic acid (JA) and ethylene (ET) signal [
9]. In the ISR system, ethylene-regulated processes are initiated by ethylene synthesis, in which S-adenosylmethionine is changed into 1-aminocyclopropane-1-carboxylic acid (ACC) by the enzyme ACC synthase (ACS). Then ACC is changed to ethylene by ACC oxidase (ACO). Following these processes, ethylene and its membrane-bound receptor proteins regulate the downstream central regulatory protein EIN2, which trigger the expression of diverse transcription factors. These factors activate downstream gene expressions, including those of some protection genes and ethylene responsive genes [
17,
18]. In
Arabidopisis plants expressing WCS417r-mediated ISR, a series of ET-and/or JA-responsive defense-related genes, such as
VSP2,
PDF1.2,
LOX1,
LOX2, PAL1,
CHIB, and
HEL, were also expressed [
19]. Presently, our comprehension about the signaling constituents in ethylene-biosynthetic downstream, the ethylene-signal pathways which change biosynthesis under disease stress, and especially ISR response mechanisms, are largely unknown. Thus, in our study BDO was applied to roots of creeping bentgrass as a new type of disease-resistance compound to induce ISR disease-resistant responses. Our objectives were: (i) to lay a foundation in genetic level for the creeping bentgrass transcriptomic analysis; (ii) to analyze ethylene-dependent signal transduction pathways involved in ISR mechanisms; and (iii) to find the intermediate substances in the signaling pathway and the relationships between the BDO-induced ISR disease-resistance genes and the response genes of the ethylene signal pathway.
3. Discussion
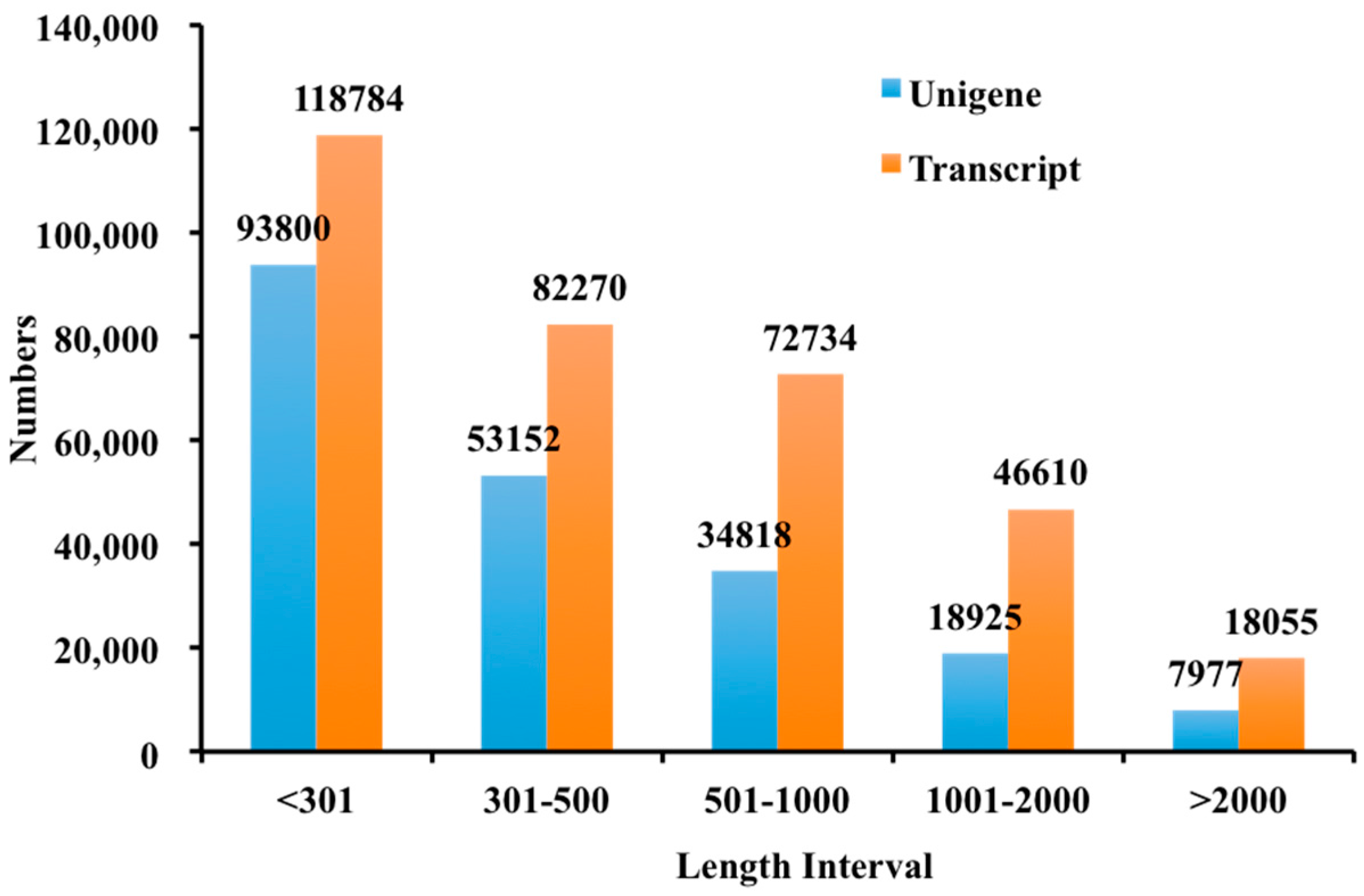
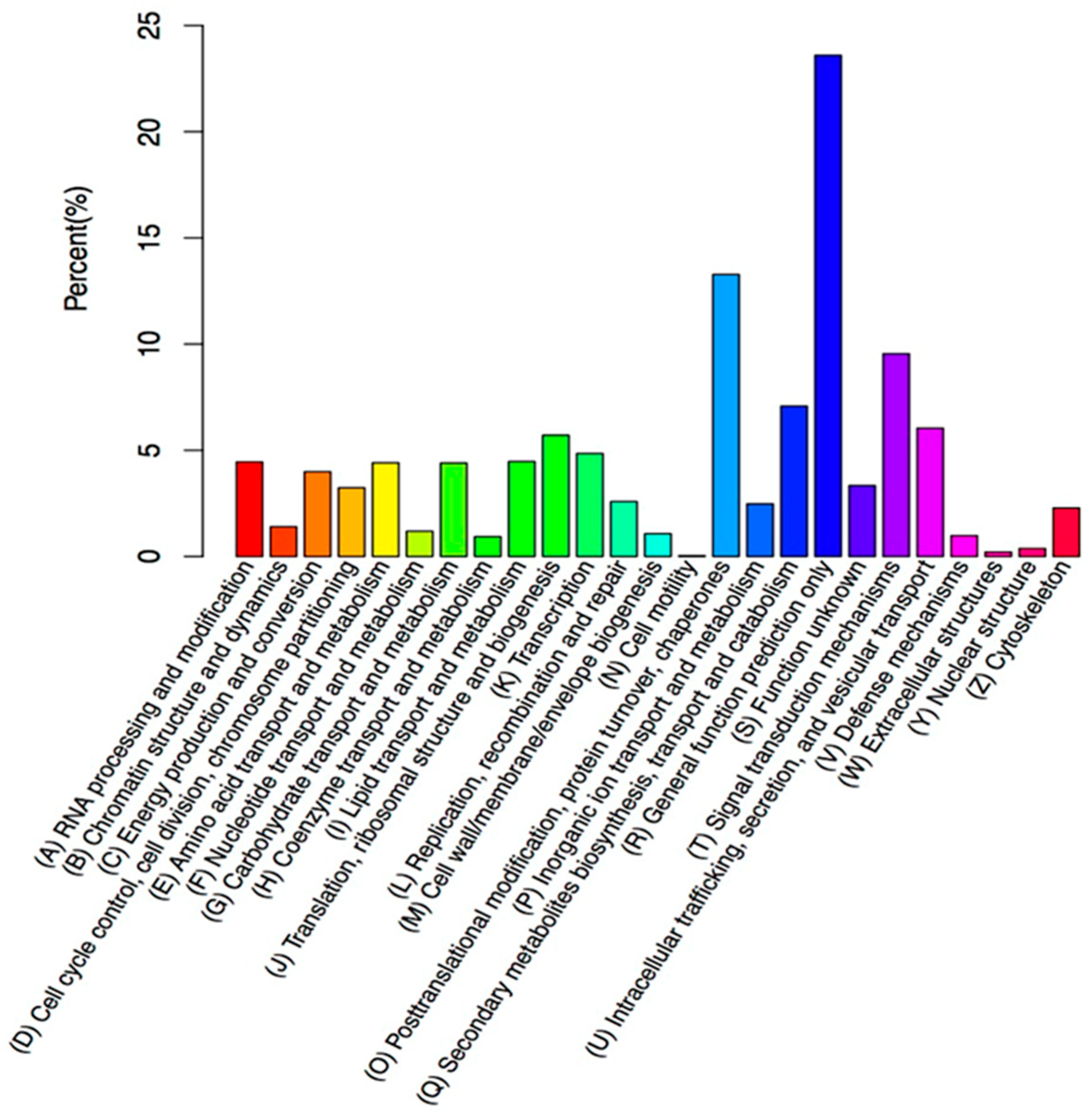
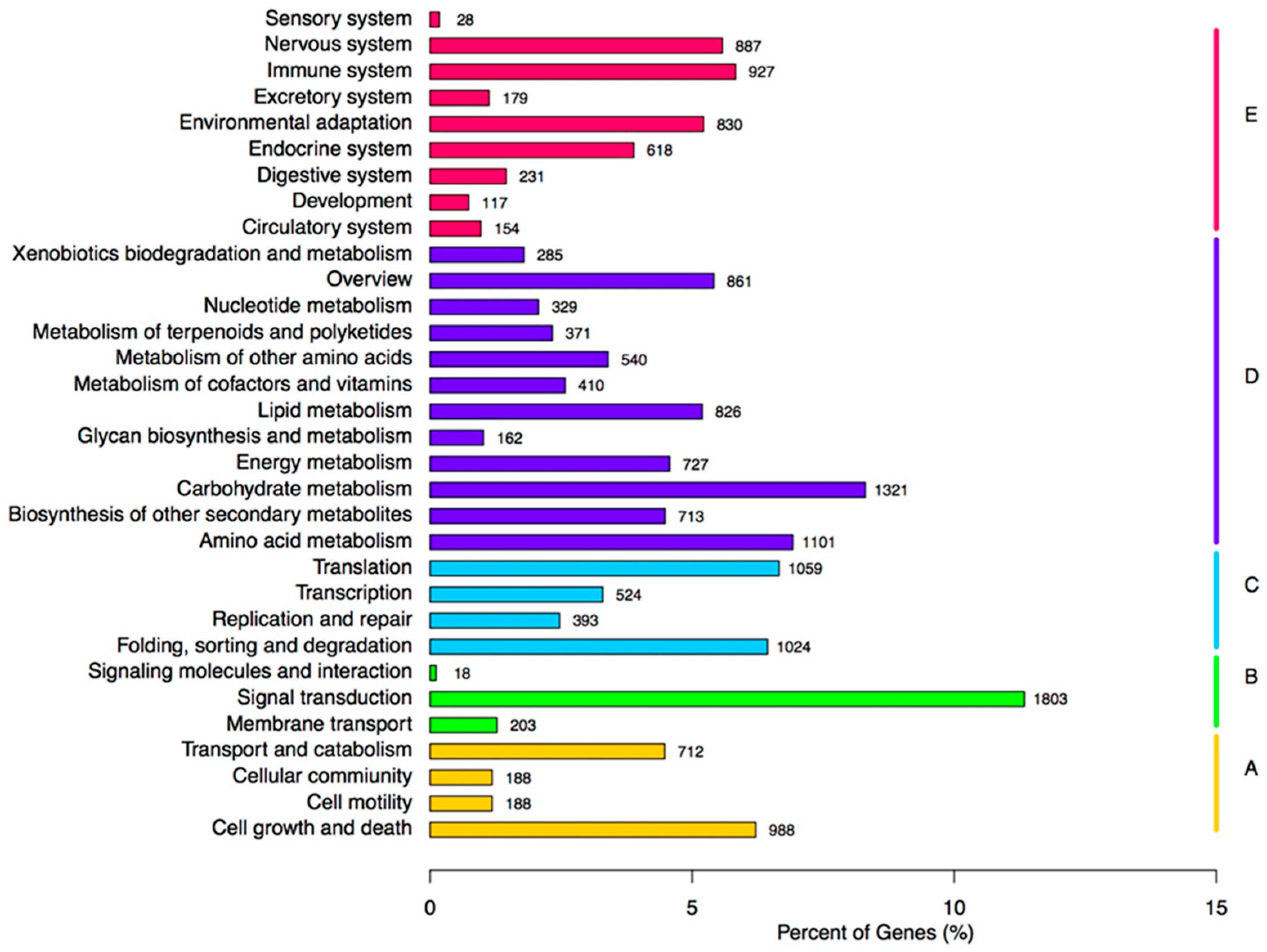
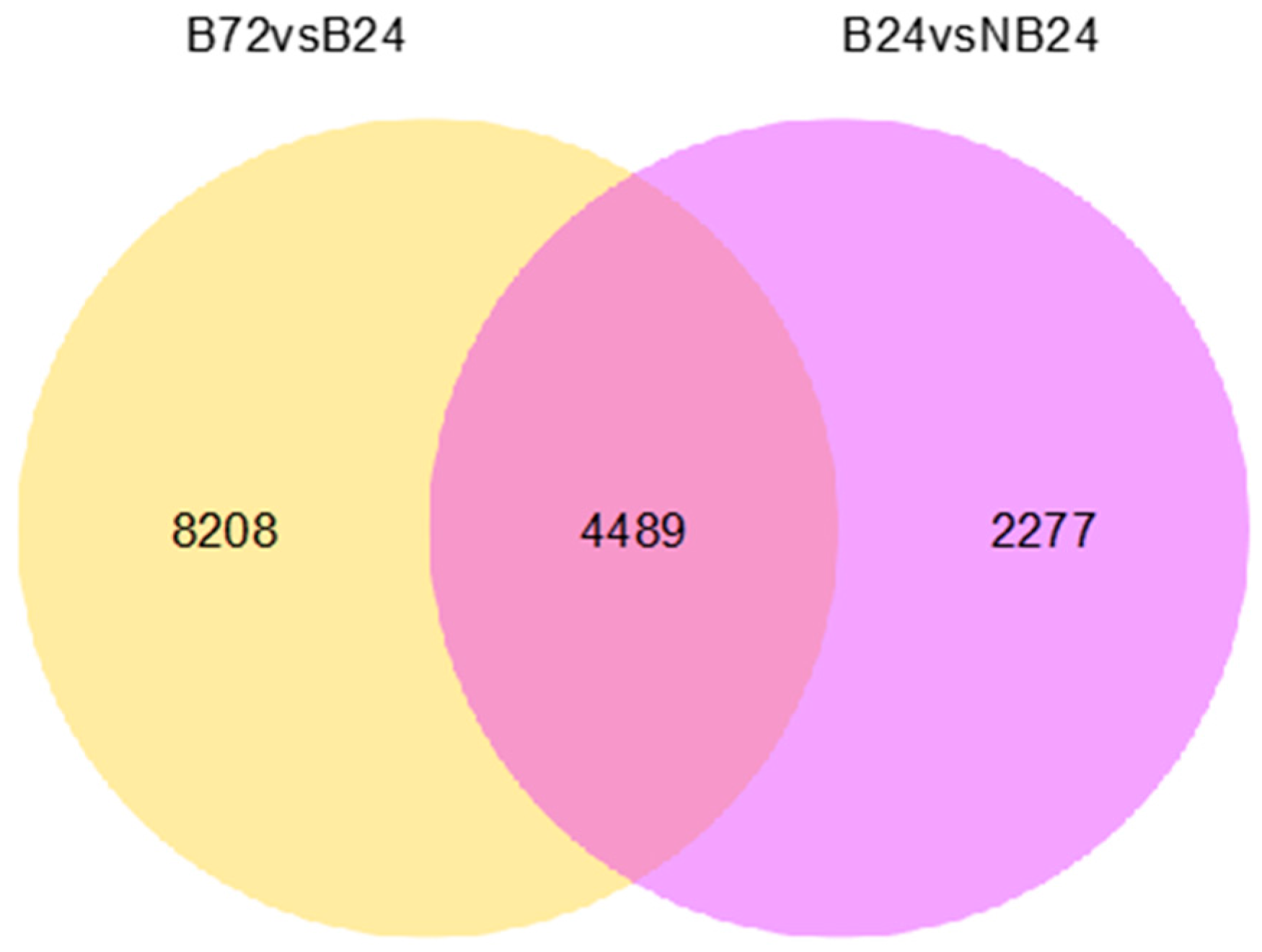
In this study, 208,672 unigenes were found for creeping bentgrass. In total, 51.90% of the transcripts had hits from a BLASTx algorithm-based search against
B. distachyon proteins by comparison with the NR database. It suggested that creeping bentgrass have close relation to Brachypodium, which is a C3 grass and usually taken as a model plant in grass studies [
21]. Additionally, creeping bentgrass had a 13% similarity with
O. sativa (
Figure 3). Thus, we hypothesized that
B. distachyon and
O. sativa are closely related species to creeping bentgrass. In further research, the gene recourses of above two plants can be used the reliable references. Additionally, we examined diseases-resistance and ISR-response genes expression differences among NB24, CKB24, B24 and B72. These factors are critical in selecting DEGs based on the GO annotations and enriched KEGG terms for signal transduction to further understand the role of the ET signal pathway in ISR disease-resistance response. In this research, 15,903 annotated unigenes were grouped into 33 reference canonical pathways in the KEGG (
Figure 6), among which, the greatest amount, 1803 unigenes, were found in ‘signal transduction’.
Besides their functions in plant growth and development, plant hormones are important signal molecules and play complex roles in the signal pathway response in plant–pathogen recognition [
22]. In our research, 31 genes were plant hormone signal transduction molecules, including in the plant-pathogen signal transduction network in the B24 vs. NB24 treatment comparison. Among these plant hormones, ethylene plays important roles in developmental and physiological processes in plants including ripening, leaf and flower senescence, seedling emergence, organ abscission, and responses to adverse abiotic and biotic stresses [
23]. Under a diversity of stress induction, such as drought, pathogen infection, wounding and oxidative stresses, ethylene production is induced and increased in some plant species [
24,
25]. Through determination and analysis, 13 genes were the ethylene-responsive genes. In the previous research, early ethylene production enhances responses to plant-pathogen interactions and positively induces the defense reactions [
26]. For example, ethylene was found to be related disease-resistance in carrot [
27]. And ethylene involvement disease resistance is mostly based on the induction of a set of defense-related genes after pathogen attack in
Arabidopsis thaliana.
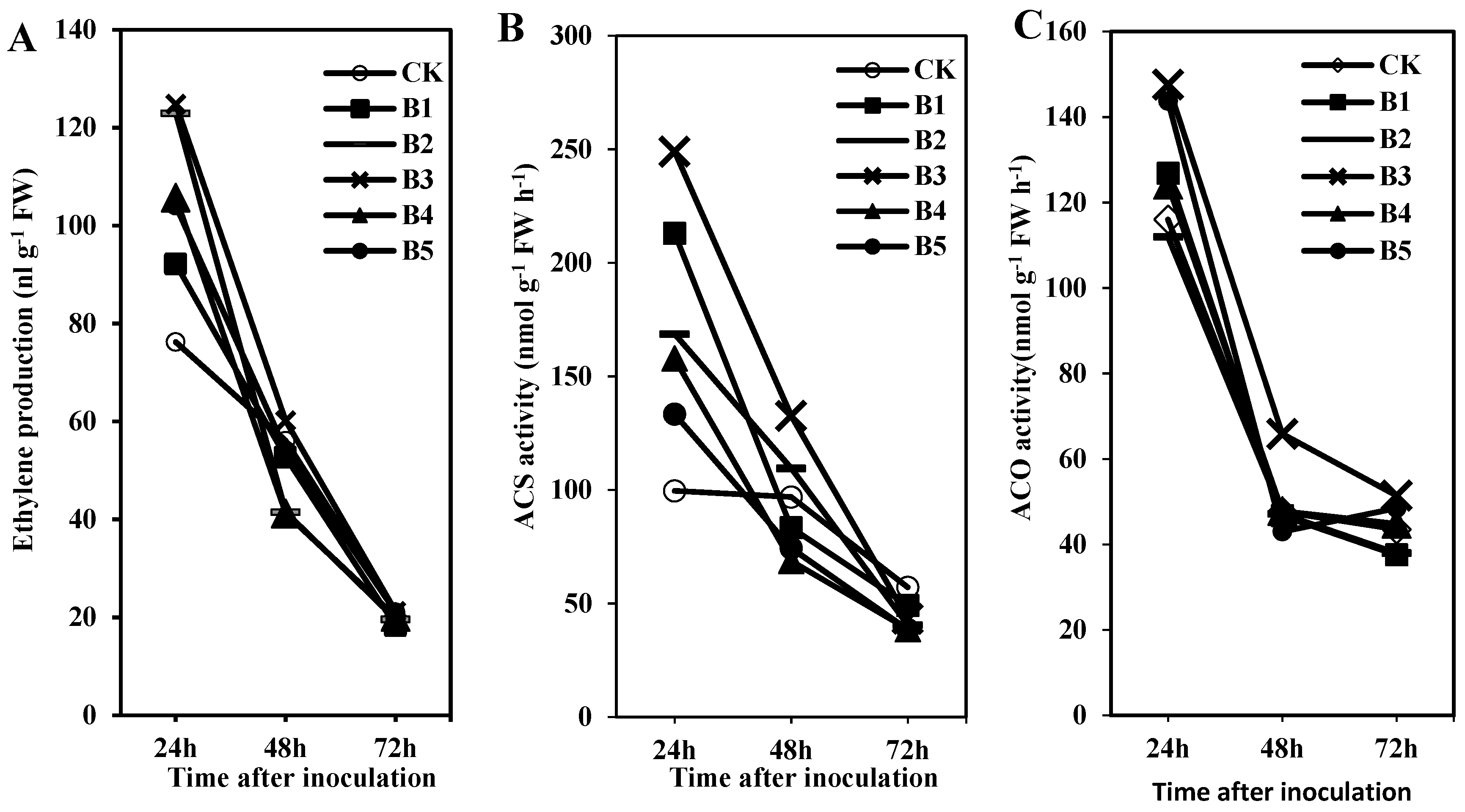
At present detailed measurements of ethylene biosynthesis and related enzymes in turfgrass resistance are lacking, especially in the ISR reaction pathway induced by BDO. In our study, we characterized changes in the important ethylene biosynthetic enzymes and ethylene concentrations after BDO treatments (
Figure 1). Our conclusion that the ethylene production and related enzymes (ACS and ACO) activities showed similar trends in ISR response induced by BDO, which gradually decreased from 24 to 72 h after
R. solani inoculation, and were greatest in the first 24 h in creeping bentgrass after BDO treatment. Based on analyses of ethylene biosynthesis, we concluded that ethylene positively influences creeping bentgrass resistance against
R. solani in a short time after inoculation, and ethylene molecule triggers ethylene signal pathway to participate in ISR response induced by BDO, which is a rapid process. It is interesting to analyze ethylene biosynthetic characters within 24 h inoculation on creeping bentgrass in future study.
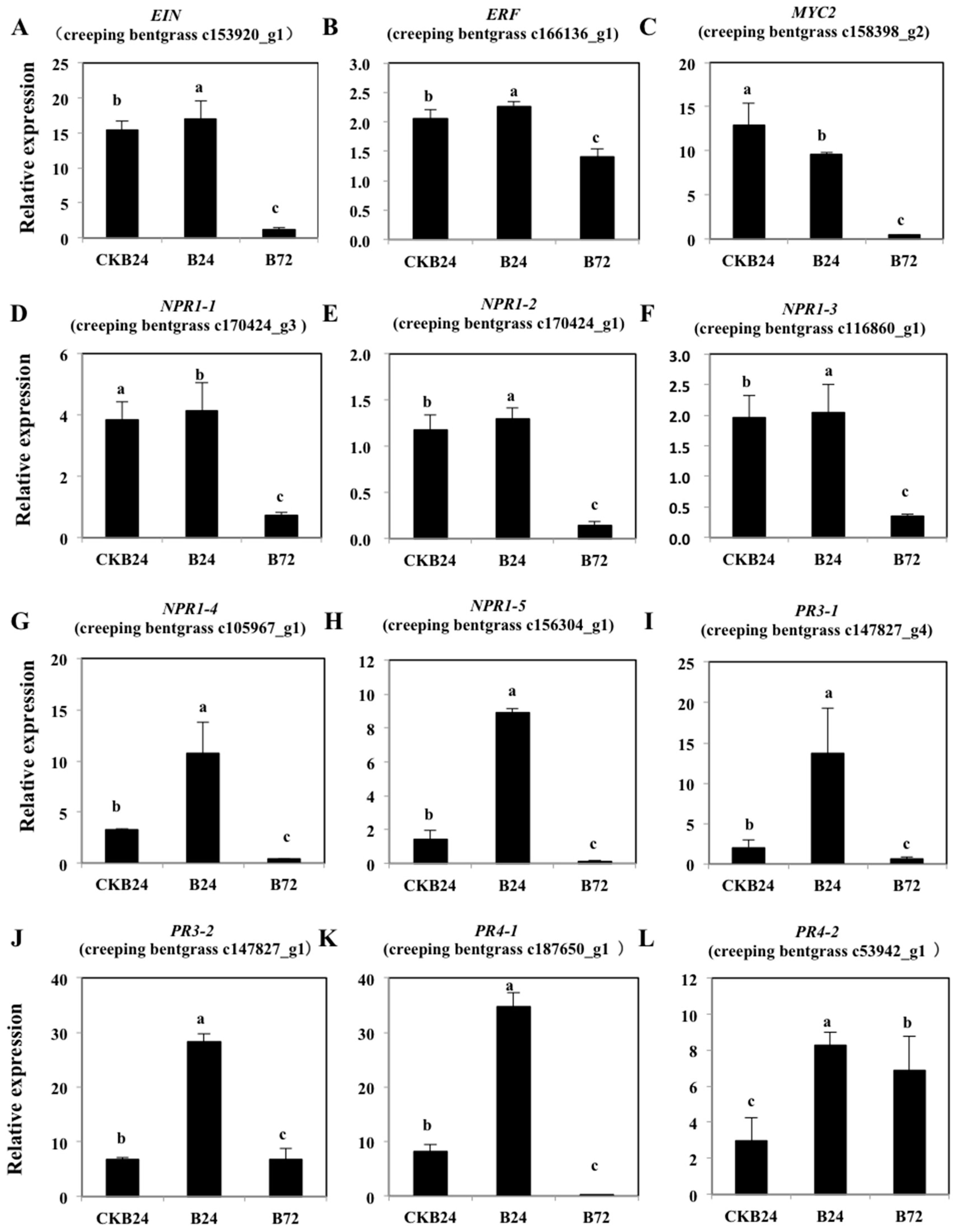
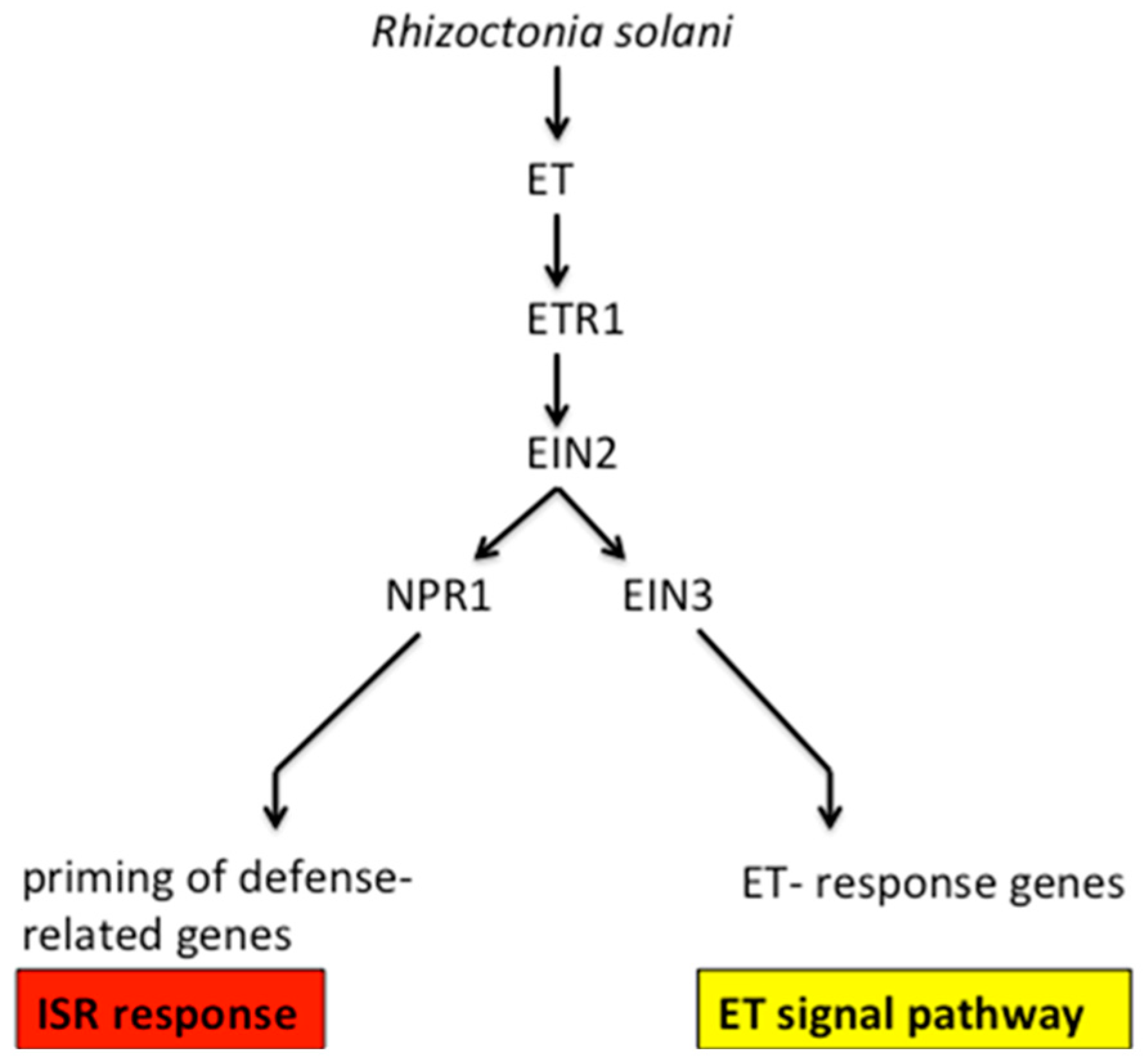
We explored the relevance of ethylene signal pathway-responsive genes and BDO-induced ISR resistance genes. The results of RT-PCR suggested that the expression of ET-responsive and defense-related genes in the BDO-induced ISR response among different treatments (
Figure 9). The
EIN and
ERF from “ET-responsive genes” are very important genes in the ethylene signal conduction pathway, and are upstream of five ethylene receptors,
ETR1,
EIN4,
ERS2,
ERS1 and
ETR2. First, ethylene binds to the membrane-bound receptor, then, receptor signaling is inactivated, allowing the downstream signaling of the central regulatory protein EIN2, and resulting in the activation of diverse transcriptional regulatory factors and defense-related genes. EIN2 has important effects in the ethylene signal pathway, and the loss of the
EIN2 gene function leaves the plant completely insensitive to ethylene [
28]. Through a transcriptional cascade reaction, EIN2 regulates EIN3 and EIL1, and then, EIN3 binds to the EIN3-binding site in the promoter of ERF1, and then activates ethylene responses [
17]. ERF, the ethylene response factor, encodes a transcriptional activator to promote several downstream ethylene-responsive genes (
Figure 10). Additionally, MYC2 and ERF1, the transcription factors, participate in the ET and JA signaling pathways and activated defense-related genes, which are responsive to both ET and JA. In our study, the expression levels of
EIN and
ERF increased and reached their maximum levels in the first 24 h after
R. solani infection in the BDO-induced ISR reaction compared with non-pathogenic treatments at 24 h (NB24) (
Figure 9A,B). This conclusion is consistent with the changes in important ethylene biosynthetic enzymes and in the ethylene concentration during the process. And it also demonstrates the short-time effect of ethylene molecule on disease-resistance of creeping bentgrass. The ethylene reaction is needed for the development and expression of ISR [
29]. For example, in ET insensitive mutants of
Arabidopsis, the ET receptor gene
etr-1 is susceptible to the non-pathogenic oomycete
Pythium sylvaticum. A similar phenomenon, in which the ET-responsive mutant
etr1-1,
ein2-1-ein7 could not express ISR when root is infected by strain WCS417, has been observed [
30]. However, several studies have demonstrated that the defense-regulating hormone jasmonic acid or salicylic acid is also required for the expression of defensive activities in different plant species [
31]. For example, in tomato or tobacco, the salicylic acid (SA) dependent defense pathway is relevant to resistance against
Botrytis cinerea and the powdery mildew fungus
Oidium neolycopersici [
32]. Thus, the plant’s induced resistance signaling is a complex crosstalk network, and the ET-dependent ISR response is also important in the induction of plant disease-resistance.
In the SA-mediated SAR reaction of
Arabidopsis, NPR1 is a crucial regulatory protein and impacts the SAR pathway downstream of the SA signal pathway. During SAR, NPRI induces the
PR gene expression, and even after inducing by SA, BTH or INA,
NPR1 expression levels increase [
33,
34]. Pieterse et al. [
29] determined that
NPR1 also participates in the jasmonate- and ethylene-dependent ISR. The
npr1 mutant was unable of generating a WCS417r-mediated ISR response. A further study of ISR responses unveiled that NPR1 is the downstream of the ET- and JA-dependent signal-transduction pathway steps (
Figure 10). Our study found that the expression levels of creeping bentgrass
NPR1 homologous genes were highly induced in the B24 treatment, but only slightly induced in B72. Additionally, the expression of
EIN and
ERF genes from “ET-responsive genes” and the changes in important ethylene biosynthetic enzymes and ethylene concentrations in the process were similar. These indicate a key role for the NPR1 protein in regulating ET-dependent defense pathways, and BDO-induced ISR was an NPR1-dependent defense response (
Figure 9).
In
Arabidopsis, ET applied exogenously activates certain series of defense-related genes and induces resistance. To further research the molecular mechanism of rhizobacteria-mediated ISR induced by BDO, we focused on the PR proteins because their accumulation may be related to induced disease resistance. The expression levels of creeping bentgrass
PR3 and
PR4 genes were highly induced in the B24 treatment but only slightly induced in B72. This conclusion is consistent with the changes in the important ethylene biosynthetic enzymes, ethylene concentrations and “ET-responsive genes” in the process. In ISR response of
Arabidopsis, the JA/ET response gene
PR4 is elicited by the JA/ET signal and expressed, and the expression product induces resistance to multiple rhizobacteria [
19,
35], and some studies have reported ET and JA co-regulate series of genes encoding PR proteins, including
PR-3,
PR-4 and
PR-1 [
36]. Cortes-Barco et al. [
5] pointed that BDO inspires the expression of the disease-resistance genes
AsAOS1,
AsO-PR4 and
AsGns5. However, some previous research showed that radish roots treated with ISR-inducing WCS417r did not accumulate PR protein, even though disease resistance was enhanced [
37]. Analogously, the expression of SAR marker genes
PR-1,
PR-2 and
PR-5 and WCS417r-mediated ISR was not active, but plants showed enhanced resistance in
Arabidopsis [
38]. In short, we preliminarily discussed the intermediate substances in the signaling pathway, as well as the relationships between the expressed BDO-induced ISR disease-resistance genes and the response genes of the ethylene signal pathway on creeping bentgrass. It must have more complex mechanism between ISR response and ethylene signal pathway on turfgrass, which needs further investigation. Our findings present a genetic basis for systemic resistance of creeping bentgrass through transcriptomic analysis and our study provides a theoretical and practical basis for the improvement of turfgrass disease resistance and quality.
4. Materials and Methods
4.1. Plant Growth Conditions and Treatments
Seeds from creeping bentgrass ‘PennA-4′ were grown by modified method of Kroes et al. [
39]. The surface of seeds was sterilized with 70% ethanol for 1 min, followed by 15% sodium hypochlorite for 5 min. After five rinses with sterile water for 10 min, seeds were dried with filter paper. Seeds were then sown in 50-mL gas chromatography (GC) vials with 10 mL of MS medium containing different concentrations of BDO (50 µmol L
−1, 75 µmol L
−1, 100 µmol L
−1, 125 µmol L
−1 and 150 µmol L
−1). Approximately 20 seedlings per vial (
Figure 11) were placed at 22 °C under continuous light (100 µE m
−2 s
−1) in a growth chamber. The control was not received a BDO treatment (CK). The experimental materials were seedlings which is twelve-day-old grown in GC vials.
Rhizoctonia solani (#3.2888 from China General Microbiological Culture Collection Center) isolates were grown in liquid potato dextrose medium (200 g L−1) in triangular flask with shaking for 4 day at 25 °C. Mycelia were removed and washed with sterile water three times, added to sterile water after grinding in a mortar. Concentration of the bacterial sample was a final OD340 of 0.8. Roots of seedlings were directly sprayed with 2 mL of the R. solani bacterial fluid under sterile conditions.
In our experiment, brown blotch symptoms were observed within 3–5 day post-inoculation on creeping bentgrass seedlings. The mycelia grew but had severe symptoms. After 24, 48 and 72 h post-inoculation, ethylene and related enzymes were measured. The 100 µmol L−1 BDO-treated materials were removed from the medium, and the leaves were cut with a sharp blade, cleaned with sterile water and dried. Leaves were placed in 5-mL tubes, frozen in liquid nitrogen and stored at −80 °C for further analysis. In this experiment, four seedling treatments, CKB24 (inoculated-rhizobacteria in MS medium without BDO for 24 h), B24 (inoculated-rhizobacteria in MS medium with 100 µmol L−1 BDO for 24 h), NB24 (no rhizobacteria in MS medium with 100 µmol L−1 BDO for 24 h) and B72 (inoculated-rhizobacteria in MS medium with 100 µmol L−1 BDO for 72 h), were performed for transcript profiling.
4.2. Ethylene Measurement
The creeping bentgrass seedlings in GC vials were inoculated with rhizobacteria and immediately capped. At the measurement times, the ethylene levels in the headspace were measured using gas chromatography (Varian 450-GC, Palo Alto, CA, USA) by the modified method of Kim et al. [
40].
4.3. 1-Aminocyclopropane-1-Carboxylic Acid Synthase (ACS) and 1-Aminocyclopropane-1-Carboxylic Acid Oxidase (ACO) Activity Assays
Leaf tissue (2 g) was homogenized at 0 °C with 100 mM Hepes extraction buffer (5 mL; 4 mM dithiothreitol, 5 mM ethylene diamine tetraacetic acid, 0.5 μM pyridoxal phosphate, 1% polyvinyl pyrrolidone and 10% glycerol, pH 8.5). The homogenates were centrifuged at 25,000×
g at 4 °C for 20 min. The obtained supernatant was desalted with 2 mM Hepes (0.1 mM dithiothreitol and 0.2 μM pyridoxal phosphate, pH 8.5) for 12 h. The dialysate was used in the ACS activity assay. ACS activity was measured by incubating 1 mL of the enzyme preparation with 0.5 mL of 60 μM S-adenosylmethionine at 30 °C for 2 h, and 0.2 mL 10 mM HgCl was added to end the reaction [
41]. Then, 1 mL of reaction mixture was moved to a 10-mL blood collection tube, then added to 200 μL 5% NaClO–saturated NaOH (2:1,
v/
v). It was whirled and shaken for 5 s, which was repeated after 3 min. Released gas (1 mL) was assayed for ethylene production. The amount of ethylene (nmol) produced per g of fresh weight (FW) per h was used to express the ACS enzyme’s activity level.
Using a previous method [
42] with some changes, leaf tissue (2 g) was homogenized at 0 °C with 0.1 M Tricine buffer (10% glycerol and 30 mM sodium ascorbate, pH 7.5). Homogenates were centrifuged at 20,000×
g at 4 °C for 20 min. Then, 3 mL obtained supernatant was moved to a 10-mL blood collection tube, 0.35 mL 0.2 M Tricine buffer (20% glycerol, 0.2 mM FeSO
4, 60 mM sodium ascorbate, 40 mM NaHCO
3, pH 7.5) was added, then 0.35 mL 1 mM ACC was added for 30 min at 30 °C, and the production of 1 mL ethylene was determined. The amount of ethylene (nmol) produced per g of FW per h was used to express the ACO activity level.
4.4. Total RNA, mRNA Isolation and Library Preparation for Transcriptome Sequencing
Creeping bentgrass leaf blades were gathered from CKB24, B24, NB24 and B72 with three repetitions to minimize transcriptional variation caused by the changes of the plant’s growth stage or physiology. Leaves were gathered for RNA extraction and frozen in liquid nitrogen. By using TRIzol reagent (Qiagen, Hilden, Germany) total RNA was extracted, and purified by using an RNeasy Plant Mini kit (Qiagen) following the manufacturer’s instructions. By using a NanoPhotometer spectrophotometer (IMPLEN, Munich, CA, USA), RNA purity was tested. An Agilent 2100 system (Agilent Technologies, Palo Alto, CA, USA) was used to test the quality and integrity of the RNA.
4.5. Library Preparation for Transcriptome Sequencing
The 2-μg RNA samples were prepared as input materials. Sequencing libraries were produced using NEBNext UltraTM RNA Library Prep Kit for Illumina (NEB, San Diego, CA, USA), and each sample attribute sequences for index codes. In brief, the poly-T oligo-attached magnetic beads were used to purify mRNA. First-strand cDNA synthesis was performed using random primers and reverse transcriptase. Second-strand cDNA was then synthesized using RNase H and DNA Polymerase I. The AMPure XP system (Beckman Coulter, Beverly, MA, USA) was used to purify the library fragments to isolate cDNA fragments in length of ~200–300 bp. Then, adaptor-ligated, size-selected cDNA was mixed with 3 μL USER Enzyme (NEB) for 15 min at 37 °C, then 5 min at 95 °C before PCR. Phusion High-Fidelity DNA polymerase, Index (X) Primer and Universal PCR primers were used to purify and enrich the products by PCR to create the final cDNA library. Using HiSeq 4000 PE Cluster Kit Box1 (Illumina, San Diego, CA, USA), the clustering of the index-coded samples was produced on a cBot Cluster Generation System. Then, on an Illumina HiSeq 4000 platform the multiplexed library preparations were sequenced and 150-bp paired-end reads were produced. RNA-seq read data were placed on the National Center for Biotechnology Information (NCBI) Sequence Read Archive under accession number SRR5658390.
4.6. Preprocessing and De Novo Assembly
Using perl scripts, raw reads in fastq format were processed. To remove a small amount of joint pollution, low-quality reads (Q < 20) and reads containing poly-N (N > 10%) were eliminated, and the qualified reads were assembled. In the meantime, the GC content and Q20, Q30 of the clean data were calculated. The high-quality clean data is used to perform all of the downstream analyses. Because there is no reference creeping bentgrass genome, using the Trinity software package (v2012-10-05
http://trinityrnaseq.github.io) clean reads were joined de novo with min_kmer_cov set to 2 by default and all other parameters set to default according to a previous description by Grabherr et al. [
43].
4.7. Quantification of Gene Expression Levels and Differential Expression Analysis
Gene expression levels were assessed using the RNA-seq by expectation-maximization alignment of each sample [
20]. Clean data were mapped back onto the collected transcriptome libraries. From the mapping results, a read count for each gene was obtained. Differential expression analyses of four treatments (two biological replicates per treatment) were done using the DESeq R program package (1.10.1). Using the Benjamini–Hochberg’s method to multiple comparisons [
44] the
p-values were assessed. By DESeq Genes with an adjusted
p-value < 0.05 were found, and assigned as differentially expressed. Using fragments per kilobase of transcript per million fragments mapped (FPKM), the levels of expression were quantified.
4.8. Unigene Annotation and Classification
Determining the gene functions of the transcripts were conducted based on NR, KOG, SwissProt, KEGG databases using the BLASTALL package (release 2.2.28) [
45] from the NCBI with a significant threshold of
E-value ≤ 10
−5. The statistical enrichment of differential expression genes (DEGs) was checked using KOBAS software in the KEGG pathways [
46]. Gene Ontology (GO) consists of three parts, cellular component, biological process, and molecular function, for the functional categorization using Blast2go v2.5 software (Biobam, Spain). The
E-value filter for the GO annotation was 1 × 10
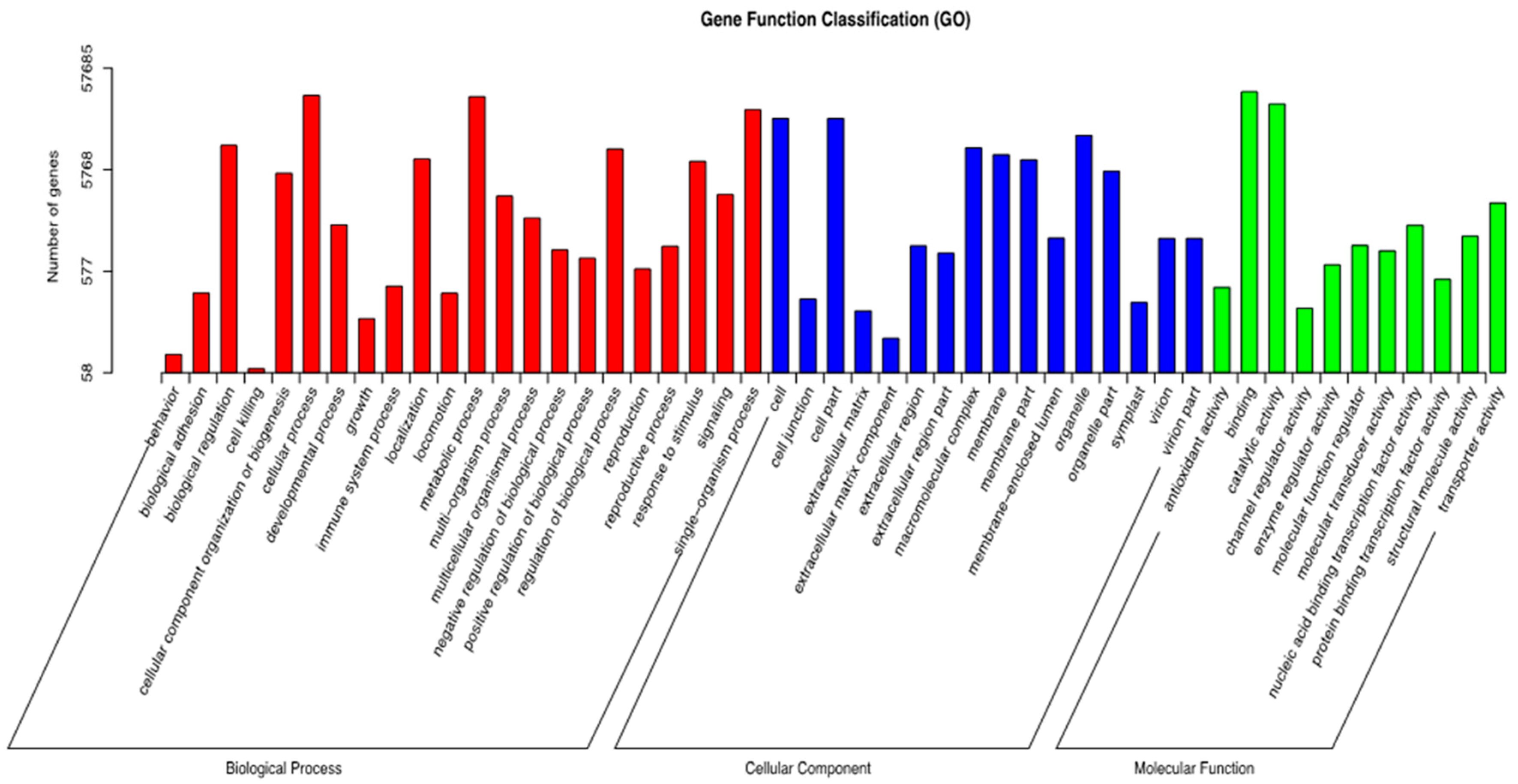
−6. Using gene IDs the corresponding GO terms in functional categories can be identified. GO classification associations, combined with statistically transcripts, are presented.
4.9. Quantitative Real-Time PCR Analysis
The differential expression levels of 12 unigenes under BDO induction of the ISR disease-resistance reaction were analyzed by real-time qRT-PCR. These genes were protective proteins, ethylene response factors and transcription factors in the ET signal pathway. Extraction of total RNA was performed using the TRIzol reagent (Qiagen). First-strand cDNA was synthesized from 500 ng of total RNA using a FastQuant RT Kit (Qiagen). The 1 µL of cDNA was used for qRT-PCR. The RT-PCR analysis was performed using SuperReal PreMix Plus (SYBR Green, Qiagen). The Actin gene was chosen as an internal standard, and the relative expression of each target gene was obtained using the 2
−ΔΔCT method. For each of four treatments, CKB24, B24, NB24 and B72, three independent biological replicates were performed. Each biological replicate included two technical replicates for the qRT-PCR analysis. The names of the unigenes and primer sequences are listed in
Supplementary Table S1.
4.10. Statistical Analysis
At least, three independent biological replicates were performed independently for transcriptomic experiments, which were at multiple time points. Three independent repetitions were performed for single time point experiments. For qRT-PCR, three independent biological replicates were performed and each biological replicate include two technical replicates. The statistical analysis was performed by using the Student’s t-test. A one-way analysis of variance (ANOVA) was performed on the experimental time point data set (p < 0.05).














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}