Computational Modeling of In Vitro Swelling of Mitochondria: A Biophysical Approach




Abstract
:1. Introduction
2. Model Description
2.1. A Basic Model of Mitochondrial Dynamics
2.2. Ca2+ Transport across the IMM
2.3. PTP Transport of Ionic and Neutral Species
2.4. Weak Acid Dissociation
- AHout ↔ A−out + H+out
- A−out ↔ A−in
- H+out ↔ H+in
- A−in + H+in ↔ AHin
2.5. Effect of Respiration (ETC Activity) on H+ Generation
2.6. Model Implementation
2.7. Mitochondrial Swelling Dynamics
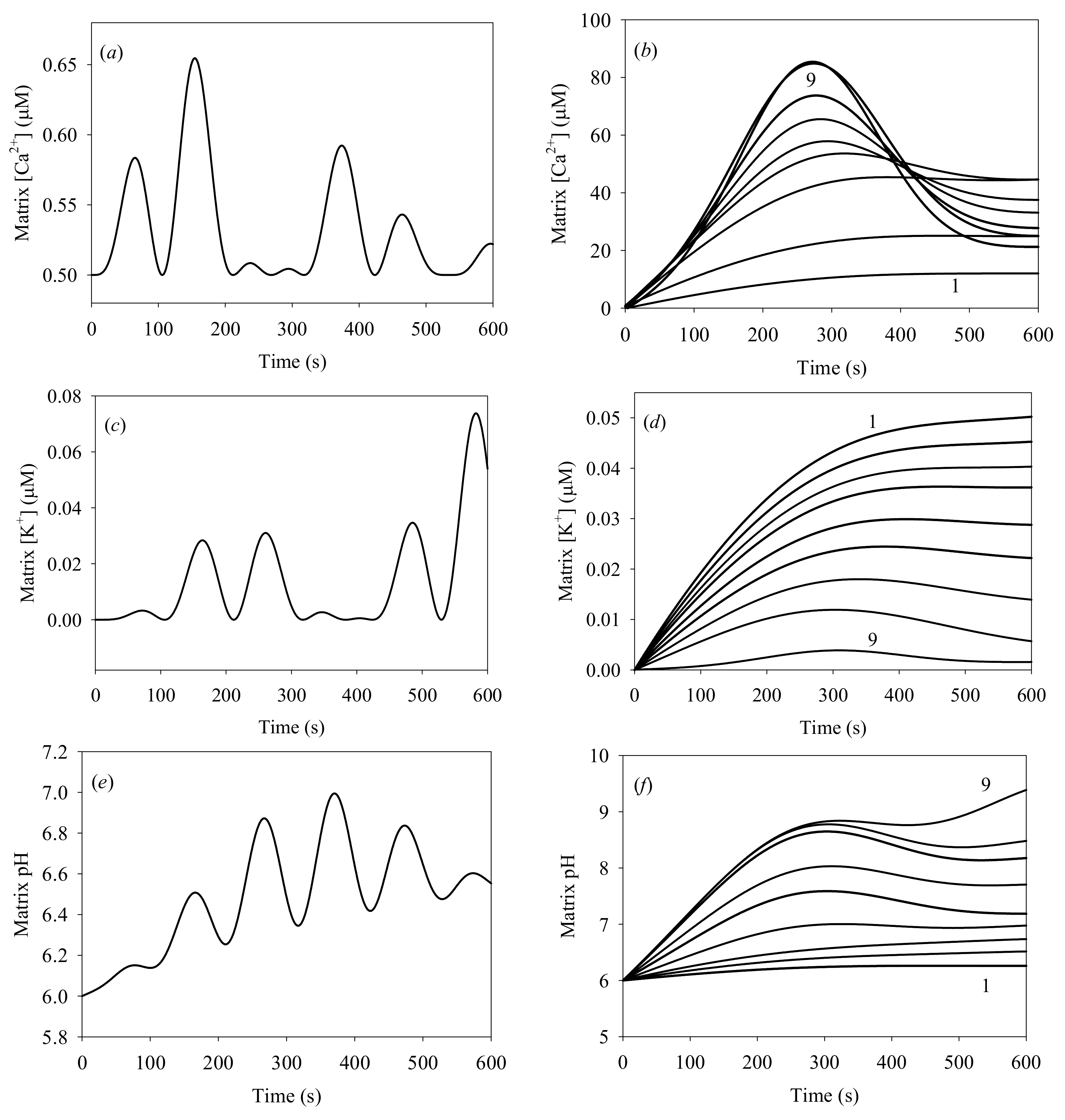
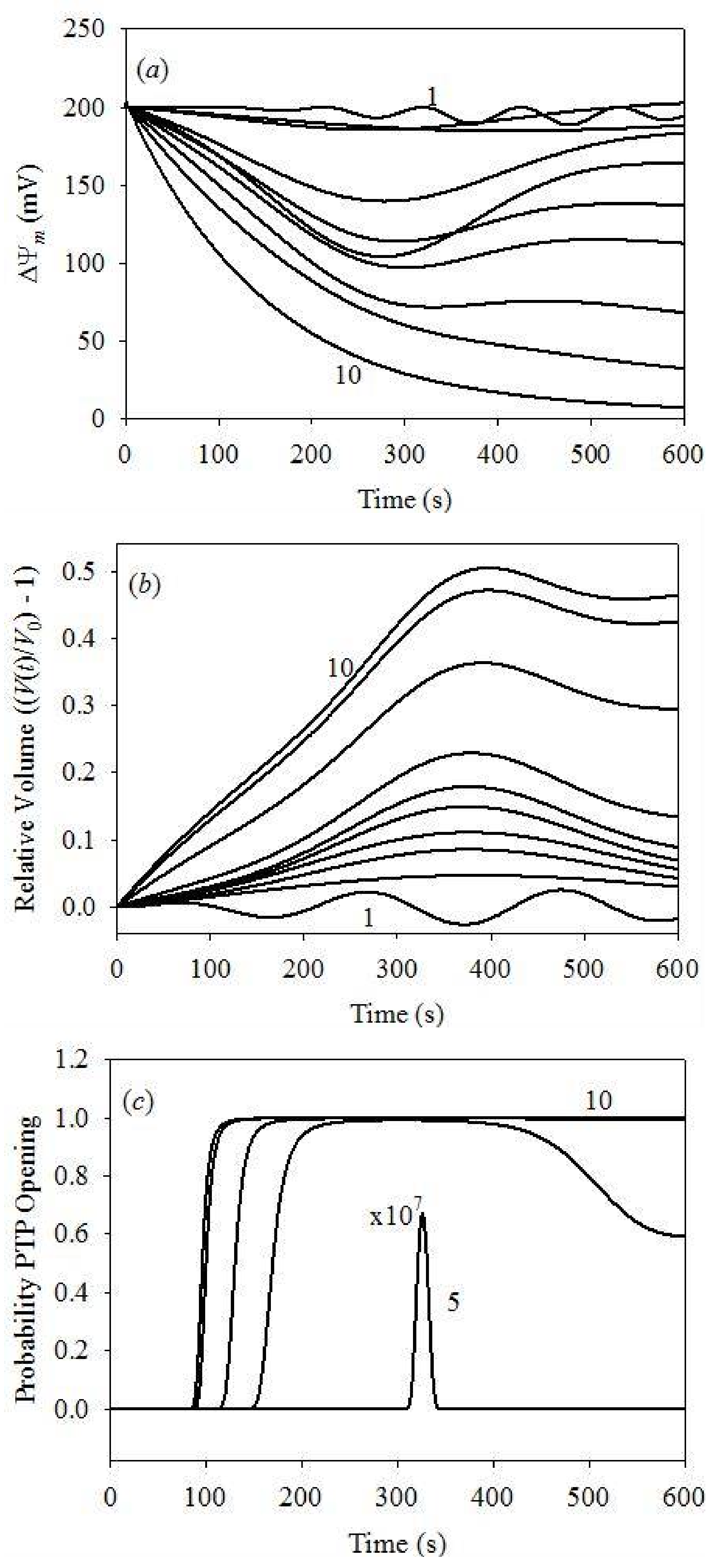
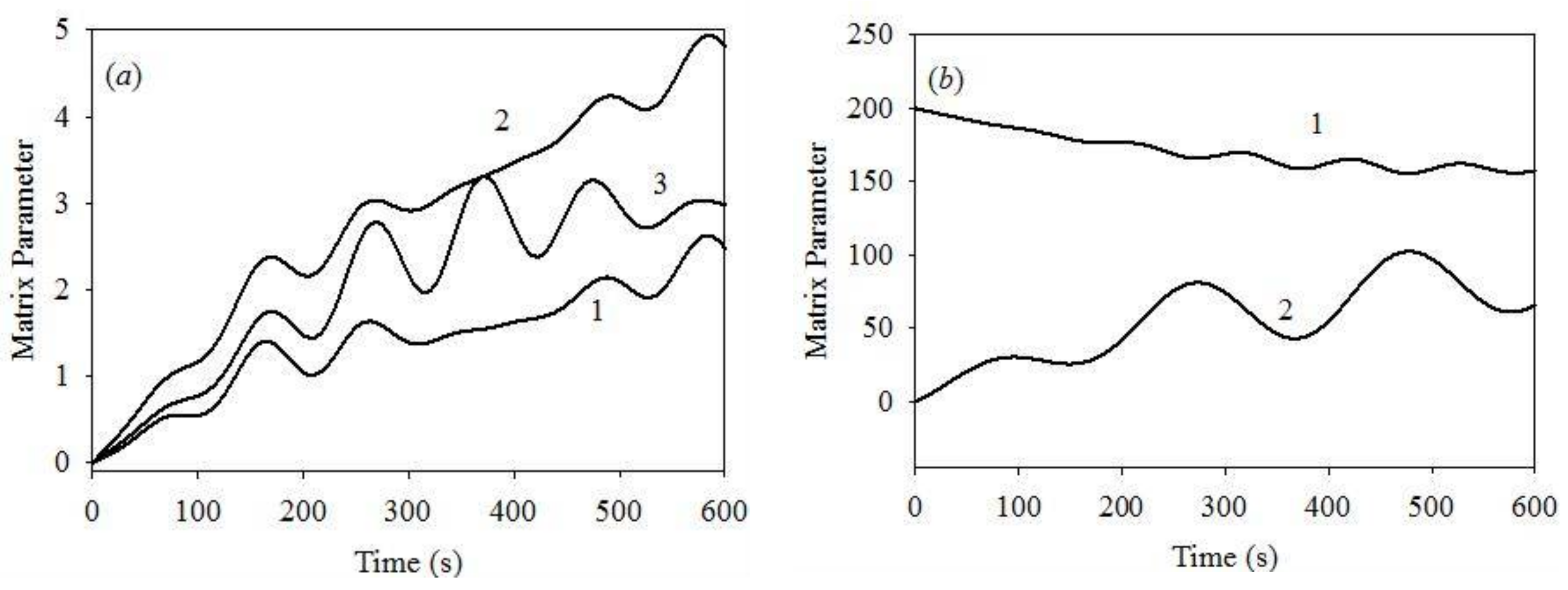
3. Numerical Experiments
4. Results and Discussion
5. Conclusions
6. Limitations of the Study
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B.; Ramza, B.M.; Marban, E. Oscillations of membrane current and excitability driven by metabolic oscillations in heart cells. Science 1994, 265, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Sagara, Y.; Liu, Y.; Maher, P.; Schubert, D. The regulation of reactive oxygen species production during programmed cell death. J. Cell Biol. 1998, 141, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Kerr, P.M.; Javadov, S.; Woodfield, K.Y. Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim. Biophys. Acta 1998, 1366, 79–94. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Parodi-Rullan, R.; Khuchua, Z.; Kuznetsov, A.V. Mitochondrial permeability transition in cardiac ischemia-reperfusion: Whether cyclophilin d is a viable target for cardioprotection? Cell. Mol. Life Sci. 2017, 74, 2795–2813. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the f1fo atp synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, V.; von Stockum, S.; Antoniel, M.; Fabbro, A.; Fogolari, F.; Forte, M.; Glick, G.D.; Petronilli, V.; Zoratti, M.; Szabo, I.; et al. Dimers of mitochondrial atp synthase form the permeability transition pore. Proc. Natl. Acad. Sci. USA 2013, 110, 5887–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the mitochondrial permeability transition in the absence of subunit c of human atp synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 3409–3414. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of atp synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 9086–9091. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Moulin, M. Physiological roles of the permeability transition pore. Circ. Res. 2012, 111, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.R.; Haworth, R.A.; Southard, J.H. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J. Biol. Chem. 1976, 251, 5069–5077. [Google Scholar] [PubMed]
- Ichas, F.; Mazat, J.P. From calcium signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta 1998, 1366, 33–50. [Google Scholar] [CrossRef]
- Ichas, F.; Jouaville, L.S.; Mazat, J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of atp production by mitochondrial Ca(2+). Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Calcium signaling in cardiac mitochondria. J. Mol. Cell. Cardiol. 2013, 58, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. Regulation of mitochondrial metabolism through changes in matrix volume. Biochem. Soc. Trans. 1994, 22, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef] [PubMed]
- Petit, P.X.; Goubern, M.; Diolez, P.; Susin, S.A.; Zamzami, N.; Kroemer, G. Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: The impact of irreversible permeability transition. FEBS Lett. 1998, 426, 111–116. [Google Scholar] [CrossRef]
- Saotome, M.; Katoh, H.; Satoh, H.; Nagasaka, S.; Yoshihara, S.; Terada, H.; Hayashi, H. Mitochondrial membrane potential modulates regulation of mitochondrial Ca2+ in rat ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1820–H1828. [Google Scholar] [CrossRef] [PubMed]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J. Mitochondrial calcium transport in the heart: Physiological and pathological roles. J. Mol. Cell. Cardiol. 2009, 46, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Bernardi, P.; Zoratti, M. Modulation of the mitochondrial megachannel by divalent cations and protons. J. Biol. Chem. 1992, 267, 2940–2946. [Google Scholar] [PubMed]
- Hoppe, U.C. Mitochondrial calcium channels. FEBS Lett. 2010, 584, 1975–1981. [Google Scholar] [CrossRef] [PubMed]
- Altschuld, R.A.; Hohl, C.M.; Castillo, L.C.; Garleb, A.A.; Starling, R.C.; Brierley, G.P. Cyclosporin inhibits mitochondrial calcium efflux in isolated adult rat ventricular cardiomyocytes. Am. J. Physiol. 1992, 262, H1699–H1704. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. Calcium-dependent opening of a non-specific pore in the mitochondrial inner membrane is inhibited at ph values below 7. Implications for the protective effect of low ph against chemical and hypoxic cell damage. Biochem. J. 1991, 278, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Nicolli, A.; Basso, E.; Petronilli, V.; Wenger, R.M.; Bernardi, P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, and cyclosporin a-sensitive channel. J. Biol. Chem. 1996, 271, 2185–2192. [Google Scholar] [CrossRef] [PubMed]
- Antoniel, M.; Jones, K.; Antonucci, S.; Spolaore, B.; Fogolari, F.; Petronilli, V.; Giorgio, V.; Carraro, M.; Di Lisa, F.; Forte, M.; et al. The unique histidine in oscp subunit of f-atp synthase mediates inhibition of the permeability transition pore by acidic ph. EMBO Rep. 2018, 19, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Kitakaze, M.; Takashima, S.; Funaya, H.; Minamino, T.; Node, K.; Shinozaki, Y.; Mori, H.; Hori, M. Temporary acidosis during reperfusion limits myocardial infarct size in dogs. Am. J. Physiol. 1997, 272, H2071–H2078. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Choi, A.; Rajapurohitam, V.; Zeidan, A.; Basnakian, A.G.; Karmazyn, M. Nhe-1 inhibition-induced cardioprotection against ischaemia/reperfusion is associated with attenuation of the mitochondrial permeability transition. Cardiovasc. Res. 2008, 77, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Zanin, C.; Vayssiere, J.L.; Petit, P.X.; Kroemer, G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med. 1995, 181, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Lartigue, L.; Kushnareva, Y.; Seong, Y.; Lin, H.; Faustin, B.; Newmeyer, D.D. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol. Biol. Cell 2009, 20, 4871–4884. [Google Scholar] [CrossRef] [PubMed]
- Barreto-Torres, G.; Hernandez, J.S.; Jang, S.; Rodriguez-Munoz, A.R.; Torres-Ramos, C.A.; Basnakian, A.G.; Javadov, S. The beneficial effects of amp kinase activation against oxidative stress are associated with prevention of pparalpha-cyclophilin d interaction in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H749–H758. [Google Scholar] [CrossRef] [PubMed]
- Chapa-Dubocq, X.; Makarov, V.; Javadov, S. Simple kinetic model of mitochondrial swelling in cardiac cells. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Selivanov, V.A.; Ichas, F.; Holmuhamedov, E.L.; Jouaville, L.S.; Evtodienko, Y.V.; Mazat, J.P. A model of mitochondrial Ca2+-induced Ca2+ release simulating the Ca2+ oscillations and spikes generated by mitochondria. Biophys. Chem. 1998, 72, 111–121. [Google Scholar] [CrossRef]
- Javadov, S.; Chapa-Dubocq, X.; Makarov, V. Different approaches to modeling analysis of mitochondrial swelling. Mitochondrion 2018, 38, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L.; Kashiwaya, Y.; King, M.T. The resting membrane potential of cells are measures of electrical work, not of ionic currents. Integr. Physiol. Behav. Sci. 1995, 30, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Massari, S. Kinetic analysis of the mitochondrial permeability transition. J. Biol. Chem. 1996, 271, 31942–31948. [Google Scholar]
- Baranov, S.V.; Stavrovskaya, I.G.; Brown, A.M.; Tyryshkin, A.M.; Kristal, B.S. Kinetic model for Ca2+-induced permeability transition in energized liver mitochondria discriminates between inhibitor mechanisms. J. Biol. Chem. 2008, 283, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Bazil, J.N.; Buzzard, G.T.; Rundell, A.E. Modeling mitochondrial bioenergetics with integrated volume dynamics. PLoS Comput. Biol. 2010, 6, e1000632. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.K.; Qi, F.; Beard, D.A. A biophysically based mathematical model for the kinetics of mitochondrial calcium uniporter. Biophys. J. 2009, 96, 1318–1332. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, S.; Tookos, F.; Hense, B.A.; Schulz, S.; Filbir, F.; Zischka, H. A mathematical model of mitochondrial swelling. BMC Res. Notes 2010, 3, 67. [Google Scholar] [CrossRef] [PubMed]
- Pokhilko, A.V.; Ataullakhanov, F.I.; Holmuhamedov, E.L. Mathematical model of mitochondrial ionic homeostasis: Three modes of Ca2+ transport. J. Theor. Biol. 2006, 243, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Barr, L. Membrane potential profiles and the goldman equation. J. Theor. Biol. 1965, 9, 351–356. [Google Scholar] [CrossRef]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochim. Biophys. Acta 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Bernardi, P.; Petronilli, V. The permeability transition pore as a mitochondrial calcium release channel: A critical appraisal. J. Bioenerg. Biomembr. 1996, 28, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.V.; Kudzina, L.; Zinchenko, V.P.; Evtodienko, Y.V. Chlortetracycline-mediated continuous Ca2+ oscillations in mitochondria of digitonin-treated tetrahymena pyriformis. Eur. J. Biochem. 1985, 153, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Fuoss, R.M.; Kraus, C.A. Properties of electrolytic solutions. XV. Thermodynamic properties of very weak electrolytes. J. Am. Chem. Soc. 1935, 57, 1–4. [Google Scholar] [CrossRef]
- Naraghi, M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium 1997, 22, 255–268. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Mitochondrial oscillations in physiology and pathophysiology. Adv. Exp. Med. Biol. 2008, 641, 98–117. [Google Scholar] [PubMed]
- Kaasik, A.; Safiulina, D.; Zharkovsky, A.; Veksler, V. Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 2007, 292, C157–C163. [Google Scholar] [CrossRef] [PubMed]
- Yugi, K.; Tomita, M. A general computational model of mitochondrial metabolism in a whole organelle scale. Bioinformatics 2004, 20, 1795–1796. [Google Scholar] [CrossRef]
- Bešter-Rogač, M.; Habe, D. Modern advances in electrical conductivity measurements of solutions. Acta Chim. Slov. 2006, 53, 391–395. [Google Scholar]
- Macey, R.I. Transport of water and urea in red blood cells. Am. J. Physiol. 1984, 246, C195–C203. [Google Scholar] [CrossRef] [PubMed]
- Gerencser, A.A.; Doczi, J.; Torocsik, B.; Bossy-Wetzel, E.; Adam-Vizi, V. Mitochondrial swelling measurement in situ by optimized spatial filtering: Astrocyte-neuron differences. Biophys. J. 2008, 95, 2583–2598. [Google Scholar] [CrossRef] [PubMed]
- Atkins, P.; de Paulo, J. Physical Chemistry, 7th ed.; W.H. Freeman and Company: New York, NY, USA, 2002; pp. 841–853. ISBN 0-7167-3539-3. [Google Scholar]
- Glaser, R. Biophysics; An Introduction; Springer: Berlin/Heidelberg, Germany, 2012; pp. 171–183. ISBN1 13978-3642252112. ISBN2 103642252117. [Google Scholar]
- Song, D.H.; Park, J.; Maurer, L.L.; Lu, W.; Philbert, M.A.; Sastry, A.M. Biophysical significance of the inner mitochondrial membrane structure on the electrochemical potential of mitochondria. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2013, 88, 062723. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Javadov, S.A.; Das, M.; Clarke, S.J.; Suleiman, M.S.; Halestrap, A.P. The effects of ischaemic preconditioning, diazoxide and 5-hydroxydecanoate on rat heart mitochondrial volume and respiration. J. Physiol. 2002, 545, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Santo-Domingo, J.; Demaurex, N. Calcium uptake mechanisms of mitochondria. Biochim. Biophys. Acta 2010, 1797, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.E.; Rottenberg, H. Membrane potential and surface potential in mitochondria. Fluorescence and binding of 1-anilinonaphthalene-8-sulfonate. J. Biol. Chem. 1983, 258, 11039–11048. [Google Scholar] [PubMed]
- De Marchi, U.; Szabo, I.; Cereghetti, G.M.; Hoxha, P.; Craigen, W.J.; Zoratti, M. A maxi-chloride channel in the inner membrane of mammalian mitochondria. Biochim. Biophys. Acta 2008, 1777, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |
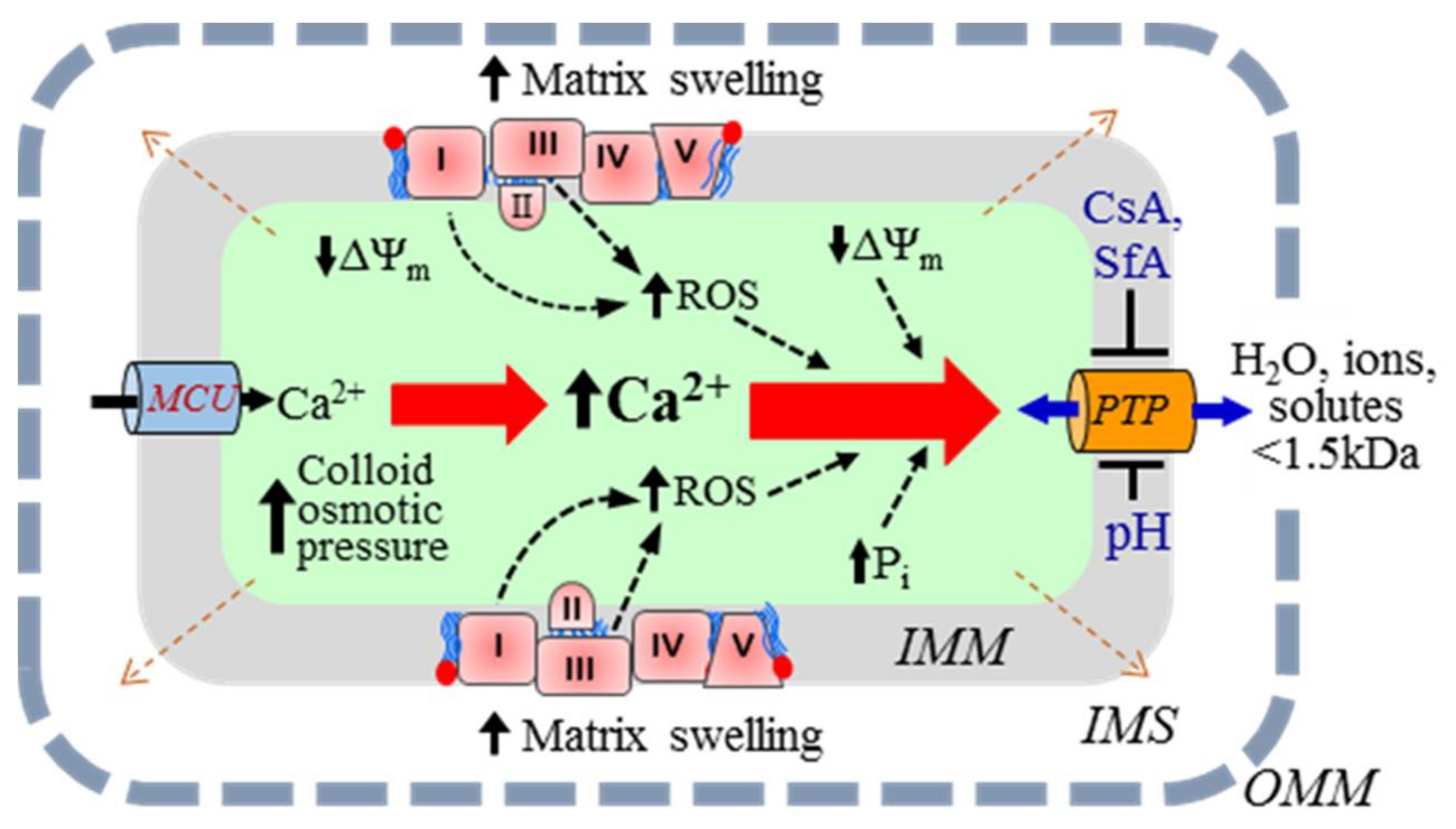






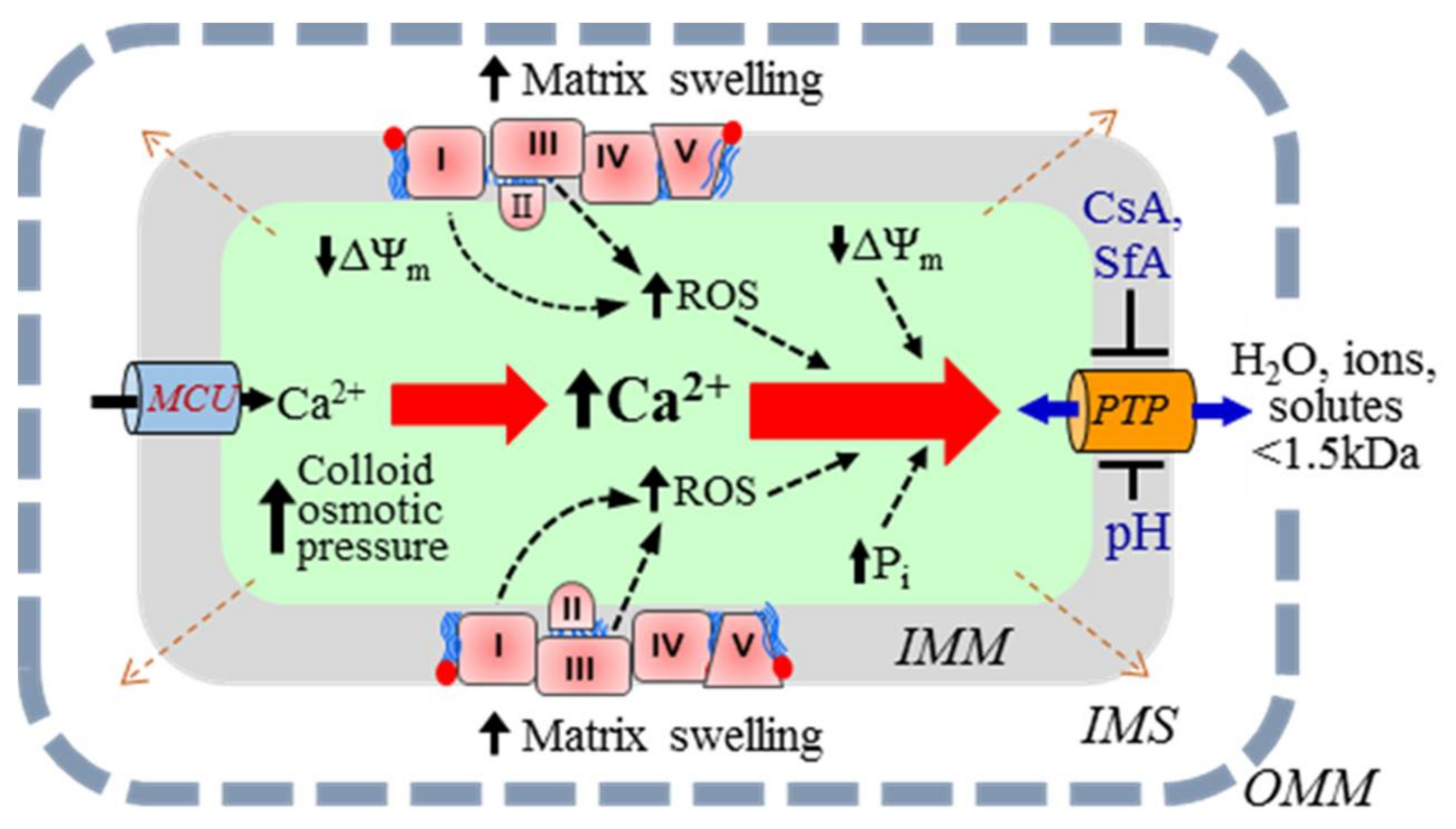{kind=link}
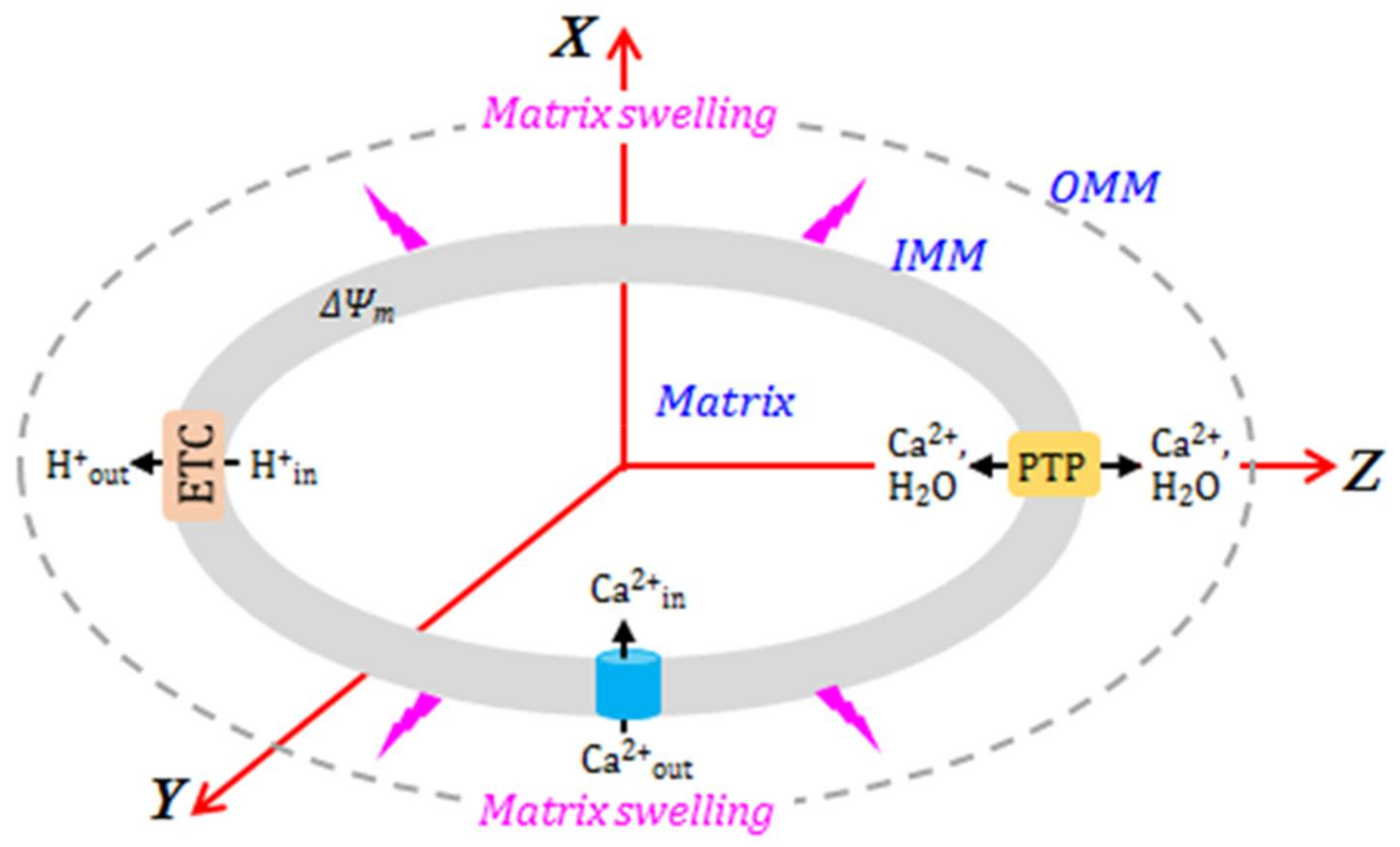{kind=link}
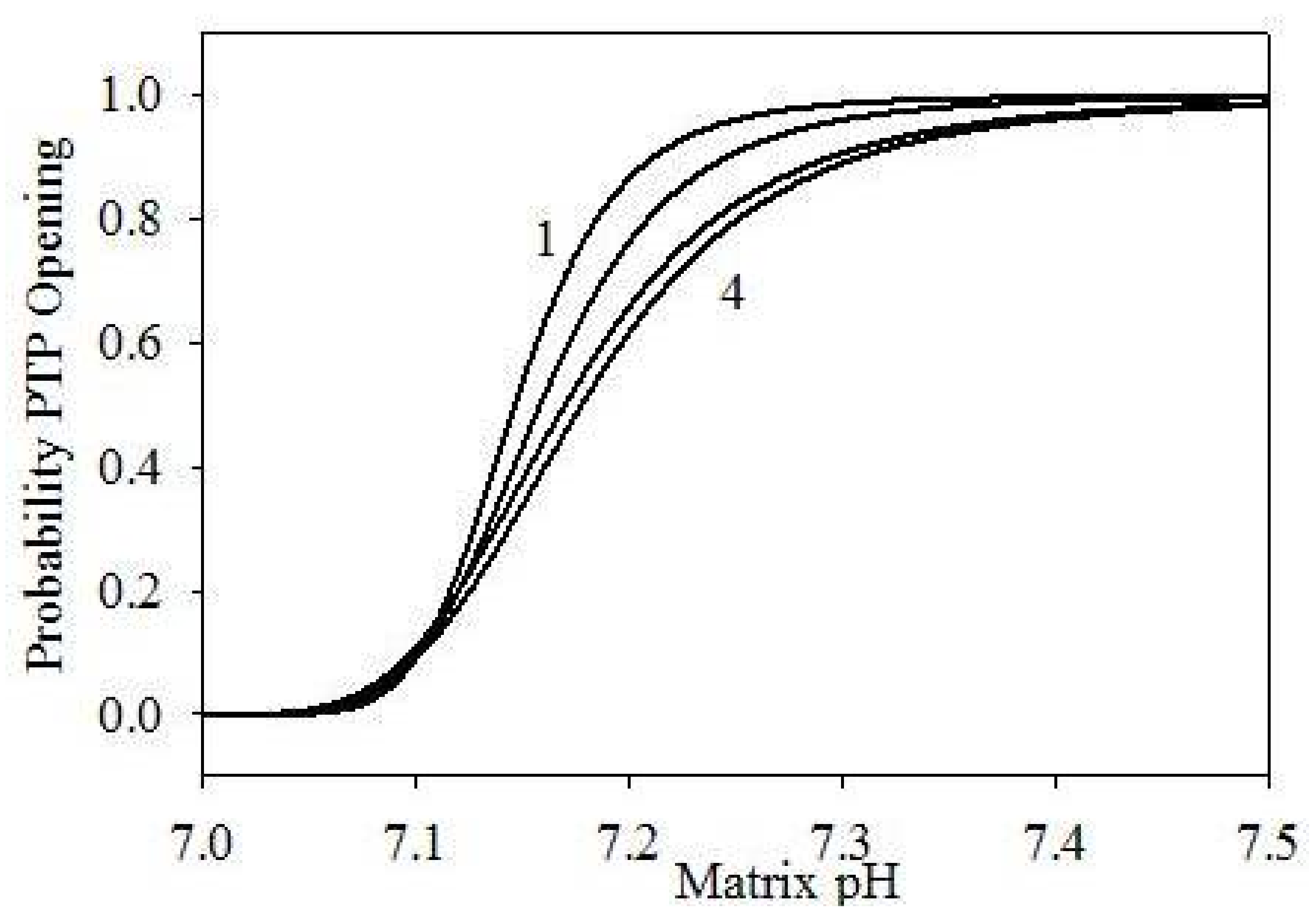{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Definition | Value | Refs |
|---|---|---|---|
| pCa,UP | Reduced permeability coefficient of the Ca2+ transport by uniporter | 6.43 × 105, min−1mV−1 (mg protein)−1 (K × esu/erg) | [18,48,49,50] |
| pK,H(0) | Reduced permeability coefficient of the K+/H+ exchange | 3.20 × 105, min−1mV−1 (mg protein)−1 (K × esu/erg) (μMH)−1 | [47] |
| PH,K(0) | Reduced permeability coefficient of the H+/K+ exchange | 3.20 × 105, min−1mV−1(mg protein)−1 (K × esu/erg) (mMK)−1 | [47] |
| pH+,PTP | Reduced permeability coefficient of the H+ transport by PTP | 4.30 × 107, min−1mV−1 (mg protein)−1 (K × esu/erg) | [47] |
| pCa2+,PTP | Reduced permeability coefficient of the Ca2+ transport by PTP | 8.61 × 107, min−1mV−1 (mg protein)−1 (K × esu/erg) | [48] |
| PK+,PTP | Reduced permeability coefficient of the K+ transport by PTP | 4.30 × 107, min−1mV−1 (mg protein)−1 (K × esu/erg) | [51] |
| pA-,PTP | Reduced permeability coefficient of the A− transport by PTP | 6.43 × 102, min−1mV−1 (mg protein)−1 (K × esu/erg) | [48] |
| pA,PTP | Reduced permeability coefficient of the A transport by PTP | 32.1, min−1 (mg protein)−1 | [48] |
| |e| | Absolute value of the electron charge | 4.80286 × 10−10 esu | [52] |
| kB | Boltzmann constant | 1.38044 × 10−16 erg/K | [52] |
| ΔΨm | IMM potential | 200 mV | [44,47,48] |
| VC/Vm(t = 0) | Ratio of adult rat heart cell volume to total volume of all cell mitochondria | 2.86 | [49] |
| α | 1.2 × 103 | |||||||||
| n | 4 | |||||||||
| ; μM | 0.10 | 1.2 | 2.3 | 3.4 | 4.5 | 5.6 | 6.7 | 7.8 | 8.9 | 10 |
| ; μM | 1.0 | 25 | 50 | 75 | 100 | 150 | 200 | 300 | 400 | 500 |
| −log() | 7.00 | |||||||||
| −log() | 6.00 | |||||||||
| ; μM | 50 | |||||||||
| δ(ΔΨm); mV | 43.42 | |||||||||
| ; μM | 10 | |||||||||
| kA; min−1 | 1 | |||||||||
| kA,H+; min−1 | 1 | |||||||||
| g00; dyn/nm | 0.017 | |||||||||
| gzz,0; dyn/nm | 0.019 | |||||||||
| β0; nm−n1 | 106 | |||||||||
| βz; nm−n1 | 107 | |||||||||
| n1 | 4 | |||||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makarov, V.I.; Khmelinskii, I.; Javadov, S. Computational Modeling of In Vitro Swelling of Mitochondria: A Biophysical Approach. Molecules 2018, 23, 783. https://doi.org/10.3390/molecules23040783
Makarov VI, Khmelinskii I, Javadov S. Computational Modeling of In Vitro Swelling of Mitochondria: A Biophysical Approach. Molecules. 2018; 23(4):783. https://doi.org/10.3390/molecules23040783
Chicago/Turabian StyleMakarov, Vladimir I., Igor Khmelinskii, and Sabzali Javadov. 2018. "Computational Modeling of In Vitro Swelling of Mitochondria: A Biophysical Approach" Molecules 23, no. 4: 783. https://doi.org/10.3390/molecules23040783
APA StyleMakarov, V. I., Khmelinskii, I., & Javadov, S. (2018). Computational Modeling of In Vitro Swelling of Mitochondria: A Biophysical Approach. Molecules, 23(4), 783. https://doi.org/10.3390/molecules23040783